

Abstract
E2F plays critical roles in cell cycle progression by regulating the expression of genes involved in nucleotide synthesis, DNA replication, and cell cycle control. We show that the combined loss of E2F1 and E2F2 in mice leads to profound cell-autonomous defects in the hematopoietic development of multiple cell lineages. E2F2 mutant mice show erythroid maturation defects that are comparable with those observed in patients with megaloblastic anemia. Importantly, hematopoietic defects observed in E2F1/E2F2 double-knockout (DKO) mice appear to result from impeded S phase progression in hematopoietic progenitor cells. During DKO B-cell maturation, differentiation beyond the large pre-BII-cell stage is defective, presumably due to failed cell cycle exit, and the cells undergo apoptosis. However, apoptosis appears to be the consequence of failed maturation, not the cause. Despite the accumulation of hematopoietic progenitor cells in S phase, the combined loss of E2F1 and E2F2 results in significantly decreased expression and activities of several E2F target genes including cyclin A2. Our results indicate specific roles for E2F1 and E2F2 in the induction of E2F target genes, which contribute to efficient expansion and maturation of hematopoietic progenitor cells. Thus, E2F1 and E2F2 play essential and redundant roles in the proper coordination of cell cycle progression with differentiation which is necessary for efficient hematopoiesis.
E2F transcriptional activity is composed of a variety of heterodimers formed by the association of one of at least six E2F family members (E2F1 to E2F6) with one of at least three DP proteins (14). The E2F family can be divided into three groups based on structure and transcriptional activity. E2F1, E2F2, and E2F3 are structurally similar, potent transcriptional activators. Overexpression of each of these E2F proteins is sufficient to drive quiescent cells to reenter the cell cycle. E2F4 and E2F5 are thought to primarily function in the active repression of E2F target genes in quiescent cells by recruiting pRB family members. Rb association with E2F not only blocks transcriptional activation by E2F but also forms an active transcriptional repressor complex at promoters that can block transcription by recruiting histone deacetylase and remodeling chromatin, which is reversed following cyclin-dependent kinase (Cdk) phosphorylation of Rb (20). In a class by itself, E2F6 functions as a transcriptional repressor through a mechanism independent of pRB family members (37, 51).
Genetic model systems have provided valuable insight into physiological roles for E2F proteins (11). In Drosophila melanogaster, mutation of the dE2F1 gene results in reduced numbers of cells entering S phase and reduced activation of E2F target genes (13, 45). dE2F2 appears to function in opposition to dE2F1, repressing E2F-dependent transcription (6, 16). Targeted E2F-knockout mice have been created to identify distinct roles for E2F family members in mouse development and physiology. Surprisingly, the disruption of E2F1 in the mouse results in the genesis of a diverse range of tumors in older adults (57). E2F1 also plays an important role in the elimination of self-reactive immature T cells during thymic negative selection and activation-induced death of mature T cells (15, 17, 34, 58). E2F2 mutant mice frequently develop autoimmunity and tumors (36, 59). Unexpectedly, E2F1 and E2F2 function redundantly to limit T-cell proliferation, as E2F1−/− E2F2−/− T cells exhibit a more rapid entry into S phase and proliferate much more extensively in response to subthreshold antigenic stimulation (59). E2F4−/− mice are stunted, demonstrate developmental craniofacial defects, and display defects in late-stage erythroid maturation (21, 41). E2F5−/− mice develop nonobstructive hydrocephalus as newborns (32). While E2F4−/− E2F5−/− mouse embryo fibroblasts (MEFs) show normal serum starvation-induced cell cycle arrest and proliferation kinetics, these MEFs are resistant to G1 arrest induced by the Cdk4-Cdk6 inhibitor p16INK4A or the inhibition of Ras activity (18).
E2F3 deficiency is partially embryonic lethal, and adult survivors die prematurely of congestive heart failure (9). G1-to-S phase progression and the induction of multiple target genes are reduced in E2F3−/− MEFs (22). The combined mutation of E2F3 with E2F2, E2F1, or both results in progressive reductions in cell cycle reentry, such that E2F1−/− E2F2−/− E2F3−/− MEFs do not detectably reenter the cell cycle, exhibit reduced induction of E2F target genes, and fail to proliferate, arresting in all stages of the cell cycle (55). While these results suggest that E2F1, E2F2, and E2F3 are redundantly required for S phase entry, the failure of these cells to enter S phase could be the result of the persistence of E2F-dependent repressors, such as Rb/E2F4. Although critical roles for E2F1, E2F2, and E2F3 in promoting the ectopic S phase entry and apoptosis resulting from Rb inactivation in mouse embryos have been demonstrated previously (46, 52, 60), the challenge will be to understand how the functions of different E2F proteins in the control of cell cycling and cell fate, as well as the specific regulation of transcription by individual E2F proteins, relate to normal development and function in mammals.
Mature blood cell types all derive from common hematopoietic stem cells (HSC) residing in the bone marrow (BM), with progressive differentiation and commitment to more restrictive lineage choices (53). The continuous production of blood cells from HSC is required throughout life as terminally differentiated blood cells have limited half-lives that may vary from hours to years. Coordination between proliferative expansion and differentiation of BM precursor cells is essential at all stages of hematopoiesis. B-cell ontogeny in the BM proceeds through alternating phases of proliferation and quiescence. Proper immunoglobulin (Ig) chain rearrangement results in proliferative expansion, followed by cell cycle exit to allow for additional rearrangements and differentiation (43). Thus, efficient hematopoiesis requires both proper cell cycling for expansion and appropriate cell cycle exit for differentiation.
In this study, we show that E2F1 and E2F2 redundantly regulate cell cycle progression during hematopoiesis. The combined loss of E2F1 and E2F2 results in decreased expression of a subset of E2F target genes and defective S phase progression in hematopoietic progenitors in mouse BM, leading to severely defective hematopoiesis. Thus, proper E2F-dependent control of the cell cycle is required for efficient hematopoiesis.
MATERIALS AND METHODS
Mice.
Mice were housed in the University of Colorado Health Sciences Center animal resource center in cages with microisolator lids. Sterile cage setups, food, and water were provided for mice postirradiation. The generation of the E2F1- and E2F2-knockout mice has been previously described (15, 31), and these mice were the kind gifts of S. Field, S. Orkin, and M. Greenberg. Green fluorescent protein (GFP) transgenic (Tg) mice were the generous gift of Brian Schaefer. Bcl2 Tg mice and BALB/c recipient mice were purchased from Jackson Labs. The Bcl2 Tg, GFP Tg, and E2F1/E2F2 mutant mice used for these studies were backcrossed three or four times into BALB/c mice (originally in the C57BL/6 background). Mice were genotyped by PCR analysis of DNA extracted from a small tail biopsy sample. The University of Colorado Health Sciences Center Animal Care and Use Committee approved all mouse experiments.
BM transplantation and cell culture.
BM cells isolated from double-knockout (DKO) mice or control mice were transferred into recipient mice by tail vein or subocular injection. Recipient mice were irradiated twice with 450 rads at 4- to 5-h intervals with a 60Co irradiator.
BM cells were cultured in 10% fetal bovine serum (FBS; HyClone) in Iscove's modified Dulbecco's medium (IMDM; Gibco BRL) ± 4% interleukin-7 (IL-7)-conditioned medium as previously described (4). Apoptosis was detected by staining cells with fluorescein isothiocyanate (FITC)-conjugated annexin V per the protocol supplied by Molecular Probes.
Flow cytometry and cell cycle analysis.
Single-cell suspensions were washed in phosphate-buffered saline (PBS) containing 5% FBS (FBS-PBS). A number of cells (106) were stained in 30 μl of antibody solution (1:100 dilution of each antibody) for 30 min on ice. Cells were washed once with 1 ml of FBS-PBS and resuspended in 400 μl of PBS. The following PharMingen antibodies were used: phycoerythrin (PE)-linked anti-CD4, Cy-Chrome-anti-CD8, allophycocyanin-linked anti-B220, PE-anti-GR-1, PE-anti-Mac-1, allophycocyanin-anti-Ter119, and PE-anti-CD43. The FITC-linked B7.6 (anti-IgM) monoclonal antibody was the kind gift of R. Benschop and J. Cambier. Fluorescence was detected with a Coulter Epics XL (Beckman Coulter) or FACSCalibur (Becton Dickinson) cytometer. Automated hematocrit analysis of peripheral tail vein blood was performed with a Bayer Advia120 hematology system.
Mice were injected intraperitoneally with bromodeoxyuridine (BrdU; 1 mg/25 g of body weight; Boehringer Mannheim) and then sacrificed after 2 h. BM cells were isolated in 10% FBS-PBS. Cells were fixed in 70% ethanol at 4°C for at least 1 h. BrdU incorporation was detected by using FITC-linked anti-BrdU (PharMingen) according to the manufacturer's protocols (HCl denaturation method). After washing, cells were resuspended in 500 μl of 10-μg/ml propidium iodide (PI; Roche; in 1× PBS) for flow cytometric analysis. The length of S phase was calculated by previously described methods (2). Briefly, changes in PI intensity of S phase cells (relative movement [RM]) during the labeling time relative to the PI positions of G1 and G2 are calculated as follows: RM = (FL − FG1)/(FG2/M − FG1), where FL is the mean red fluorescence (PI) of BrdU-positive (BrdUPos) cells and F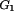 and F
and F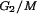 are the mean PI fluorescence values for G1 and G2/M cells, respectively. The formula for calculating Ts (DNA synthesis time) is as follows: Ts = [0.5/(RM − 0.5)] × t, where t is the sampling time (2-h in vivo labeling in our experiments).
are the mean PI fluorescence values for G1 and G2/M cells, respectively. The formula for calculating Ts (DNA synthesis time) is as follows: Ts = [0.5/(RM − 0.5)] × t, where t is the sampling time (2-h in vivo labeling in our experiments).
RPA.
RNA samples were prepared from cells by using Trizol reagent (Gibco BRL) according to the manufacturer's instructions. Levels of cell cycle regulator mRNAs were measured by using the RiboQuant multiprobe RNase protection assay (RPA) system with a custom template set.
Kinase assays and Western blotting.
Kinase assays were performed as previously described (12). Ter119+ BM cells were purified by magnetically activated cell sorting (Miltenyi; per the manufacturer's instructions) and resuspended in kinase lysis buffer, and 100 μg of cell lysate was used for each immunoprecipitation. Protein A/G beads and antibodies (anti-cyclin A2 [CycA2] sc-751, anti-CycB1 sc-245, and anti-CycE1 sc-481) were purchased from Santa Cruz Biotechnology. Histone H1 was purchased from Boehringer Mannheim. Western blotting was performed per Pierce instructions except that 0.2% Tween 20 was included in the antibody solutions and washes. Antibodies used were diluted 1:500 including anti-Rb (PharMingen, 14001A, and Santa Cruz, sc-102, IF-8), anti-CycA2 (sc-751, H432), anti-CycE1 (sc-481, M20), anti-E2F3 (sc-878, C-18), and antiactin (sc-1616, I19). Secondary antibodies used were anti-mouse-horseradish peroxidase or anti-rabbit-horseradish peroxidase (1:2,000; Bio-Rad Laboratories), and blots were developed by using Pierce SuperSignal Femto chemiluminescence detection.
RESULTS
The combined loss of E2F1 and E2F2 results in decreased hematopoietic cellularity.
It was previously reported that the loss of E2F1 and E2F2 results in increased proliferative capacity of peripheral T cells in response to antigen stimulation (59). Surprisingly, E2F1−/− E2F2−/− (DKO) adult mice and in some cases E2F2 mutant mice, demonstrated marked reductions in the cellularity of all examined hematopoietic compartments including BM, thymus, lymph nodes, and spleen (Fig. 1A), as well as significant reductions in the numbers of red blood cells (RBC), lymphocytes, and monocytes in the peripheral blood (Fig. 1B). Mouse genotypes in this paper are designated as E2F1 genotype/E2F2 genotype, with genotypes indicated as wild type (Wt), heterozygous (Ht), and mutant (Mt). For example, Ht/Mt mice are heterozygous for the E2F1 mutation and homozygous mutant for E2F2. E2F1+/− E2F2+/− (Ht/Ht) mice exhibit relatively normal hematopoiesis and are used as littermate (LM) controls in our experiments. RBC deficiencies are primarily attributable to E2F2 loss, as the combined loss of one or both E2F1 alleles does not result in further reductions in RBC numbers, revealing a specific function for E2F2 in RBC maturation. In fact, the loss of E2F1 alone slightly increases the cellularity of the spleen, lymph nodes, and thymus, consistent with a previous report (15). However, when combined with the E2F2 null mutation, the loss of either one or both E2F1 alleles further decreases lymphocyte cellularity in the peripheral blood (to about 60 and 30% of Wt/Mt values for Ht/Mt and Mt/Mt, respectively [Fig. 1B]). Thus, while the loss of either E2F1 or E2F2 alone generally results in opposite effects on hematopoietic cellularity, the combined disruption of these E2F proteins reveals their redundant roles in hematopoiesis.
FIG. 1.

Hematopoietic deficiencies in E2F1 and E2F2 mutant mice. (A) E2F1/E2F2 mutant mice exhibit reduced cellularity of all hematopoietic compartments. Sets of LMs from 6- to 12-week-old E2F1/E2F2 mutant mice were sacrificed, and the cellularity of the indicated tissues was determined by cell counting with a hemocytometer. BM cells were isolated from the femurs and tibias of both hind legs. The calculated cellularities are graphed ± standard errors (note that the error indicated in all figures represents standard error). A plus sign indicates heterozygosity for the E2F allele, and a minus sign indicates a null mutant. Statistical comparisons are with Ht/Ht mice, unless indicated otherwise. The number of mice analyzed is indicated above each bar. *, P < 0.05; **, P < 0.01; and ***, P < 0.001 (all figures). (B) Loss of E2F1 and E2F2 reduces the numbers of RBC, lymphocytes, and monocytes in the peripheral blood from adult mice (5 to 14 weeks). Peripheral blood was isolated from tail veins of sets of E2F1/E2F2 mutant LMs, and cellularities were determined by automated hematocrit analysis (average cell counts are shown below each genotype). The significance of decreased cellularities relative to Wt/Wt mice, unless otherwise indicated, is shown. The number of mice of each genotype analyzed is indicated above the RBC graph.
E2F2 mutant and DKO mice exhibit erythropoietic defects similar to those of humans with megaloblastic anemia.
In red BM pulp, the maturation and differentiation of committed erythroid precursors are tightly coupled with DNA synthesis and cell division. As erythroid cells mature, their cell size decreases and their nuclei become increasingly condensed (Fig. 2A). Eventually RBC precursor cells in the BM discard their nuclei and mature into enucleated cells called reticulocytes, which normally stay in the BM for a few days before they migrate to peripheral blood and become RBC. In the peripheral blood of E2F2 mutant and DKO mice, significant decreases in reticulocyte cell numbers were observed (Fig. 2B). However the ratio of reticulocytes to mature RBC was not significantly different from that for control mice (data not shown). These results indicate that the decreased RBC count in peripheral blood is due to decreased reticulocyte production from BM erythroid precursors and is not due to increased turnover rates or other hemolytic types of anemia that would result in increased percentages of reticulocytes relative to RBC. While E2F1 mutant mice have no obvious defects in RBC maturation, average RBC volumes in E2F2 mutant and DKO mice are substantially increased relative to normal RBC (Fig. 2C and D). In fact, E2F2 and DKO RBC show a much wider range of cell sizes than do Wt mice, with many cells exhibiting substantially larger volumes (Fig. 2C). Interestingly, the anemia, RBC maturation defects, and large RBC size that we observe for E2F2 mutant and DKO mice are similar to those observed for humans with megaloblastic anemia, which is caused by DNA synthesis defects due to nutritional or genetic deficiencies (including dihydrofolate reductase mutation), resulting in insufficient nucleotide levels (28). These patients exhibit reduced maturation of multiple hematopoietic lineages with a prevalence of large, early-stage precursor cells in the BM. Increased percentages of cells in S phase are also observed, together with evidence of the induction of a DNA damage response (27). RBC precursors and other hematopoietic progenitors in DKO mice also exhibit increased percentages of cells in S phase (59).
FIG. 2.

Erythroid defects in E2F2 mutant and DKO mice are similar to those in the human disease megaloblastic anemia. (A) Differentiation pathway of committed erythroid precursor cells in the BM. (B) E2F2 Mt and DKO mice exhibit significantly decreased reticulocyte cell numbers in peripheral blood. (C and D) RBC size is substantially increased in E2F2 Mt and DKO mice, as observed both by light microscopy (C) and by automated hematocrit analysis (D). Reticulocyte numbers and RBC size were determined by automated hematocrit analysis of peripheral blood from E2F1/E2F2 mutants and LM controls between 5 and 9 weeks of age.
DKO BM is defective in the reconstitution of hematopoiesis in irradiated recipient mice.
Many factors such as hormones and cytokines influence hematopoietic efficiency, and these factors are derived from both hematopoietic and nonhematopoietic cells. BM transplantation (BMT) experiments can determine whether hematopoietic defects observed for DKO mice are autonomous to the hematopoietic system by examining the capacity of DKO BM to reconstitute hematopoiesis in lethally irradiated recipient mice that provide an otherwise normal environment. Wt or DKO BM cells were transferred into lethally irradiated BALB/c recipient mice. At 8 weeks posttransplantation, the analysis of peripheral blood from recipients revealed abnormally low RBC, white blood cell (WBC), and lymphocyte counts in recipients of DKO mouse BM (Fig. 3A). Similarly decreased blood cell counts were observed for long-term reconstituted recipient mice more than 4 months posttransfer (data not shown). Secondary transplantation with BM isolated from primary recipient mice showed similar hematopoietic deficiencies in recipients of BM from primary DKO-transferred mice (data not shown). Thus, defective hematopoiesis in DKO mice is due to the loss of E2F2 and E2F1 in BM cells and is therefore autonomous to the hematopoietic system. Notably, DKO BM cells provide long-term reconstitution of hematopoiesis to the extent that lethally irradiated recipient mice survived for at least 15 months posttransfer, similar to recipients of Wt mouse BM (data not shown). Thus, while long-term HSC functions do not appear to be severely defective in DKO mice, recipients of DKO BM have hematopoietic defects that mirror the phenotypes seen for the original DKO mice.
FIG. 3.

DKO hematopoietic defects are severe and cell autonomous. (A) BM cells (2 × 106) isolated from Wt or E2F1/E2F2 DKO mice were transferred into lethally irradiated BALB/c mice (5 to 8 weeks old). Blood samples (each point represents data from an individual recipient mouse) were isolated from recipient mice 8 weeks post-BMT. WBCs represent all nucleated cells in the peripheral blood. Similar results were obtained in three other BMT experiments. (B) Competitive BMTs were performed with BM cells from GFP Tg mice as Wt competitors. BM mixtures (0:1, GFP only, or 4:1, E2F1/E2F2 to GFP; 2 × 106 total cells) were transferred into BALB/c mice. Recipients were sacrificed 8 weeks post-BMT, and the expression of GFP and lineage markers was determined in all hematopoietic compartments by flow cytometry. Representative results from the BM are shown in the top panels, and examples of results from the thymus, spleen, and peripheral blood are shown in the bottom panels. Lineage markers include GR-1 (myeloid lineage), B220 (B-cell lineage), Ter119 (erythroid lineage), CD4 and CD8 (immature thymocytes), and Mac-1 (granulocytes, monocytes, and macrophages). Similar results were observed for three individual recipient mice for each group.
DKO BM precursors compete poorly with Wt precursors during hematopoiesis.
In order to estimate the production efficiency of DKO hematopoietic precursor cells, we measured the ability of DKO BM cells to proliferate and differentiate in competition with Wt BM cells. As a highly quantitative approach, we performed competitive BMT experiments using GFP Tg mouse BM cells as Wt competitors. The GFP transgene is expressed from the ubiquitin C promoter, and GFP appears to be expressed in all cells (47). We bred the GFP transgene into the BALB/c background (major histocompatibility complex H-2d/d verified), in order to avoid graft-versus-graft and graft-versus-host immune reactions. BM from Ht/Ht or DKO mice mixed with GFP Tg competitor cells (0:1 or 4:1 ratio of E2F1/E2F2 to GFP) was transferred into lethally irradiated BALB/c recipients. Recipients were sacrificed 8 weeks posttransplantation, and hematopoietic cells were analyzed for the expression of GFP and lineage markers. The percentages of GFP+ versus GFP− cells of each lineage represent the relative contribution of GFP Wt BM cells versus DKO or Ht/Ht BM cells. As expected, almost all cells in the BM, thymus, spleen, and peripheral blood of recipients of GFP BM cells (0:1; “GFP only”) are GFP+, regardless of hematopoietic lineage (>97.8% [Fig. 3B]). At 4:1 ratios of Ht/Ht to GFP BM, 13 to 65% of hematopoietic cells of various lineages and stages of development were derived from GFP BM, indicating that Ht/Ht BM effectively competed with GFP BM in contributing to various blood lineages. It is unclear whether deviations from the expected 80:20 ratio for 4:1 Ht/Ht to GFP recipients reveal negative effects of heterozygosity for E2F1 and E2F2 on hematopoiesis or non-E2F genetic differences from GFP Tg mice. Importantly, even at a 4:1 ratio, DKO BM cells fail to compete with GFP BM cells to reconstitute hematopoiesis in recipient mice in any lineage (Fig. 3B). For example, B-cell lineage cells (B220+) in the BM of these recipients are 98% GFP+, which is similar to the 98.8% contribution of GFP+ cells to B220+ BM cells seen when no DKO competitor BM is transplanted. Similarly, DKO BM cells fail to substantially contribute to the myeloid (GR-1+ and Mac-1+), RBC (Ter119+), and T-cell (represented by CD4+ CD8+ thymocytes) lineages when in competition with Wt (GFP+) BM, even though four times as much DKO BM was transplanted. A small amount (less than 3%) of contribution of DKO BM cells to some lineages may occur (Fig. 3B). We also analyzed the contribution of GFP+ cells to various specific stages of B-cell and T-cell maturation in the BM and thymus, respectively, and DKO BM cells failed to compete at every stage (data not shown).
We performed similar competitive BMT experiments to determine the relative ability of BM cells from Ht/Mt and Mt/Ht mice to compete with Wt GFP+ BM during hematopoiesis. Interestingly, even the disruption of three of four E2F1/E2F2 alleles did not substantially impede hematopoiesis, as Ht/Mt and Mt/Ht cells competed similarly to Ht/Ht BM cells in reconstituting hematopoiesis (Fig. 4A). E2F2 deficiency does appear to partially affect the reconstitution of B220+ cells in the BM, but not mature B cells in the spleen. Again, DKO hematopoietic cells failed to compete with GFP Tg cells, as a 4:1 ratio of DKO to GFP BM resulted in virtually all GFP+ cells in all hematopoietic lineages, similar to the transplantation of GFP Tg BM alone. Clearly, DKO precursor cells are more compromised than even Mt/Ht or Ht/Mt cells in reconstituting hematopoiesis, providing physiological evidence for functional redundancy between E2F1 and E2F2. Given that E2F2 mutant mice (Wt/Mt or Ht/Mt) show erythroid defects that are similar to defects in DKO mice, we were surprised that Ht/Mt BM competed so effectively against Wt (GFP Tg) BM, particularly in the Ter119+ RBC lineage. As also presented above in Fig. 2, Ht/Mt and DKO mice display anemia and megaloblastosis (increased RBC volume) in peripheral blood (Fig. 4B, top row). As expected, cotransplantation of GFP Tg BM with DKO BM restored normal RBC size and improved RBC numbers (Fig. 4B, bottom right), which is not surprising given that hematopoiesis is effectively all Wt (GFP) in these mice. Surprisingly, cotransplantation of GFP Tg BM with Ht/Mt BM did not significantly affect either anemia or megaloblastosis. We currently do not understand why Ht/Mt hematopoietic cells have what appears to be a dominant-negative effect on erythropoiesis, preventing Wt progenitors from filling the erythropoietic void.
FIG. 4.

Competitive BMTs show essential and redundant roles of E2F1 and E2F2 in hematopoiesis. (A) Competitive BMTs with BM cells from donor mice with indicated E2F1/E2F2 genotypes were performed as described for Fig. 3B. BM from two or three donors for each genotype was separately transplanted in competition with GFP Tg BM into at least two BALB/c recipients per donor. The average percentages of GFP+ cells in the myeloid (GR-1+ and Mac-1+), lymphoid (B220+), and erythroid (Ter119+) lineages from BM, spleen, and blood are shown. (B) RBC size and hemoglobin content were determined by automated hematocrit analysis with blood samples from mice of the indicated E2F1/E2F2 genotypes (top panels) or from recipient mice that received E2F1/E2F2 mutant and GFP BM (4:1; bottom panels). Representative plots are shown. RBC numbers (106/μl) and average cell size (fL) (averages of multiple experiments) are indicated in the upper left of each panel.
These results indicate that hematopoietic deficiencies in DKO mice are cell autonomous, since the Wt hematopoietic cells in the competitive reconstitution should provide any necessary hematopoietic cell-derived transacting factors. Notably, RBC cellularity is reduced in DKO mice to only about 70% of controls (Fig. 1B), indicating that the severe defects in RBC maturation that are evident in competitive experiments (see Ter119+ cells in Fig. 3B) are largely compensated for by homeostatic regulatory mechanisms in the original DKO mice. Thus, competitive transplantation experiments, by providing an alternative source of progenitor cells, reveal the full extent of developmental defects that are otherwise masked in the original gene-deficient mice by homeostatic mechanisms. We speculate that anemia in DKO mice signals increased erythropoietin production, which increases erythropoiesis, albeit at a low production efficiency. In summary, these data reveal severe pan-hematopoietic defects due to the cell-autonomous loss of E2F1 and E2F2.
S phase progression is defective in E2F2 KO and DKO BM cells.
Considering the megaloblastic anemia phenotype in E2F2 mutant mice and the critical roles for E2F in cell cycle regulation and DNA synthesis, we explored the mechanism underlying hematopoietic phenotypes, focusing on potential roles for E2F2 (and, redundantly, E2F1) in regulating S phase progression. Indeed, our cell cycle analysis reveals a striking requirement for E2F1 and E2F2 (particularly E2F2) in S phase progression in hematopoietic progenitors. DKO mice and LM controls were injected with the thymidine analog BrdU. Two hours postinjection, BM cells were isolated and immunostained to detect BrdU incorporation into DNA. The PI staining DNA profile for DKO BM cells revealed a significant increase (ca. twofold) in the percentage of cells with S phase DNA content (Fig. 5A and B). For Wt and Ht/Ht mice, most BM cells with S phase DNA content were also BrdUPos, as expected, indicating that DNA synthesis was in progress. Surprisingly, a substantial fraction of DKO BM cells possessed S phase DNA content but did not incorporate BrdU. Ht/Mt mice also exhibited increased percentages of BrdU-negative (BrdUNeg) BM cells with S phase DNA content, while Mt/Ht mice did not (Fig. 5B), correlating with hematopoietic phenotypes observed in mice of these E2F1/E2F2 genotypes (Fig. 1). These results indicate that substantial numbers of hematopoietic precursor cells in S phase of the cell cycle from DKO and Ht/Mt BM are not actively replicating their DNA over prolonged periods of time. The failure of DKO BM cells in S phase to incorporate BrdU was not due to the unavailability of BrdU, as these BrdUNeg S phase cells were still present following culture of DKO BM cells ex vivo in the presence of 10 μM BrdU for up to 6 h (data not shown).
FIG. 5.

S phase is stalled and prolonged in DKO hematopoietic progenitors. (A) Many DKO and Ht/Mt BM cells with S phase DNA content fail to incorporate BrdU. Mice of the indicated genotypes were injected with BrdU, and BM was harvested after 2 h. BrdU incorporation, together with PI staining to determine DNA content, was measured by flow cytometry. Representative flow cytometric profiles are shown with an arrow indicating BrdUNeg S phase cells. (B) Quantitation of multiple experiments performed as for panel A. PI staining intensity in both BrdUPos and BrdUNeg cells was measured in order to determine the fractions of cells with S phase DNA content that incorporated BrdU (% S, BrdUPos) or failed to incorporate sufficient BrdU for detection (% S, BrdUNeg). Both the increased percentage of cells in S phase and the increased percentage of BrdUNeg S phase cells in DKO BM are statistically significant relative to Wt and Ht/Ht controls. (C) DNA synthesis time in DKO BM cells is substantially prolonged. DNA synthesis time is calculated as described in Materials and Methods. (D and E) DKO BM cells in S phase progress poorly to G2/M phase. Mice of the indicated E2F1/E2F2 genotypes were injected with BrdU. BM cells were harvested 2 h later and either immediately fixed in 70% ethanol (t = 0) or cultured with BrdU (10 μM) for 16 h in the presence of nocodazole (0.25 ng/ml; +Noc 16 h). BrdU incorporation and PI staining were determined by flow cytometry. Cells gated as BrdUPos were then analyzed for PI staining intensity. Accumulation of BrdUPos cells with 4N DNA content is used to determine S phase progression to G2. Representative flow cytometric profiles of the PI intensity of BrdUPos cells and quantitation of multiple experiments are shown in panels D and E, respectively. DKO BM cells show significant decreases in the fraction of BrdUPos cells that accumulate with G2/M DNA (+Noc) relative to Wt and Ht/Ht BM.
We next asked whether the DKO BM cells that did incorporate BrdU displayed altered S phase progression. In vivo BrdU labeling together with the intensity of PI staining for DNA content can be used to calculate the length of S phase in cycling BM cells by the methods and calculations described by Begg et al. (2). Following injection of BrdU (a pulse, given the short in vivo half-life), BrdU-labeled cells chase through S phase and into G2/M. DNA synthesis of pulse-labeled BrdUPos BM cells can be tracked by the increase in PI intensity during the period between BrdU injection and BM cell isolation. While our calculations indicated an average DNA synthesis time of 9.9 h for Ht/Ht BM, we calculated an average DNA synthesis time for DKO BM cells of 26 h (Fig. 5C). Previous calculations for the length of S phase for Wt mouse BM cells estimated a length of 5 to 8 h (2). Thus, even DKO BM cells that incorporate sufficient BrdU to allow for immunodetection progress through S phase at a very low rate. In summary, E2F2 mutant and DKO progenitor cells accumulate in S phase, with some cells showing no significant movement through S phase and the remainder progressing at about one-third the normal rate.
As an additional measure of the rate of S phase progression, BM cells labeled for 2 h in vivo were harvested and cultured in vitro for 16 h in the presence of nocodazole, a microtubule depolymerizing drug that causes cell cycle arrest in metaphase of mitosis. We restricted our analysis to BM cells that had incorporated BrdU in order to eliminate noncycling cells. While BrdUPos Wt, Ht/Ht, and E2F1-null (Mt/Ht) BM cells accumulated in M phase, BrdUPos E2F2-null (Ht/Mt) and DKO BM cells exhibited greatly reduced accumulation in M phase, indicating arrested or substantially delayed progression through S phase (Fig. 5D and E). Notably, many BrdUPos DKO BM cells accumulate with DNA content that is close to G1, indicating cell cycle arrest in early S phase (Fig. 5D; see also BrdUNeg population in Fig. 5A). Thus, our calculations may actually underestimate the fraction of DKO BM cells in S phase, as cells stalled in very early S phase have an essentially G1 content of DNA. In summary, by various criteria, E2F2 mutation, and to a greater extent the combined mutation of E2F2 and E2F1, results in substantial defects in S phase progression in hematopoietic progenitor cells.
B-cell deficiencies in E2F1/E2F2 mutant mice appear to result from reduced maturation from progenitors.
B-cell maturation is intimately linked to the cell cycle (3, 43). The earliest B cells are characterized by expression of B220, c-Kit, and CD43, with either germ line Ig genes or rearranged DJ heavy (H) chains (pro-B and pre-BI [Fig. 6A ]). Proper H chain rearrangement (V to DJ) in pre-BI cells results in pre-B-cell receptor (pre-BCR) expression, which signals proliferation and differentiation to large pre-BII cells. Proliferation results in dilution of the surrogate light (L) chain, leading to cell cycle exit (now small pre-BII), L chain rearrangement, and, with appropriate expression of surface IgM, differentiation to immature B cells (43). Cell cycle exit appears necessary for Rag-mediated recombination, as Rag activity is antagonized by Cdk activity (30). Nonself-reactive immature B cells then migrate to peripheral lymphoid tissues wherein they become mature B cells. From pro-B to pre-BII cells, the failure to properly rearrange Ig chains to allow for the expression of functional pre-BCR or BCR prevents proliferation and leads to apoptosis.
FIG. 6.

(A) Cells committed to the B-cell lineage express B220 at all stages of development. BM cells from 5- to 9-week-old sets of LMs of the indicated E2F1/E2F2 genotypes were harvested and stained with fluorescently labeled anti-B220, anti-CD43, and anti-IgM. Cells were analyzed by flow cytometry. The relative numbers of B220+ CD43+ IgM−, B220+ CD43− IgM−, and B220+ CD43− IgM+ cells in Mt/Ht, Ht/Mt, and DKO mice relative to Ht/Ht control LMs in five independent experiments are shown. For Wt and Ht/Ht mice, these three B-cell stages are usually present in BM at a ratio of about 1:2:2. The diagram of B-cell development incorporates information from reference 3. (B) Mixtures (1:1) of GFP Tg and DKO (or Wt/Ht LM control) BM cells (4 × 106 cells/ml) were cocultured with IMDM-10% FCS and 4% IL-7 for 2 or 7 days. The fraction of B220+ cells expressing GFP was determined by flow cytometry. While DKO and GFP Tg B220+ cells were present at similar numbers following 2 days of culture, GFP Tg pre-B cells predominate after 7 days of culture. In contrast, Wt/Ht pre-B cells expand similarly to GFP Tg cells in cultures.
It was previously reported that DKO mice exhibit defective B-cell maturation (59), and we have extended this analysis and included statistical evaluation (Fig. 6A). While the cellularity of the more immature progenitors (pro-BI and pre-BI) was not significantly reduced, at later stages of B-cell development substantial reductions in cellularity were evident in DKO BM and, to a lesser extent, in Ht/Mt mice. Mt/Ht mice did not show decreased B-cell maturation. The maturation block in DKO mice is most evident during the large-pre-BII-to-small-pre-BII transition. We consistently observed a significant fraction of large DKO (and E2F2 mutant) pre-B cells, while such larger cells are a smaller percentage of Ht/Ht and Wt pre-B cells (Fig. 7A). In fact, when pre-BII cells (B220+ CD43− IgM−) were gated for cell size, we observed a substantial reduction in the percentage of small pre-BII cells, but not large pre-BII cells, in Ht/Mt and to a greater extent DKO mice (Fig. 7B). Cell sorting and PI staining demonstrated that large pre-BII cells are predominantly in S phase (data not shown). Similarly, larger cell sizes among myeloid and B220+ populations were also microscopically evident in DKO BM (Fig. 8). Given that reduced cellularity is observed in DKO BM at the pre-BII large- to small-cell transition, these data suggest that S phase delay at the pre-BII stage prevents cell cycle exit to the small pre-BII stage, which is necessary for IgL rearrangement and differentiation.
FIG. 7.

The combined loss of E2F2 and E2F1 results in substantial inhibition of B-cell development at the pre-BII-cell stage. (A) DKO BM cells exhibit increased percentages of large B-cell precursors. BM cells from Wt and DKO mice were analyzed by flow cytometry for the expression of B220 and forward scatter (an indicator of cell size). Pre/pro-B- and B-cell populations, distinguishable by the level of B220 expression, are indicated. (B) Preferential reductions of small pre-BII cells in DKO mice. The percentages of large CD43− pre-BII cells versus small CD43− pre-BII cells (relative to total B220+ cells) in BM from mice of the indicated E2F1/E2F2 genotypes are graphed. The percentage of small pre-BII cells is significantly decreased in DKO BM versus Ht/Wt and Ht/Ht BM (combined and labeled “CON”). (C) BM cells (including stroma) were cultured with IMDM-10% FCS and 4% IL-7 for 7 days, washed with PBS, and then replated in IMDM-10% FCS without IL-7 for an additional 24 h. Apoptosis in B-lineage cells was determined by flow cytometric analysis of B220 expression together with the determination of DNA content with PI. Cells with sub-G1 DNA content are considered apoptotic. (D) Cultures both before (+IL-7) or 24 h after (−IL-7) IL-7 withdrawal were stained with annexin V-FITC and PI. Annexin V+ PI− cells are considered early apoptotic cells. Annexin V+ PI+ cells are either late apoptotic or necrotic.
FIG. 8.

Purified B220+ and Mac-1 BM cells (by magnetically activated cell sorting) from LM Ht/Ht and DKO mice were immobilized on slides by cytospinning and stained (May-Grunwald method). Microscopic images of DKO BM cells show increased cell size in both lineages.
B-cell maturation defects in DKO mice can be recapitulated in vitro. IL-7 promotes the proliferation and limits the differentiation of pre-BI/II cells in vitro. Withdrawal of IL-7 results in eventual cell cycle exit, Rag-mediated IgL chain rearrangement, and differentiation to IgM+ immature B cells (42, 44). We cultured BM from mice of different E2F1/E2F2 genotypes with IL-7. By coculture of either Ht/Ht or DKO BM cells with GFP Tg BM cells, decreased cell-autonomous expansion of DKO pre-B cells was evident (Fig. 6B). We did not observe any differences in apoptosis dependent on E2F1/E2F2 in the IL-7-supplemented cultures (Fig. 7D, upper panels). However, upon withdrawal of IL-7 for 1 day, increased apoptosis was observed in DKO pre-B cultures, but not in Wt cultures (Fig. 7C and D). IL-7 withdrawal resulted in the efficient differentiation of Wt pre-B cells to IgM+ immature B cells (data not shown). Notably, the few surviving DKO B220+ cells in the cultures (−IL-7) were also IgM+ (data not shown), suggesting that differentiation per se was not blocked but that the fraction of pre-B cells with the capacity to differentiate was severely reduced. The reduced production of IgM+ immature B cells in DKO cultures is similar to what we observe in vivo. We hypothesize that S phase arrest-delay in DKO pre-B cells is incompatible with Rag-mediated recombination and differentiation, resulting in apoptosis in the absence of IL-7. Apoptosis could result either from the failure to express BCRs, which provide survival signals, or from differentiation signals that are in conflict with E2F deregulation.
Bcl2 expression fails to rescue lymphopoiesis defects in DKO mice.
We addressed whether increased apoptosis during B-cell differentiation is the cause of reduced maturation or whether S phase defects, even without apoptosis, are the primary causes. We bred the Eμ-Bcl2 WEHI 36 Tg line, which expresses the antiapoptotic Bcl2 protein throughout the development of both the B and T lineages (50), into E2F1/E2F2 mutant mice. Tg Bcl2 expression substantially decreases some but not all apoptosis that normally occurs during B- and T-cell maturation (25), which together with reduced turnover results in the accumulation of three- to fivefold-increased numbers of T and B cells. Indeed, we observed a more-than-twofold increase in peripheral blood lymphocyte numbers in Wt/Wt, Ht/Ht, and Mt/Ht Bcl2 Tg mice (Fig. 9A). All of our Ht/Mt Bcl2 Tg mice died within a few weeks of birth (for unknown reasons), preventing an analysis of their peripheral blood. Strikingly, the expression of the Bcl2 Tg in DKO lymphoid lineage cells did not improve lymphopoiesis, as mature lymphocyte cellularity was indistinguishable between DKO and Bcl2 Tg/DKO mice (Fig. 9A). Importantly, Bcl2 expression did effectively prevent the increased pre-B-cell apoptosis resulting from IL-7 withdrawal in DKO cultures (Fig. 9B). In summary, Bcl2 expression facilitates the survival but not the differentiation of DKO pre-B cells. Therefore, we believe that apoptosis is the consequence, rather than the cause, of decreased B-cell maturation. Both pre-B-cell expansion and cell cycle exit to allow for maturation appear to be incompatible with S phase stalling, even if apoptosis is blocked.
FIG. 9.

Transgenic Bcl2 expression does not rescue lymphopoiesis in DKO mice. (A) Peripheral blood from Wt and Ht/Ht (data combined), Mt/Ht, or DKO mice, transgenic for Eμ-Bcl2 or not, was analyzed by automated hematocrit for lymphocyte numbers. (B) Transgenic Bcl2 expression prevents the apoptosis of DKO pre-B cells in response to culture without IL-7. BM cells from Bcl2 Tg Ht/Wt and DKO LMs were cultured, and apoptosis was determined as for Fig. 7D. (C) DKO BM cells with and those without transgenic Bcl2 expression show similar BrdU incorporation defects. Freshly isolated BM cells of the indicated E2F1/E2F2 and Bcl2 genotypes were cultured in vitro in the presence of 10 μM BrdU for 2 h. BrdU incorporation, together with PI staining to determine DNA content, was measured by flow cytometry. Representative flow cytometric plots are shown. Numbers represent the averages of BrdUPos cells (gate B) and BrdUNeg S/G2 phase cells (gate A) from two and seven experiments with Bcl2 Tg and non-Tg sets of LMs, respectively.
Consistent with previous studies that showed Bcl2-mediated antagonism of G1-to-S phase progression (38), Bcl2 expression decreased the percentages of Ht/Ht BM cells incorporating BrdU ex vivo (Fig. 9C, left panels). However, Bcl2 expression did not significantly decrease BrdU incorporation in DKO BM cells, suggesting that E2F1/E2F2 loss positively affects G1-to-S phase progression in the BM. A similar S phase arrest-delay was observed in both DKO and Bcl2 Tg/DKO BM cells, as indicated by the accumulation of BrdUNeg cells with S/G2 DNA content (Fig. 9C, right panels). As shown above, the percentage of BM cells that incorporate BrdU in vivo was only modestly increased by E2F1/E2F2 mutation (Fig. 5A and B). Still, we consistently observed significantly greater percentages of DKO BM cells incorporating BrdU during ex vivo culture (Fig. 9C), consistent with our previous observations (59). Given that BrdU has an in vivo half-life of less than 30 min (40), BrdU should be primarily incorporated into cells already in S phase in vivo. In contrast, during labeling (2 h) in vitro, BrdU incorporation also labels cells that enter S from G1 phase, which may explain increased BrdU incorporation in DKO BM cells in vitro (particularly evident in comparing Bcl2 Tg cells). Given the previous observations that the loss of E2F1 and E2F2 accelerates G1-to-S phase progression (59), DKO BM cells appear to progress efficiently from G1 into S phase, but then many cells appear to become stalled in S phase.
Expression of specific E2F transcriptional target genes is reduced in DKO BM cells.
Given established roles for CycA2 in the cell cycle (23), we examined whether the regulation of this E2F target gene is dependent on E2F1 and E2F2 in BM. By RPA, CycA2 mRNA levels were consistently reduced in the BM of DKO mice relative to Ht/Ht and Wt controls (Fig. 10B), despite a ca. twofold-increased percentage of DKO cells in S phase (Fig. 5B). We observe similarly reduced CycA2 mRNA levels in the thymus and in purified B-lineage, erythroid, and myeloid cells from the BM (Fig. 10A and data not shown). Since even purified B220+ cells are heterogeneous with respect to maturation stage, CycA2 expression may be dependent on E2F1/E2F2 at some stages but not others. We observe a similar decrease in CycA2 protein levels in DKO and Ht/Mt BM, while E2F3 protein levels are unaffected by E2F1/E2F2 loss (Fig. 10C). Statistically significant reductions in the expression of Cdc2 mRNA levels are also observed in DKO BM (Fig. 10B). Interestingly, the expression of several other E2F target genes such as CycE1 and Cdk2 is not significantly changed in DKO BM (Fig. 10B to D and data not shown). Notably, in two separate oligonucleotide array experiments combining Ter119+ BM cells from more than eight DKO and eight Ht/Ht mice, the expression of dihydrofolate reductase was not significantly different between the two genotypes (data not shown), in contrast to significant reductions in CycA2 (average, 1.6-fold) and Cdc2 (average, 2.0-fold). Furthermore, the expression levels and phosphorylation state of the Rb protein are not affected by E2F1/E2F2 loss (Fig. 10D). Note that, in Ter119+ BM cells, three forms of the Rb protein are evident, with a prominent form of intermediate migration that probably corresponds to partially hyperphosphorylated Rb. The upper form comigrates with hyperphosphorylated Rb observed in activated lymphocytes. Regardless, the modest variability in the amount of hyperphosphorylated Rb between E2F1/E2F2 genotypes shown in this figure is not consistently observed in other experiments. In summary, E2F1 and E2F2 are required for the regulation of some E2F target genes, but not others, in BM cells.
FIG. 10.

Reduced expression and activities of E2F transcriptional target genes in DKO hematopoietic precursors. (A and B) RNA was isolated from thymocytes (A) or BM cells (B) from mice of the indicated genotypes. Two micrograms of total RNA was used to determine RNA levels of the indicated genes by RPA. CycA2 and Cdc2 levels were reduced an average of 1.7- and 1.5-fold, respectively, in the BM of DKO mice (n = 8; P < 0.005 and P < 0.05, respectively), relative to Ht/Ht and Wt controls. The mRNA levels of CycE1, CycG1, and CycD3 in DKO and control BM were not significantly different (three experiments). (C) Levels of CycA2 and Rb protein in three experiments with three sets of LMs of the indicated E2F1/E2F2 genotypes were determined by Western blotting. Ponceau S staining indicated similar total protein loading within each experiment. Note that, in this Western analysis, we did not resolve different phosphorylation forms of Rb. E2F3 protein levels in BM cells of the indicated E2F1/E2F2 genotypes were also determined in comparison to concanavalin A (mitogen)-stimulated spleen cells. (D) CycE1 and Rb protein levels were determined in Ter119+ purified BM cells of the indicated E2F1/E2F2 genotypes. The migration of hypophosphorylated and hyperphosphorylated Rb was determined by comparison with the migration of Rb in control (quiescent) and concanavalin A-stimulated spleen cells. (E) Ter119+ cells were purified from freshly isolated BM, lysed in kinase lysis buffer, and immunoprecipitated with antibodies against the indicated cyclins, and kinase activity was determined with histone H1 as the substrate and [γ-32P]ATP. Data are representative of three experiments.
DKO erythroid lineage cells exhibit substantially reduced CycE1, CycA2, and CycB1-associated kinase activities (two- to fourfold decrease in different experiments [Fig. 10E]), correlating with reduced expression of CycA2 and Cdc2. Given that the expression of CycE1 and Cdk2 is not decreased in DKO BM, it is not clear why associated kinase activity is decreased, but the stalling of DKO cells in S phase may indirectly decrease this G1/S kinase activity. We also cannot rule out that S phase arrest-stalling in DKO cells contributes indirectly to reduced CycB1-associated kinase activity by preventing progression into G2. However, given that CycA2-associated kinase activity peaks in S phase and that DKO BM contains at least twice the normal percentage of cells in S phase, the reduction in CycA2-Cdk activity that we observe in DKO cells is actually greater on a per-cell-in-S phase basis. In summary, we show that loss of E2F1 and E2F2 specifically reduces the expression of a subset of E2F transcriptional target genes, which may underlie reduced S phase progression and defective hematopoietic maturation.
DISCUSSION
Specific roles for E2F2 and E2F1 in promoting S phase progression in blood cell progenitors.
We have demonstrated that the loss of both E2F2 and E2F1 results in impeded S phase progression in hematopoietic progenitors, increased apoptosis during B-cell maturation, and severely impaired hematopoiesis. We hypothesize that DKO B-cell progenitors may mature poorly due to defective cell expansion resulting from impeded S phase, as well as defective Rag-mediated recombination and differentiation due to the inability to exit S and M into G1 (Fig. 11). In particular, the loss of E2F1 and E2F2 may impede the maturation of large pre-BII cells to small pre-BII cells by preventing cell cycle exit. Although this differentiation failure results in increased apoptosis, we demonstrate that apoptosis is the secondary consequence of blocked maturation. Our data suggest that S phase defects also account for failed expansion and maturation of other hematopoietic lineages. Thus, the failure to properly coordinate the cell cycle with differentiation impedes blood cell maturation.
FIG. 11.

Model showing that E2F1 and E2F2 play essential roles in the coordination of cell cycle progression and differentiation. E2F1/E2F2 loss results in impaired S phase progression, decreased differentiation, and inefficient hematopoiesis.
Given our demonstration that the loss of E2F1 and E2F2 results in significantly reduced expression of several genes, including CycA2, in hematopoietic progenitors and required roles for CycA2 in S phase (10, 23), E2F1/E2F2-dependent regulation of CycA2 expression may be required for proper progression through S phase and efficient hematopoiesis. Blocking CycA2 inhibits S phase in mammalian cells (19, 39, 61), and CycA2 localizes to sites of DNA replication (5). Furthermore, roles for Rb-dependent control of CycA2 expression in regulating S phase progression have been demonstrated. In addition to G1 arrest, the expression of active Rb (phosphorylation site mutant) blocks cells in S phase, which can be bypassed by ectopic E2F activity (26). Active Rb in S phase cells disrupts chromatin binding by PCNA, but not components of the prereplication complex (48). Ectopic expression of CycA2 restores PCNA tethering and partially restores S phase progression, suggesting that other Rb targets also mediate the S phase arrest. Finally, the expression of phosphorylation site mutant Drosophila Rb during the second mitotic wave in eye disk development results in an S phase delay phenotype that cannot be rescued by ectopic CycE expression (56). Nonetheless, it is not clear whether the reduction in CycA2 levels is sufficient to account for S phase defects in DKO BM cells or whether restoration of CycA2 expression could rescue hematopoiesis. Histone and/or nucleotide depletion, due to reduced expression of other E2F targets, may also contribute to impeded S phase progression. S phase stalling in DKO hematopoietic progenitors may also either activate or result from the activation of an S phase checkpoint. In budding yeast, deregulation of Swi4, which like E2F activates late G1 regulatory genes, results in precocious S phase entry and stalled S phase progression, the latter of which appears to activate a Rad53-dependent checkpoint (49). However, p53 protein levels and activity do not appear increased in E2F1/E2F2 mutant BM cells (data not shown). Finally, it is also possible that the loss of E2F1 and E2F2 impedes S phase progression in BM cells due to direct roles played by these E2F proteins in controlling origin firing independent of transcriptional functions (6).
E2F2 mutant and DKO mice exhibit hematopoietic defects that are very similar to those exhibited by patients with megaloblastic anemia, including anemia, larger RBC size, defective pan-hematopoietic maturation, and inhibited S phase progression in BM progenitors (1). In humans, megaloblastic anemia can be caused by vitamin B12 and folate deficiencies, which result in decreased thymidine synthesis, resulting in impaired DNA synthesis and misincorporation of UTP during DNA synthesis, leading to DNA damage. Antineoplastic drugs that inhibit nucleotide synthesis, such as methotrexate, can also result in megaloblastic anemia. Although we have observed selective reductions in several E2F target genes important for S phase in DKO BM progenitors, we have not addressed whether nucleotide synthesis is defective in these cells.
An obvious question is why we observe increased proliferation of mature DKO T cells in response to antigen (59) but decreased cell cycle progression of developing lymphocytes. The increased fraction of DKO BM cells in S phase was previously interpreted as indicative of hyperproliferation (59), which we now show is actually due to S phase arrest-delay. We speculate that E2F3 is able to compensate for the loss of E2F1 and E2F2 in promoting cell cycle progression in antigen-activated T cells, thus revealing negative roles for E2F1 and E2F2 in regulating G1-to-S progression. Indeed, we observe indistinguishable induction of the expression of many E2F target genes, including CycA2, in either DKO or Ht/Ht T cells following antigenic stimulation (data not shown). We do not understand why E2F3 or other E2F proteins are unable to compensate for the loss of E2F1/E2F2 in the BM. Furthermore, while we clearly observe accelerated G1-to-S phase progression in DKO peripheral T cells in response to antigen, we suspect that G1-to-S phase progression is also accelerated in DKO BM, albeit followed by poor S phase progression. Finally, our results reveal functional specificity for E2F1 and E2F2, with the loss of either E2F protein having opposing effects on lymphocyte maturation, as well as functional redundancy, with the combined mutation of E2F1 and E2F2 most dramatically impeding hematopoiesis.
Cell cycle control of hematopoiesis.
Rb mutant mice exhibit decreased erythroid maturation, and embryonic death in part results from severe anemia (8, 24, 29). Deregulated E2F activity contributes to erythroid defects in Rb-null embryos, as the combined mutation of Rb with either E2F1 or E2F3 alleviates at least some of the erythroid defects, resulting in increased percentages of enucleated erythrocytes (52, 60). While deregulated E2F activity can contribute to increased apoptosis and defective erythropoiesis in Rb-null embryos, our data indicate that the loss of E2F2 impedes erythropoiesis by preventing cell cycle progression, suggesting that the proper balance of E2F activity is critical for the appropriate cell cycle and differentiation control required for RBC maturation. Notably, however, erythropoiesis defects in Rb-null mice appear to be largely non-cell autonomous (33, 35, 54).
E2F4−/− mice display defects in late-stage erythroid maturation that appear autonomous to the hematopoietic system (21, 41), although the issue of cell autonomy has not been addressed. As in E2F2 KO and DKO mice, RBC in E2F4−/− embryos and mice exhibit greater cell size variability and an overall increase in cell volume. E2F4−/− embryos exhibit increased percentages of reticulocytes in peripheral blood, suggesting that these defects are not due to insufficient erythropoiesis but are instead indicative of stressed erythropoiesis. In contrast, E2F2 KO and DKO mice exhibit decreased reticulocyte numbers in adults, indicating reduced erythropoiesis. Decreased maturation of other hematopoietic lineages, including reduced thymocyte numbers, is also evident in E2F4−/− mice, coincident with increased apoptotic rates in BM cells. Thus far, it is not clear how hematopoietic phenotypes relate to E2F4 function as a transcription factor or as a regulator of cell cycle progression. Finally, studies of mice disrupted for the p27KIP1 Cdk inhibitor demonstrate that p27 restrains hematopoietic progenitor cell proliferation (7). In summary, the proper regulation of hematopoiesis requires precise regulation of the cell cycle machinery, with disruption of cell cycle control leading to defective blood cell maturation.
Relationship between the promotion of hematopoiesis and tumor suppression by E2F1 and E2F2.
It might be difficult to envision how defective cell cycle progression and increased B-cell progenitor apoptosis might contribute to the hematopoietic malignancies that we observe in E2F2 mutant mice (59). An intriguing, but entirely speculative, possibility is that rare survivors of Rag-mediated recombination in S phase-stalled cells could show increased rates of inappropriate translocations involving the Ig-T-cell receptor loci and oncogenes such as Myc, which could contribute to lymphomagenesis in E2F2 mutant mice. In addition, highly inefficient blood cell maturation combined with increased homeostatic demands (such as erythropoietin-driven erythropoiesis) may contribute to an increased error rate during DNA replication, allowing for the accumulation of mutations that promote tumorigenesis. In other words, in order to produce a given number of blood cells, E2F1/E2F2 mutant hematopoietic progenitors must undergo a much greater number of “attempts” at maturation, with most attempts resulting in S phase arrest and aborted maturation. More attempts should result in more mistakes. Furthermore, mutations that improve cell cycle progression should easily provide a selective advantage over the poor competition offered by E2F1/E2F2 mutant progenitors. Alternatively, E2F1/E2F2-dependent roles in limiting G1/S phase progression, in promoting apoptosis, or in maintaining genomic stability may contribute to tumor suppression (11).
Acknowledgments
J.D. is supported by grants from the NIH (CA77314) and the ACS (RSG LIB-101051), and by a Scholar Award from the Leukemia and Lymphoma Society. C.J.H. is supported by NIH/NHLBI 2 RO1 HL61382-04.
We thank D. Johnson, G. Shapiro, D. DeRyckere, and J. Hagman for critical review of the manuscript and B. Schaefer, S. Field, S. Orkin, and M. Greenberg for mice. We also thank K. Helm and M. Ashton of the Cancer Center Flow Cytometry Core (supported by grant 2 P30 CA 46934-09).
REFERENCES
- 1.Antony, A. C. 1999. Megaloblastic anemias, p. 446-483. In R. E. A. Hoffman (ed.), Hematology: basic principles and practice, 3rd ed. Churchill Livingstone, New York, N.Y.
- 2.Begg, A. C., N. J. McNally, D. C. Shrieve, and H. Karcher. 1985. A method to measure the duration of DNA synthesis and the potential doubling time from a single sample. Cytometry 6:620-626. [DOI] [PubMed] [Google Scholar]
- 3.Benschop, R. J., and J. C. Cambier. 1999. B cell development: signal transduction by antigen receptors and their surrogates. Curr. Opin. Immunol. 11:143-151. [DOI] [PubMed] [Google Scholar]
- 4.Benschop, R. J., D. Melamed, D. Nemazee, and J. C. Cambier. 1999. Distinct signal thresholds for the unique antigen receptor-linked gene expression programs in mature and immature B cells. J. Exp. Med. 190:749-756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cardoso, M. C., H. Leonhardt, and B. Nadal-Ginard. 1993. Reversal of terminal differentiation and control of DNA replication: cyclin A and cdk2 specifically localize at subnuclear sites of DNA replication. Cell 74:979-992. [DOI] [PubMed] [Google Scholar]
- 6.Cayirlioglu, P., P. C. Bonnette, M. R. Dickson, and R. J. Duronio. 2001. Drosophila E2f2 promotes the conversion from genomic DNA replication to gene amplification in ovarian follicle cells. Development 128:5085-5098. [DOI] [PubMed] [Google Scholar]
- 7.Cheng, T., N. Rodrigues, D. Dombkowski, S. Stier, and D. T. Scadden. 2000. Stem cell repopulation efficiency but not pool size is governed by p27(kip1). Nat. Med. 6:1235-1240. [DOI] [PubMed] [Google Scholar]
- 8.Clarke, A. R., E. R. Maandag, M. van Roon, N. M. T. van der Lugt, M. van der Valk, M. L. Hooper, A. Berns, and H. te Riele. 1992. Requirement for a functional Rb-1 gene in murine development. Nature 359:328-333. [DOI] [PubMed] [Google Scholar]
- 9.Cloud, J. E., C. Rogers, T. L. Reza, U. Ziebold, J. R. Stone, M. H. Picard, A. M. Caron, R. T. Bronson, and J. A. Lees. 2002. Mutant mouse models reveal the relative roles of E2F1 and E2F3 in vivo. Mol. Cell. Biol. 22:2663-2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Coverly, D., H. Laman, and R. A. Laskey. 2002. Distinct roles for cyclins E and A during DNA replication complex assembly and activation. Nat. Cell Biol. 4:523-528. [DOI] [PubMed] [Google Scholar]
- 11.DeGregori, J. 2002. The genetics of the E2F family of transcription factors: shared functions and unique roles. Biochim. Biophys. Acta 1602:131-150. [DOI] [PubMed] [Google Scholar]
- 12.DeGregori, J., G. Leone, K. Ohtani, A. Miron, and J. R. Nevins. 1995. E2F1 accumulation bypasses a G1 arrest resulting from the inhibition of G1 cyclin-dependent kinase activity. Genes Dev. 9:2873-2887. [DOI] [PubMed] [Google Scholar]
- 13.Duronio, R. J., P. H. O'Farrell, J.-E. Xie, A. Brook, and N. Dyson. 1995. The transcription factor E2F is required for S phase during Drosophila embryogenesis. Genes Dev. 9:1445-1455. [DOI] [PubMed] [Google Scholar]
- 14.Dyson, N. 1998. The regulation of E2F by pRB-family proteins. Genes Dev. 12:2245-2262. [DOI] [PubMed] [Google Scholar]
- 15.Field, S. J., F.-Y. Tsai, F. Kuo, A. M. Zubiaga, W. G. Kaelin, Jr., D. M. Livingston, S. H. Orkin, and M. E. Greenberg. 1996. E2F-1 functions in mice to promote apoptosis and suppress proliferation. Cell 85:549-561. [DOI] [PubMed] [Google Scholar]
- 16.Frolov, M. V., D. S. Huen, O. Stevaux, D. Dimova, K. Balczarek-Strang, M. Elsdon, and N. J. Dyson. 2001. Functional antagonism between E2F family members. Genes Dev. 15:2146-2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Garcia, I., M. Murga, A. Vicario, S. J. Field, and A. M. Zubiaga. 2000. A role for E2F1 in the induction of apoptosis during thymic negative selection. Cell Growth Differ. 11:91-98. [PubMed] [Google Scholar]
- 18.Gaubatz, S., G. J. Lindeman, S. Ishida, L. Jakoi, J. R. Nevins, D. M. Livingston, and R. E. Rempel. 2000. E2F4 and E2F5 play an essential role in pocket protein-mediated G1 control. Mol. Cell 6:729-735. [DOI] [PubMed] [Google Scholar]
- 19.Girard, F., U. Strausfeld, A. Fernandez, and N. J. C. Lamb. 1991. Cyclin A is required for the onset of DNA replication in mammalian fibroblasts. Cell 67:1169-1179. [DOI] [PubMed] [Google Scholar]
- 20.Harbour, J. W., and D. C. Dean. 2000. The Rb/E2F pathway: expanding roles and emerging paradigms. Genes Dev. 14:2393-2409. [DOI] [PubMed] [Google Scholar]
- 21.Humbert, P. O., C. Rogers, S. Ganiatsas, R. L. Landsberg, J. M. Trimarchi, S. Dandapani, C. Brugnara, S. Erdman, M. Schrenzel, R. T. Bronson, and J. A. Lees. 2000. E2F4 is essential for normal erythrocyte maturation and neonatal viability. Mol. Cell 6:281-291. [DOI] [PubMed] [Google Scholar]
- 22.Humbert, P. O., R. Verona, J. M. Trimarchi, C. Rogers, S. Dandapani, and J. A. Lees. 2000. E2f3 is critical for normal cellular proliferation. Genes Dev. 14:690-703. [PMC free article] [PubMed] [Google Scholar]
- 23.Hunter, T., and J. Pines. 1994. Cyclins and cancer II: cyclin D and CDK inhibitors come of age. Cell 79:573-582. [DOI] [PubMed] [Google Scholar]
- 24.Jacks, T., A. Fazeli, E. M. Schmitt, R. T. Bronson, M. A. Goodell, and R. A. Weinberg. 1992. Effects of an Rb mutation in the mouse. Nature 359:295-300. [DOI] [PubMed] [Google Scholar]
- 25.Janani, R., A. W. Harris, A. Strasser, S. Dhanoa, R. Plyam, and D. G. Osmond. 1998. Effect of a Bcl2 transgene on production and localization of precursor B cells in mouse bone marrow. Exp. Hematol. 26:982-990. [PubMed] [Google Scholar]
- 26.Knudsen, E. S., C. Buckmaster, T. T. Chen, J. R. Feramisco, and J. Y. Wang. 1998. Inhibition of DNA synthesis by RB: effects on G1/S transition and S-phase progression. Genes Dev. 12:2278-2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koury, M. J., D. W. Horne, Z. A. Brown, J. A. Pietenpol, B. C. Blount, B. N. Ames, R. Hard, and S. T. Koury. 1997. Apoptosis of late-stage erythroblasts in megaloblastic anemia: association with DNA damage and macrocyte production. Blood 89:4617-4623. [PubMed] [Google Scholar]
- 28.Koury, M. J., J. O. Price, and G. G. Hicks. 2000. Apoptosis in megaloblastic anemia occurs during DNA synthesis by a p53-independent, nucleoside-reversible mechanism. Blood 96:3249-3255. [PubMed] [Google Scholar]
- 29.Lee, E. Y.-H. P., C.-Y. Chang, N. Hu, Y.-C. J. Wang, C.-C. Lai, K. Herrup, W.-H. Lee, and A. Bradley. 1992. Mice deficient for Rb are nonviable and show defects in neurogenesis and haematopoiesis. Nature 359:288-294. [DOI] [PubMed] [Google Scholar]
- 30.Lee, J., and S. Desiderio. 1999. Cyclin A/CDK2 regulates V(D)J recombination by coordinating RAG-2 accumulation and DNA repair. Immunity 11:771-781. [DOI] [PubMed] [Google Scholar]
- 31.Leone, G., R. Sears, E. Huang, R. Rempel, F. Nuckolls, C.-H. Park, P. Giangrande, L. Wu, H. I. Saavedra, S. J. Field, M. A. Thompson, H. Yang, Y. Fujiwara, M. E. Greenberg, S. Orkin, C. Smith, and J. R. Nevins. 2001. Myc requires distinct E2F activities to induce S phase and apoptosis. Mol. Cell 8:105-114. [DOI] [PubMed] [Google Scholar]
- 32.Lindeman, G. J., L. Dagnino, S. Gaubatz, Y. Xu, R. T. Bronson, H. B. Warren, and D. M. Livingston. 1998. A specific, nonproliferative role for E2F-5 in choroid plexus function revealed by gene targeting. Genes Dev. 12:1092-1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lipinski, M. M., K. F. Macleod, B. O. Williams, T. L. Mullaney, D. Crowley, and T. Jacks. 2001. Cell-autonomous and non-cell-autonomous functions of the Rb tumor suppressor in developing central nervous system. EMBO J. 20:3402-3413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lissy, N. A., P. K. Davis, M. Irwin, W. G. Kaelin, and S. F. Dowdy. 2000. A common E2F-1 and p73 pathway mediates cell death induced by TCR activation. Nature 407:642-645. [DOI] [PubMed] [Google Scholar]
- 35.Maandag, E. C., M. van der Valk, M. Vlaar, C. Feltkamp, J. O'Brien, M. van Roon, N. van der Lugt, A. Berns, and H. te Riele. 1994. Developmental rescue of an embryonic-lethal mutation in the retinoblastoma gene in chimeric mice. EMBO J. 13:4260-4268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Murga, M., O. Fernandez-Capetillo, S. J. Field, B. Moreno, L. R. Borlado, Y. Fujiwara, D. Balomenos, A. Vicario, A. C. Carrera, S. H. Orkin, M. E. Greenberg, and A. M. Zubiaga. 2001. Mutation of E2F2 in mice causes enhanced T lymphocyte proliferation, leading to the development of autoimmunity. Immunity 15:959-970. [DOI] [PubMed] [Google Scholar]
- 37.Ogawa, H., K. Ishiguro, S. Gaubatz, D. M. Livingston, and Y. Nakatani. 2002. A complex with chromatin modifiers that occupies E2F- and Myc-responsive genes in G0 cells. Science 296:1132-1136. [DOI] [PubMed] [Google Scholar]
- 38.O'Reilly, L. A., A. W. Harris, D. M. Tarlinton, L. M. Corcoran, and A. Strasser. 1997. Expression of a Bcl2 transgene reduces proliferation and slows turnover of developing B lymphocytes in vivo. J. Immunol. 159:2301-2311. [PubMed] [Google Scholar]
- 39.Pagano, M., R. Pepperkok, F. Verde, W. Ansorge, and G. Draetta. 1992. Cyclin A is required at two points in the human cell cycle. EMBO J. 11:961-971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Penit, C. 1986. In vivo thymocyte maturation. BUdR labeling of cycling thymocytes and phenotypic analysis of their progeny support the single lineage model. J. Immunol. 137:2115-2121. [PubMed] [Google Scholar]
- 41.Rempel, R. E., M. T. Saenz-Robles, R. Storms, S. Morham, S. Ishida, A. Engel, L. Jakoi, M. F. Melhem, J. M. Pipas, C. Smith, and J. R. Nevins. 2000. Loss of E2F4 activity leads to abnormal development of multiple cellular lineages. Mol. Cell 6:293-306. [DOI] [PubMed] [Google Scholar]
- 42.Rolink, A., A. Kudo, H. Karasuyama, Y. Kikuchi, and F. Melchers. 1991. Long-term proliferating early pre-B cell lines and clones with the potential to develop to surface Ig-positive, mitogen reactive B cells in vitro and in vivo. EMBO J. 10:327-336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rolink, A. G., E. ten Boekel, T. Yamagami, R. Ceredig, J. Andersson, and F. Melchers. 1999. B cell development in the mouse from early progenitors to mature B cells. Immunol. Lett. 68:89-93. [DOI] [PubMed] [Google Scholar]
- 44.Rolink, A. G., T. Winkler, F. Melchers, and J. Andersson. 2000. Precursor B cell receptor-dependent B cell proliferation and differentiation does not require the bone marrow or fetal liver environment. J. Exp. Med. 191:23-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Royzman, I., A. J. Whittaker, and T. L. Orr-Weaver. 1997. Mutations in Drosophila DP and E2F distinguish G1-S progression from an associated transcriptional program. Genes Dev. 11:1999-2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Saavedra, H. I., L. Wu, A. de Bruin, C. Timmers, T. J. Rosol, M. Weinstein, M. L. Robinson, and G. Leone. 2002. Specificity of E2F1, E2F2, and E2F3 in mediating phenotypes induced by loss of Rb. Cell Growth Differ. 13:215-225. [PubMed] [Google Scholar]
- 47.Schaefer, B. C., M. L. Schaefer, J. W. Kappler, P. Marrack, and R. M. Kedl. 2001. Observation of antigen-dependent CD8+ T-cell/dendritic cell interactions in vivo. Cell. Immunol. 214:110-122. [DOI] [PubMed] [Google Scholar]
- 48.Sever-Chroneos, Z., S. P. Angus, A. F. Fribourg, H. Wan, I. Todorov, K. E. Knudsen, and E. S. Knudsen. 2001. Retinoblastoma tumor suppressor protein signals through inhibition of cyclin-dependent kinase 2 activity to disrupt PCNA function in S phase. Mol. Cell. Biol. 21:4032-4045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sidorova, J. M., and L. L. Breeden. 2002. Precocious S-phase entry in budding yeast prolongs replicative state and increases dependence upon rad53 for viability. Genetics 160:123-136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Strasser, A., A. W. Harris, and S. Cory. 1991. Bcl2 transgene inhibits T cell death and perturbs thymic self-censorship. Cell 67:889-899. [DOI] [PubMed] [Google Scholar]
- 51.Trimarchi, J. M., B. Fairchild, J. Wen, and J. A. Lees. 2001. The E2F6 transcription factor is a component of the mammalian Bmi1-containing polycomb complex. Proc. Natl. Acad. Sci. USA 98:1519-1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tsai, K. Y., Y. Hu, K. F. Macleod, D. Crowley, L. Yamasaki, and T. Jacks. 1998. Mutation of E2f-1 suppresses apoptosis and inappropriate S phase entry and extends survival of Rb-deficient mouse embryos. Mol. Cell 2:293-304. [DOI] [PubMed] [Google Scholar]
- 53.Weissman, I. L. 2000. Stem cells: units of development, units of regeneration, and units in evolution. Cell 100:157-168. [DOI] [PubMed] [Google Scholar]
- 54.Williams, B. O., E. M. Schmitt, L. Remington, R. T. Bronson, D. M. Albert, R. A. Weinberg, and T. Jacks. 1994. Extensive contribution of Rb-deficient cells to adult chimeric mice with limited histopathological consequences. EMBO J. 13:4251-4259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wu, L., C. Timmers, B. Maiti, H. I. Saavedra, L. Sang, G. T. Chong, F. Nuckolls, P. Giangrande, F. A. Wright, S. J. Field, M. E. Greenberg, S. Orkin, J. R. Nevins, M. L. Robinson, and G. Leone. 2001. The E2F1-3 transcription factors are essential for cellular proliferation. Nature 414:457-462. [DOI] [PubMed] [Google Scholar]
- 56.Xin, S., L. Weng, J. Xu, and W. Du. 2002. The role of RBF in developmentally regulated cell proliferation in the eye disc and in cyclin D/Cdk4 induced cellular growth. Development 129:1345-1356. [DOI] [PubMed] [Google Scholar]
- 57.Yamasaki, L., T. Jacks, R. Bronson, E. Goillot, E. Harlow, and N. J. Dyson. 1996. Tumor induction and tissue atrophy in mice lacking E2F-1. Cell 85:537-548. [DOI] [PubMed] [Google Scholar]
- 58.Zhu, J. W., D. DeRyckere, F. X. Li, Y. Y. Wan, and J. DeGregori. 1999. A role for E2F1 in the induction of ARF, p53, and apoptosis during thymic negative selection. Cell Growth Differ. 10:829-838. [PubMed] [Google Scholar]
- 59.Zhu, J. W., S. J. Field, L. Gore, M. Thompson, H. Yang, Y. Fujiwara, R. D. Cardiff, M. Greenberg, S. H. Orkin, and J. DeGregori. 2001. E2F1 and E2F2 determine thresholds for antigen-induced T-cell proliferation and suppress tumorigenesis. Mol. Cell. Biol. 21:8547-8564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ziebold, U., T. Reza, A. Caron, and J. A. Lees. 2001. E2F3 contributes both to the inappropriate proliferation and to the apoptosis arising in Rb mutant embryos. Genes Dev. 15:386-391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zindy, F., E. Lamas, X. Chenivesse, J. Sobczak, J. Wang, D. Fesquet, B. Henglein, and C. Brechot. 1992. Cyclin A is required in S phase in normal epithelial cells. Biochem. Biophy. Res. Commun. 182:1144-1154. [DOI] [PubMed] [Google Scholar]