

Abstract
Hepatitis B virus (HBV) X protein (HBx) plays an essential role in viral replication and in the development of hepatocellular carcinoma. HBx has the ability to transactivate the expression of all HBV proteins, including the viral core protein HBc. Consistent with its regulatory role, HBx is relatively unstable and is present at low levels in the cell. We report here that the level of HBx was significantly reduced by the coexpression of HBc in cultured human hepatoma cells, whereas the level of HBx mRNA was unaffected. The repression of HBx by HBc was relieved by treating cells with the proteasome inhibitor MG132, indicating that HBc acts by stimulating the proteasome-mediated degradation of HBx. Moreover, the inhibitory effect of HBc was specific to HBx and did not affect other proteins, including p53, a known target of the proteasome. Although no direct physical interaction between HBc and HBx could be demonstrated, mutational analysis indicated that the C-terminal half of HBc is responsible for its inhibitory effect. These results suggest that HBc functions as a novel regulator of the HBV life cycle and of hepatocellular carcinogenesis through control of the HBx level via an inhibitory feedback type of mechanism.
Hepatitis B virus (HBV) is a small, enveloped virus with a 3.2-kb-long, partially double-stranded circular DNA genome. HBV is a causative agent of chronic and acute hepatitis and is associated with the development of hepatocellular carcinoma. The HBV genome contains four viral promoters referred to as the C, pre-S1, S, and X promoters, the activities of which are regulated by two enhancer elements, EnI and EnII (9, 13, 33, 41). HBx, a product of the X gene, transactivates the expression of all HBV proteins through these two enhancers (6, 32, 35, 40). In particular, HBx increases the expression of the viral core protein HBc in vitro and in vivo by transactivating the C promoter (26, 39).
HBx consists of 154 amino acids and has a molecular mass of approximately 17 kDa. It is a multifunctional protein which is known to affect gene transcription, intracellular signal transduction, genotoxic stress response, protein degradation, cell cycle control, apoptotic cell death, and carcinogenesis (23, 40). With its gene conserved in all mammalian hepadnaviruses, HBx is believed to be essential for viral replication, as woodchuck hepatitis virus HBx was shown to be required for natural viral infection in woodchucks (5, 42). Several studies on HBx transgenic mice have investigated the hepatocarcinogenic effects of HBx. Initial reports showed that mice harboring HBx develop progressive features that are characteristic of the malignant transformation of liver cells (18, 36). However, not all HBx transgenic mice develop hepatocellular carcinoma, and some reports indicate that expression of HBx above a certain threshold is necessary for hepatocyte transformation (19, 20). Thus, the regulation of HBx level may be important in viral replication and hepatocellular carcinoma development.
HBx is maintained at a very low intracellular level because it is rapidly degraded, with a half-life of about 30 min (29). The instability at the protein level had been reported to be due to rapid proteolysis by the ubiquitin-proteasome pathway (16). The proteasome is the major cellular protease system for the removal of damaged or abnormal proteins, for the regulation of short-lived regulatory proteins, and for the processing of immunogenic proteins (8). The associations between HBx and proteasome components and the regulation of HBx transactivation activity by rapid turnover have been reported previously (10, 17, 31).
For many viruses, viral gene expression is sequentially regulated at the transcriptional level by gene products encoded in its genome. Examples are provided by polyomaviruses, adenoviruses, herpesviruses, reoviruses, and the influenza viruses. For example, gene expression in herpes simplex virus type 1-infected cells follows a series of inductions and repressions which occur in three phases, referred to as alpha, beta, and gamma. The alpha gene products induce the transcription of beta genes, which inhibit the expression of alpha genes and induce the transcription of gamma gene products, which in turn inhibit the expression of beta genes (37). Such a regulatory mechanism has not been reported for HBV, although X mRNA seems to disappear faster than the other viral mRNAs in transiently transfected cells (38).
In this report, we show that the intracellular level of HBx can be downregulated by HBc. We demonstrate that this regulation is achieved via a novel mechanism involving the activation of the proteasome-mediated degradation of HBx. We hypothesize that HBx activates the synthesis of HBc during the early stage of viral replication and that HBc in turn functions as an effective downregulator of HBx in a feedback-type inhibitory manner.
MATERIALS AND METHODS
Plasmid constructs.
DNA coding for HBc and HBx was amplified by PCR from the serum of a chronic hepatitis patient. Sequence analysis indicated that the amino acid sequence of the HBc used in this study was identical to that of the published core sequence of the adw2 subtype (3). The sequence for HBx differed only at the fifth and sixth amino acid residues, which were Met and Cys in our HBx compared with Lys and Tyr in the adw2 sequence.
The core protein expression plasmid pHBcHA was constructed in three steps. First, the core gene was amplified by PCR with primers HBcF7 (5′-ATG GAC ATT GAC CCG TAT) and HBcHA-R (5′-TCA GTA GTC GGG GAC GTC GTA AGG CTC GAG ACA TTG AGA TTC). The latter primer contains sequences for the influenza virus hemagglutinin (HA) epitope and an XhoI site (italic). The 0.6-kb DNA amplified was initially inserted into the pT7Blue vector (Novagen), and then the HBc gene sequence flanked by BamHI and XbaI sites was subcloned into the BamHI and XbaI sites of the expression vector pcDNA3 (Invitrogen). To construct pHBxHA, the 0.6-kb DNA containing the X gene and the sequence upstream was amplified with primers HBF3 (5′-TCA GTT ATA TGG ATG ATG) and HBx-R (5′-GGG CTC GAG GGC AGA GGT GAA AAA GTT GCA, XhoI site italic). After digestion of the amplified DNA with NcoI near the initiation codon of X, treatment with Klenow enzyme, and digestion with XhoI, the 0.45-kb DNA was inserted into the BamHI (blunted by the Klenow enzyme) and XhoI sites of pHBcHA, replacing the HBc sequence. As a result, HA epitope-tagged HBc and HBx were placed under the control of promoters derived from cytomegalovirus and bacteriophage T7. To express HBx without the HA tag, plasmid pHBx was constructed by PCR with primers HBF3 and HBR2 (5′-GTA TGT AAA TAA TGC CTA). After digestion of the amplified DNA with NcoI near the initiation codon of X, treatment with Klenow, and digestion with BglII, the 0.45-kb DNA was inserted into the HindIII (blunted by Klenow enzyme) and BamHI sites of pcDNA3.
The frameshift mutation pHBc-FS and substitution mutation pHBc-NIS were constructed by site-directed mutagenesis with the Quick-change mutagenesis kit (Stratagene) as instructed. Briefly, PCR was performed with pHBc as the template and a primer pair carrying the intended sequence. An extra cytosine nucleotide was inserted (TTCT) into the ninth codon for Phe (TTT) of wild-type HBc with primers C-FS-F (5′-AT AAA GAA TTC TGG AGC TAC T) and C-FS-R (5′-A GTA GCT CCA GAA TTC TTT AT). The PCR product was used to transform Escherichia coli, and several colonies were screened by digestion of the resultant plasmids with EcoRI, which would cut at the newly generated site (the target sequence included in the PCR primer). The mutation was confirmed by sequencing.
Substitution for the two initiation codons within HBc (at the 66th and 92nd positions) was performed by a two-step PCR to make plasmid pHBc-NIS, which had no internal start codon. First, the 66th codon (5′-ATG) was changed to Ile (5′-ATA) with the PCR primers C-2mut-F (5′-GGG AAT TGA TAT CTC TAG CTA C) and C-2mut-R (5′-GTA GCT AGA GAT ATC AAT TCC C). The Thr codon (5′-ACT) at the 67th position was also changed to a Ser codon (5′-TCT) in this process. Second, the initiation codon at the 92nd position was replaced with a Pro codon (5′-CCG) by PCR with primers C-3mut-F (5′-TAA TAC TAA CCC GGG TTT AAA G) and C-3mut-R (5′-CTT TAA ACC CGG GTT AGT ATT A). Finally, the plasmid with all three mutations, pHBc-FS-NIS, was constructed likewise with pHBc-FS as the PCR template.
Deletion mutants were constructed by PCR and substituted for the wild-type HBc sequence located between the BamHI and XhoI sites of pHBcHA. Primers CN-F (5′-ACC GGA TCC ATG GAC ATT GAC CCT TAT) and CN-R (5′-ACC CTC GAG TAA ACC CAT GTT AGT ATT) were used to construct pHBc-N, which contained the amino-terminal 95 residues of HBc. To construct pHBc-f2, which started at the second initiation codon (66th codon), primers C-f2 (5′-ACC GGA TCC ATG ACT CTA GCT ACC TGG) and CC-R (5′-ACC CTC GAG ACA TTG AGA TTC CCG AGA) were used, and primers C-f3 (5′-ACC GGA TCC ATG GGT TTA AAG ATC AGG) and CC-R were used to construct pHBc-f3, which started from the third initiation codon (the 92nd codon) of HBc.
Expression of HBV proteins.
The expression of HBc and HBx in cultured cells was driven in most experiments by bacteriophage T7 RNA polymerase, supplied in the recombinant vaccinia virus vTF7-3 (11). Huh7 human hepatoma or 143B human osteosarcoma cells (ATCC CRL-8303) were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS). When cells had grown to 70 to 90% confluency, they were infected at a multiplicity of infection of 3. The medium was aspirated after 1 h, and the cells were incubated with 10 ml of DMEM containing 10% FBS. Three hours later, cells were transfected with plasmid DNA by the standard calcium phosphate method, and the medium was replaced with fresh DMEM containing 10% FBS 12 h after the transfection. The cells were then incubated for an additional 36 h, collected, and analyzed. In some experiments, Lipofectamine Plus (Invitrogen) was used to transfect cells with plasmid DNA without vaccinia virus infection.
Immunoblotting.
Cells were harvested and washed once with Dulbecco's phosphate-buffered saline (PBS; 136.8 mM NaCl, 2.5 mM KCl, 0.8 mM Na2HPO4, 1.47 mM KH2PO4, pH 7.4), lysed in 1× sample loading buffer (100 mM Tris-Cl [pH 6.8], 2% sodium dodecyl sulfate [SDS], 10% glycerol, 0.02% bromophenol blue, 5% β-mercaptoethanol) and boiled for 30 min. The proteins obtained from about 105 cells were separated in a 12% polyacrylamide gel under denaturing conditions and then electrically transferred to a nitrocellulose membrane at 250 mA for 3 h. The membrane was blocked with 5% skim milk in TNT buffer (10 mM Tris-Cl [pH 8.0], 150 mM NaCl, 0.05% Tween 20) for 5 h with gentle agitation and incubated with anti-HA rabbit polyclonal antibody (Santa Cruz Biotechnology) at 200 ng/ml for 5 h. Blots were probed with anti-HBc (Dako Corp.) or anti-HBx (a generous gift from Y. Yun) rabbit serum or anti-p53 monoclonal antibody Ab-1 (Calbiochem). The membrane was then washed three times for 10 min with TNT buffer, incubated with horseradish peroxidase-conjugated, donkey anti-rabbit immunoglobulin or sheep anti-mouse immunoglobulin antibody (Amersham) diluted 1:3,000 with blocking solution for 30 min, and finally washed three times with TNT. Bands were visualized on X-ray film with an enhanced chemiluminescence system (ECL; Amersham) and quantified by densitometry with TINA 2.10e (Raytest GmbH).
Semiquantitative RT-PCR.
Total RNA from transfected cells was isolated with Trizol LS reagent (Gibco-BRL) as instructed by the manufacturer. The amount of RNA template and number of PCR cycles required to generate amplified DNA within the proportional range were determined empirically as follows. First, 50, 250, or 500 ng of total RNA from HBx-expressing cells was used as the template for reverse transcription (RT). The RNA sample was mixed with 2 pmol of primer HBx-R, and cDNA was synthesized with the RNase H-negative reverse transcriptase Superscript II (Gibco-BRL) as instructed by the manufacturer. One tenth of the synthesized cDNA was used as a template for PCR with HBF6 (5′-ACATAAGAGGACTCTTGG) and HBx-R as primers. To estimate the amount of DNA amplified, an aliquot of the reaction was collected after every fifth cycle and analyzed by electrophoresis in a 1% agarose gel. Based on the intensity of ethidium bromide staining, a 0.2-kb-long DNA produced after 25 cycles appeared to be proportional in amount to the initially added total RNA. A relative quantity of DNA was made from RNA ranging from 50 to 250 ng in amount.
RNase protection assay.
StuI-digested pHBxHA DNA containing the 130-bp 3′-terminal sequence of the HBx gene under the control of the SP6 promoter was used as a template for in vitro transcription of the antisense RNA. Digoxigenin RNA labeling mix (Roche) and SP6 RNA polymerase (Ambion) were used as instructed by the manufacturers. Five micrograms of total cellular RNA, as described above, and 500 ng of the antisense probe RNA were coprecipitated with ethanol and analyzed with an RNase protection kit (Roche), as instructed by the manufacturer. Digestion products were subjected to electrophoresis on a 4% polyacrylamide-7 M urea gel in 1× TBE (90 mM Tris-borate, 1 mM EDTA, pH 8.0) buffer and then transferred to a nylon membrane. The membrane was incubated with alkaline phosphatase-conjugated antidigoxigenin antibody (Roche), and the signal was visualized on X-ray film with CDP-star reagent (Roche).
Detection of ubiquitinated HBx.
Cells were cotransfected with pHBxHA and another plasmid coding for hexahistidine-fused ubiquitin and then infected with recombinant vaccinia virus vTF7-3 as described above. The proteasome inhibitor MG132 (Calbiochem) was added 48 h after transfection to a final concentration of 25 μM. After further incubation for 6 h, the cells were collected, washed once with PBS, resuspended in lysis buffer (50 mM Tris-Cl [pH 7.4], 150 mM NaCl, 0.5% Triton X-100, 1 μg of leupeptin per ml, 1 μg of aprotinin per ml, 1 mM phenylmethylsulfonyl fluoride, 1 μM MG132) supplemented with 1% (wt/vol) SDS and 1% (vol/vol) beta-mercaptoethanol, and then boiled for 30 min to denature cellular proteins. The denatured lysate was diluted 1:5 with lysis buffer and centrifuged at 12,000 × g for 10 min. Prewashed Talon resin (Clontech, 50% in lysis buffer) was added to the supernatant and mixed for 30 min at room temperature, and the resin was washed twice with lysis buffer. Bound proteins were eluted with 50 μl of 0.1 M EDTA (pH 8.0), separated in a 12% polyacrylamide gel, and immunoblotted with anti-HA antibody.
Determination of HBx half-life.
Twenty-four hours after transfection, cells were starved for 30 min in methionine-free DMEM. The cells were then labeled for 20 min in methionine-free DMEM containing 0.2 mCi of [35S]methionine (New England Nuclear). The labeling medium was replaced with regular DMEM, and the cells were harvested after various chase periods. The cells were washed once with PBS, resuspended in lysis buffer (50 mM Tris-Cl [pH 7.4], 150 mM NaCl, 0.5% Triton X-100, 1 μg of leupeptin per ml, 1 μg of aprotinin per ml, 1 mM phenylmethylsulfonyl fluoride, 1 μM MG132) supplemented with 1% SDS and 1% beta-mercaptoethanol, and boiled for 30 min to denature the cellular proteins. The denatured lysates were diluted 1:5 with lysis buffer and centrifuged at 12,000 × g for 10 min. Cell lysates were incubated with anti-HA antibody for 12 h at 4°C, protein A-Sepharose (Pharmacia, 50% in lysis buffer) was added to the lysates, and the mixture was further incubated for 8 h at 4°C. The protein A-Sepharose beads were separated by centrifugation, washed twice with lysis buffer, and resuspended in the gel loading buffer. After boiling for 10 min, the eluted proteins were separated by electrophoresis in a 12% polyacrylamide gel. An autofluorograph obtained was scanned and quantified with a BAS phosphoimage analyzer (FujiXerox). Results are presented as percentages of the HBx signal obtained at the beginning of the chase. The decay curve was drawn with Sigmaplot 2000 (SPSS Science).
RESULTS
Coexpression of HBc reduces the intracellular level of HBx.
Studies with cultured cells suggested that HBx functions as a transcriptional transactivator of a variety of genes, including the HBV genes (7, 32). The ability of HBx to transactivate viral core gene expression has also been demonstrated in transgenic mice (26, 39). In herpesviruses, viral gene expression is temporally regulated; i.e., expression of one gene class is induced by gene products made in the preceding stage and inhibited by gene products synthesized in the subsequent stage (37). Such a regulatory mechanism has not been reported for HBV. To determine the existence of a relationship between HBc and HBx at the protein level, cultured human hepatoma Huh-7 cells were transfected with the two HBV genes under the control of the T7 promoter. Their expression was driven by infecting the cell with recombinant vaccinia virus carrying the T7 RNA polymerase gene (Fig. 1A). Both HBV genes were tagged with the HA epitope at their C termini to allow them to be detected easily by immunoblotting. In some experiments, HBx was expressed without the HA tag and detected with anti-HBx antibody.
FIG. 1.

HBx attenuation upon HBc coexpression. (A) Expression plasmids for HBc and HBx containing the T7 promoter and the HA tag. Numbering is that of the HBV genome according to convention (22), beginning at the unique EcoRI site. (B and C) HBc and HBx were coexpressed in Huh-7 cells by infection with recombinant vaccinia virus carrying T7 RNA polymerase followed by DNA transfection. Proteins were immunoblotted with anti-HA antibody 48 h after transfection. The amount of DNA used for transfection is indicated in micrograms above each lane. Vp35 is a vaccinia virus protein used as a control. The relative intensities of HBx and Vp35 are indicated as percentages at the bottom. (D) HBc and HBx were detected with an anti-HBc and an anti-HBx antibody, respectively. The two minor forms of HBc with asterisks are described in the text. (E) HBx and HBc were coexpressed without vaccinia virus infection. Lipofectamine Plus was used for cell transfection. Plasmid pcDNA3 was used to keep the total amount of transfected DNA constant. Immunoblots were probed with anti-HA antibody. (F) Quantification of HBx mRNA by RT-PCR. The 0.2-kb amplified DNA was separated on a 1% agarose gel and visualized with ethidium bromide. Lanes 1 to 3, 25-cycle test run PCR performed with 50, 250, and 500 ng of total RNA derived from the HBx-expressing cells in B, lane 1. Lanes 4 to 6,250 ng of RNA from the cells cotransfected with HBc and HBx (B, lanes 1 to 3, respectively) was amplified under the same conditions. (G) RNase protection assay. Total RNA was isolated from cells equivalent to those in panel B, lanes 1 to 3, except for the pHBcHA control. Five micrograms of total RNA was hybridized with 500 ng of digoxigenin-labeled antisense RNA and treated according to the manufacturer's instructions. Denaturing gel electrophoresis and detection are described in Materials and Methods. Transfection efficiency, measured by cotransfection of the reporter plasmid pCITEbetaGal (Novagen), is indicated.
Immunoblotting indicated that HBc and HBx were successfully expressed in this system as proteins of the expected sizes (21.5 and 17 kDa, respectively) (Fig. 1B, lanes 1 to 3). Interestingly, the level of HBx was significantly reduced when coexpressed with HBc, in a dose-dependent manner. The inhibitory effect of HBc was specific to HBx, as the vaccinia virus protein Vp35 (25), unrelated to HBx or HBc, remained largely unchanged by HBc coexpression (Fig. 1B, lanes 4 to 6). This result also excludes the possibility that the reduction of HBx by HBc was due to a shortage of the cellular protein synthesis machinery. In contrast, the amount of HBc protein was not significantly affected by cotransfection with increasing amount of HBx DNA (Fig. 1C). Since the expression of both genes in this system depends on the vaccinia virus-encoded T7 RNA polymerase, the transactivation activity of HBx is not expected to be operative in this system. We obtained similar results with 143B human osteosarcoma cells (data not shown).
To rule out the possibility that the HA tag might have caused HBx downregulation, we repeated the experiment with an HBx construct lacking the HA tag. Immunoblotting with anti-HBx antibody showed that the expression of untagged HBx was also reduced by HBc coexpression (Fig. 1D). An anti-HBc antibody was used in this experiment to confirm the expression of HBc. In addition to the HBc, we noted the presence of two minor proteins estimated to be 17 and 15 kDa (Fig. 1D, asterisk). The nature of these proteins will be discussed later in this paper.
Although the attenuating effect of HBc on HBx was convincing, the possibility of an artifactual effect due to the vaccinia virus on the expression of HBV proteins remained a concern. Therefore, we repeated the experiment without vaccinia virus infection. Data compatible with those obtained with the vaccinia virus-based expression were obtained by transfection of the cells with the plasmids alone (Fig. 1E), confirming the validity of the data obtained with the vaccinia virus system.
Inhibitory effect of HBc on HBx occurs at the posttranscriptional level.
To understand the mechanism of HBc-induced HBx attenuation, we first determined the HBx mRNA level by semiquantitative RT-PCR and RNase protection methods. Total RNA was isolated from cells prepared in the same way as those used in the immunoblotting (Fig. 1B, lanes 1 to 3). The number of PCR cycles required to produce DNA in an amount proportional to the amount of RNA initially present in the sample was determined empirically, as described in Materials and Methods; 25 cycles appeared to be satisfactory (Fig. 1F, lanes 1 to 3). With this assay, no discernible differences in the amounts of RT-PCR products were found between cell preparations that showed significant differences in HBx protein level (Fig. 1F, lanes 4 to 6, corresponding to the cells in lanes 1 to 3, respectively, of Fig. 1B). A similar result was obtained with the RNase protection assay (Fig. 1G). These results suggest that the inhibition of HBx by HBc occurs at the posttranscriptional level.
HBc stimulates degradation of HBx.
One interesting feature of HBx is that it undergoes rapid turnover in cells (29). Since our results showed that HBc reduces the intracellular level of HBx at a posttranscriptional step, we questioned whether HBc is involved in the regulation of HBx degradation. To address this issue, we studied the stability of HBx by pulse-chase labeling. In agreement with the results reported by other researchers (16, 29), we observed rapid degradation of HBx, with an estimated half-life of 40 min (Fig. 2A and 2B). More importantly, the coexpression of HBc shortened the half-life of HBx to approximately 20 min, which indicates that HBc stimulates the degradation of HBx. We noticed that in addition to the change in the half-life, the amount of HBx at the beginning of the chase was also reduced by about 50% in the presence of HBc (Fig. 2A, compare lanes 1 and 5), indicating that the facilitated degradation had already occurred during the pulse period.
FIG. 2.

Intracellular stability of HBx. (A) Cells expressing HBx with or without HBc were starved for 30 min in methionine-free medium, labeled for 20 min with [35S]methionine, and chased for the indicated periods. HBx was immunoprecipitated with anti-HA antibody and separated on a 12% polyacrylamide gel. An autofluorograph is shown. (B) The autofluorograph was scanned and quantified with a BAS phosphoimage analyzer (FujiXerox), and results are presented as percentages of the signal obtained at the beginning of the chase. The decay curves, with (○) and without (•) HBc, were drawn with Sigmaplot 2000 (SPSS Science).
HBc protein, not its mRNA, is responsible for accelerated degradation of HBx.
To exclude the possibility of any regulatory role played by core mRNA, a frameshift mutation was generated by introducing a cytidine nucleotide into the ninth codon of HBc (Fig. 3A, upper panel). Contrary to expectation, the frameshift mutant construct HBc-FS still stimulated HBx degradation (Fig. 3B, lanes 4 to 6). Since there was no apparent HBc expression by this construct, this finding initially suggested regulation by core mRNA. However, longer exposure of the immunoblot revealed two additional bands below HBx with sizes estimated to be 17 and 15 kDa (Fig. 3B, right). According to the core gene sequence, polypeptides of 157 or 131 amino acids can be synthesized if translation initiates from either of the two internal, in-frame start codons located at the 66th and 92nd codon positions. Such polypeptides would be similar in size to the bands observed.
FIG. 3.

HBc protein is responsible for degradation of HBx. (A) Diagram of HBc mutants. Upper panel: HBc-FS had an additional nucleotide in codon 9, as indicated by the asterisk, preventing the sequence downstream from translating in frame. In HBc-NIS, two internal in-frame start codons at positions 66 and 92 were replaced by codons for Ile and Pro, respectively, as indicated by the inverted triangles. HBc-FS-NIS had a frameshift insertion and the two substitutions. Lower panel: HBc-N contained the N-terminal part of HBc from codons 1 to 95. HBc-f2 started with a Met codon at the 66th position and ended at the C terminus. Another Met codon at the 92nd position was not changed in this construct. HBc-f3 started with a Met codon at the 92nd position and ended at the C terminus. All constructs had an HA epitope at the C terminus. (B and C) Coexpression of HBx with HBc mutant proteins. The amount of transfected DNAs is indicated in micrograms above each lane. Lanes 4 to 6, longer exposure of the autoradiograph shown in B, lanes 4 to 6. The two smaller forms of HBc, very similar in size to HBx, are indicated by arrowheads.
Although the existence of such proteins has not been reported previously, the synthesis of minor amounts under physiological conditions cannot be excluded. These two proteins may have been produced at a detectable level by the vaccinia virus-based expression system in this experiment. In fact, we detected the two minor proteins with an anti-HBc antibody as well as the anti-HA antibody (Fig. 1D, upper panel). In order to prevent complicating effects from these minor proteins, HBc-NIS with no internal start codon was constructed by substituting Ile and Pro for the two internal start codons at the 66th and 92nd positions, respectively (Fig. 3A, upper panel). While HBc-NIS functioned like wild-type HBc in terms of downregulating HBx expression (Fig. 3B, lanes 7 to 9), HBc-FS-NIS, which contained all three mutations, showed no effect on HBx expression (Fig. 3B, lanes 10 to 12). These results indicate that the inhibitory effect of HBc resides in its protein rather than in its mRNA.
C-terminal half of HBc is responsible for stimulatory effect.
As the data described above suggested that the C-terminal half of HBc is responsible for HBx degradation, three more deletion constructs of HBc were made to further confirm the result. HBc-N contained the N-terminal half of HBc, and HBc-f2 and HBc-f3 coded for the C-terminal portion starting from the second (66th) and third (92th) start codons of the core gene, respectively (Fig. 3A, lower panel). When tested for an effect on HBx, all three constructs that contained the C-terminal half (whole HBc, HBc-f2, and HBc-f3) stimulated HBx degradation (Fig. 3C, lanes 1 to 3 and 7 to 12), whereas HBc-N had no effect on HBx (Fig. 3C, lanes 4 to 6). These results, together with those with the frameshift mutant described above, suggest that the inhibitory effect of HBc on HBx resides in the C terminus of HBc.
HBc stimulates proteasome-mediated degradation of HBx.
Hu and others (16) showed that the rapid turnover of HBx was partially blocked by treatment with chemical proteasome inhibitors, such as MG132 and lactacystin. It was shown in the same study that HBx is polyubiquitinated. Thus, if HBx is a substrate of the cellular ubiquitin-proteasome pathway, we presumed that the inhibitory effect of HBc on HBx was probably mediated via the ubiquitin-proteasome pathway. To address this hypothesis, the effect of MG132 on HBx in the presence of HBc was examined. Transfected cells were treated with MG132, and proteins were analyzed by immunoblotting. MG132 treatment blocked the inhibitory effect of HBc and led to a greater than threefold increase in the HBx level (Fig. 4A). Because MG132 had a similar effect in the absence of HBc (Fig. 4B), we conclude that the effect of HBc on HBx can be completely blocked by inhibiting proteasome activity.
FIG. 4.

Effect of MG132 on HBx in the presence (A) and absence (B) of HBc. Cells transfected with HBx or HBc were treated with MG132, and proteins were analyzed by immunoblotting. The intensity of HBx bands is indicated at the bottom. The amount of transfected DNA is shown in micrograms above each lane.
Most cellular proteins degraded by proteasomes are covalently linked with multimers of ubiquitin. The attachment of five or more ubiquitin moieties leads to recognition and hydrolysis by the cellular 26S proteasome (8). To confirm that the HBx expressed in our system is ubiquitinated, as reported previously (16), the gene coding for ubiquitin with a hexahistidine tag and the HA-tagged X gene construct were used to cotransfect Huh-7 cells to allow efficient detection of HBx ubiquitination. After treating the transfected cells with MG132, proteins conjugated with hexahistidine-tagged ubiquitin were isolated by affinity chromatography and analyzed for HBx by immunoblotting with anti-HA antibody. Typically, ubiquitinated proteins vary in size depending on the number of conjugated ubiquitin units. Ubiquitinated HBx appeared as multiple, differently sized bands (Fig. 5). The smallest band, approximately 29 kDa in size, corresponded to the conjugation of one ubiquitin moiety of 12 kDa to HBx. The sizes of other bands varied in increments of 12 kDa, indicating that different numbers of ubiquitin moieties were attached. Proteins larger than 65 kDa were also observed as a smear in the blot, indicating that they were probably conjugated to more than four ubiquitin units. The ubiquitination of HBx, together with its prolonged stability in the presence of MG132, demonstrated that it is regulated by the proteasome-mediated degradation.
FIG. 5.
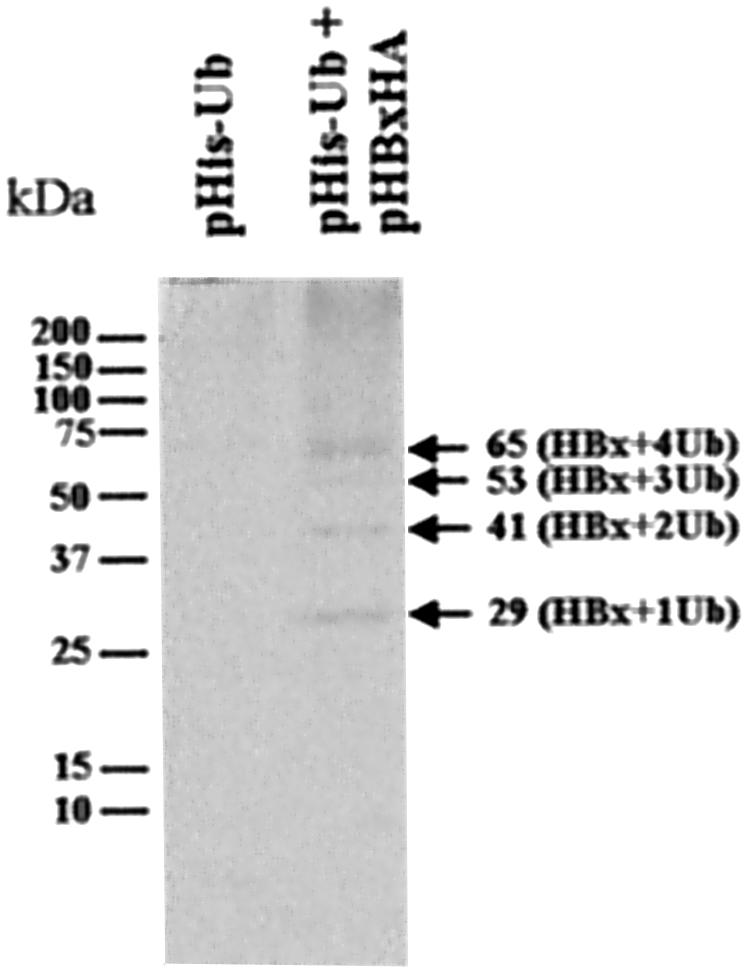
Detection of ubiquitinated HBx. Cells were cotransfected with plasmids coding for hexahistidine-tagged ubiquitin and HBx. Ubiquitinated proteins, isolated by affinity chromatography, were analyzed for HBx by immunoblotting with anti-HA antibody. The estimated number of ubiquitin moieties conjugated to HBx is shown on the right. Shown on the left are size markers (in kilodaltons).
Conjugation of ubiquitin to a protein substrate proceeds in three steps. Initially, the common enzyme E1 activates ubiquitin. One of several ubiquitin-conjugating enzymes E2 then transfers the activated ubiquitin moiety from E1 to a Lys residue of the substrate that is specifically bound to a member of the ubiquitin-protein ligase family, or E3. Thus, E3 serves as a specific recognition factor of the system. To further explore the inhibitory effect of HBc on proteasome-mediated degradation, the effect of HBc on the well-known proteasome substrate p53 was tested (14, 21). Unlike HBx, the amount of p53 was unaffected by HBc (Fig. 6), suggesting that the effect of HBc on HBx is specific. Therefore, it appears that HBc has no influence on the common steps of ubiquitin-proteasome pathway, such as ubiquitin activation by E1 or proteolysis by the 26S proteasome.
FIG. 6.

HBc has no effect on p53. Cells cotransfected with HBc and p53 were analyzed by immunoblotting with anti-HA (for HBc) or anti-p53 antibodies. The amount of transfected DNA is indicated in micrograms above each lane.
DISCUSSION
We report here that the HBV core protein HBc stimulates the proteasome-mediated degradation of the viral regulatory protein HBx. In polyomaviruses, adenoviruses, herpesviruses, reoviruses, influenza viruses, and other viruses, the expression of viral genes is regulated by interactions among the viral gene products. Our data suggest that such a feedback mechanism also operates in HBV gene regulation (Fig. 7). According to this model, HBx transactivates transcription from all four viral promoters (C, pre-S1, S, and X promoters) at the early stage of infection. When the viral proteins reach a level sufficient for viral replication, HBc stimulates the degradation of HBx. As a result, further trans-activation of the viral promoters by HBx ceases. One example analogous to the proposed relationship between HBx and HBc is provided by p53 and Mdm2. The short-lived p53 activates the expression of Mdm2, which in turn promotes the rapid degradation of p53 through enhanced proteasome-dependent proteolysis (14, 21).
FIG. 7.
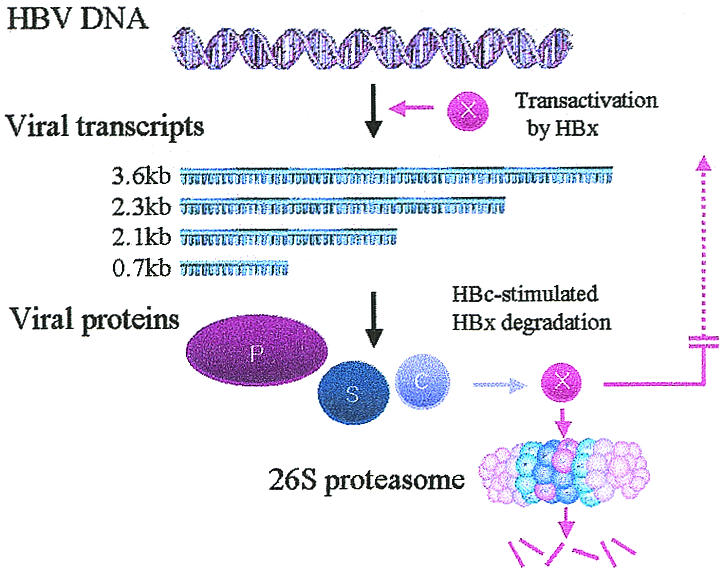
Schematic diagram of a feedback inhibition model. During the early stage of infection, HBx transactivates the transcription of the four major transcripts through EnI and EnII. When the viral proteins reach a threshold levels, HBc stimulates proteasome-mediated HBx degradation.
Using a replication-competent, longer-than-genome-length viral DNA, we attempted to confirm the attenuating effect of HBc on HBx in the context of viral replication. However, we were unable to detect HBx in this system, probably due to the low level of expression and limitation in the analysis of the untagged HBx. The use of the HA-tagged HBx construct along with the replication-competent viral DNA also did not show any effect on the HBx level (data not shown). We believe that the negative result was due to the low level of HBc expressed from the viral DNA, which may not have been sufficient to have any significant effect on HBx provided in trans and in quantity. In fact, in the immunoblot we could not detect any HBc protein expressed from the genomic construct, while in the control experiment HBc protein encoded in the cytomegalovirus promoter-based plasmid was clearly detected (and thus the reduction in HBx). This result is consistent with what we have shown repeatedly (Fig. 1B, 1D, 1E, 3B, and 3C), i.e., the effect of the core depends on the level of core protein and the ratio of the two proteins is important in this molecular interplay. We assume that in the real stage of viral replication, HBc exists in a much higher level than HBx, while in the experiment described above, this ratio was reversed.
Currently, we do not understand the mechanism by which HBc stimulates HBx degradation. Since the well-known proteasome substrate p53 was unaffected by HBc, we believe that HBc does not affect the total proteolytic activity of proteasomes. Coimmunoprecipitation and yeast two-hybrid experiments did not show any evidence of a direct interaction between HBc and HBx (data not shown). Thus, HBc may interact with an as yet unidentified HBx-specific ubiquitin ligase (E3) or with ancillary proteins involved in the proteolysis of HBx. Alternatively, HBc may counteract the effect of any HBx-stabilizing protein. One such target protein may be the DDB1 subunit of the UV-damaged DNA-binding protein complex, which was shown recently to bind and protect HBx and woodchuck hepatitis virus X protein from proteasome-mediated degradation (2).
Although we have shown in this study that the minor forms of HBc can be as effective as full-size HBc in the inhibition of HBx, there is no evidence that these proteins exist under physiological conditions. Rather, these results suggest that the effective domain of HBc resides at its carboxyl terminus. The effect of minor forms is intriguing, however, as our results show that a very small amount of the minor forms has an effect similar to that of full-length HBc. One postulation is that the effect of HBc is mediated by another protein, as discussed above, and if such a protein is present in limited amounts, the inhibitory effect of HBc may not be directly proportional to its amount.
The importance of proteasome activity on the regulation of HBV replication has also recently been addressed in a study on the antiviral activity of interferons (27). This study showed that proteasome activity is required for the cytokines to effectively inhibit HBV replication. Although a number of events could explain the proteasome dependence of the interferon-mediated inhibition of HBV replication, one possible mechanism is that interferon stimulates proteolytic degradation of a key regulatory protein, such as HBx, which is critical for viral replication. It remains to be seen whether interferon affects the enhanced degradation of HBx by HBc.
Chronic HBV infection is a major cofactor in the development of hepatocellular carcinoma, and chronic HBV carriers have a risk of developing liver cancer that is 100 times greater than normal. Several mechanisms of HBV-related tumorigenesis have been proposed, including viral genome integration, gene mutation, transcriptional activation of growth-regulatory genes by viral proteins, and the effects of viral proteins on cell apoptosis, cell signaling, and DNA repair (1). HBV DNA integration into the genome of hepatocytes has been detected in a high portion of HBV-related hepatocellular carcinomas (4, 30, 34). Because the integration of the hepadnaviral DNA into the host genome is not an orderly event, intact and complete viral genomic DNA has seldom been demonstrated in the integrants; deletion of at least some viral sequences has usually been found, and no two HBV integrants described are the same. In this regard, our result also points to a possible mechanism of HBV-related hepatocellular carcinoma. Integration of an incomplete viral genome may result in deletion or truncation of the structural or regulatory sequence for HBc. In such a situation, HBc may not be expressed sufficiently. As a result, HBx would be maintained at levels far above the normal level, leading to cellular transformation. For example, the expression of HBx but not HBc was observed in a nude mouse tumor experimentally induced by HBV DNA (15).
Proteasomes degrade ubiquitinated proteins. Nevertheless, examples of ubiquitin-independent proteolysis by the 26S proteasome have also been reported. One such example was provided by ornithine decarboxylase, the rate-limiting enzyme in the polyamine synthesis pathway (24). When ornithine decarboxylase binds to a second protein called the antizyme, it becomes a good substrate for proteasomes. HsN3 has also been shown to be involved in targeting of the p105 NF-κB subunit for proteasome processing (28). Furthermore, both ubiquitin-dependent and ubiquitin-independent proteasome pathways have been suggested in the degradation of Smad1 (12). Although HBx has been shown to be efficiently ubiquitinated, the possibility of a ubiquitin-independent pathway in HBx degradation and on HBc stimulation, through a member of such a pathway, cannot be excluded.
In conclusion, our study establishes a novel aspect of HBc function in the HBV life cycle and possibly in the development of liver cancer through the regulation of HBx levels. Future study will be directed toward identifying the molecular mechanism and understanding its implications.
Acknowledgments
We are grateful to Benedict Yen for helpful discussions and critical reading of the manuscript.
This work was supported by a Basic Science Research Fund from the Korean Research Foundation. J.-H. Kim was supported by a BK21 scholarship.
REFERENCES
- 1.Arbutohnot, P., and M. Kew. 2001. Hepatitis B virus and hepatocellular carcinoma. Int. J. Exp. Pathol. 82:77-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bergametti, F., D. Sitterlin, and C. Transy. 2002. Turnover of hepatitis B virus X protein is regulated by damaged DNA-binding complex. J. Virol. 76:6495-6501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bock, C. T., N. P. Malek, H. L. Tillmann, M. P. Manns, and C. Trautwein. 2000. The enhancer I core region contributes to the replication level of hepatitis B virus in vivo and in vitro. J. Virol. 74:2193-2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brechot, C., M. Hadchouel, and J. Scotto. 1981. State of hepatitis B virus DNA in hepatocytes of patients with hepatitis B surface antigen-positive and negative liver disease. Proc. Natl. Acad. Sci. USA 78:3906-3910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen, H.-S., S. Kaneko, and R. Girones, R. W. Anderson, W. E. Hornbuckle, B. C. Tennant, P. J. Cote, J. L. Gerin, R. H. Purcell, and R. H. Miller. 1993. The woodchuck hepatitis virus X gene is important for establishment of virus infection in woodchucks. J. Virol. 67:1218-1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Choi, B.-H., G.-T. Park, and H.-M. Rho. 1999. Interaction of hepatitis B viral X protein and CCAAT/enhancer binding protein á synergistically activates the hepatitis B viral enhancer II/pregenomic promoter. J. Biol. Chem. 274:2858-2865. [DOI] [PubMed] [Google Scholar]
- 7.Colgrove, R., G. Simon, and D. Ganem. 1989. Transcriptional activation of homologous and heterologous genes by the hepatitis B virus X gene product in cells permissive for viral replication. J. Virol. 63:4019-4026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.DeMartino, G. N., and C. A. Slaughter. 1999. The proteasome, a novel protease regulated by multiple mechanisms. J. Biol. Chem. 274:22123-22126. [DOI] [PubMed] [Google Scholar]
- 9.Dikstein, R., O. Faktor, R. Ben-Levy, and Y. Shaul. 1990. Functional organization of the hepatitis B virus enhancer. Mol. Cell. Biol. 10:3683-3689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fischer, M., L. Runkel, and H. Schaller. 1995. HBx protein of hepatitis B virus interacts with the C-terminal portion of a novel human proteasome alpha-subunit. Virus Genes 10:99-102. [DOI] [PubMed] [Google Scholar]
- 11.Fuerst, T. R., E. G. Niles, F. W. Studier, and B. Moss. 1986. Eukaryotic transient-expression system based on recombinant vaccinia virus virus that synthesizes bacteriophage T7 RNA polymerase. Proc. Natl. Acad. Sci. USA 83:8122-8126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gruendler, C., Y. Lin, J. Farley, and T. Wang. 2001. Proteasomal degradation of Smad1 induced by bone morphogenetic proteins. J. Biol. Chem. 276:46533-46543. [DOI] [PubMed] [Google Scholar]
- 13.Guo, W., K. D. Bell, and J. H. Ou. 1991. Characterization of the hepatitis B virus EnhI enhancer and X promoter complex. J. Virol. 65:6686-6692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haupt, Y., R. Maya, A. Kazaz, and M. Oren. 1997. Mdm2 promotes the rapid degradation of p53. Nature 387:296-299. [DOI] [PubMed] [Google Scholar]
- 15.Hohne, M., S. Schaefer, M. Seifer, M. A. Feitelson, D. Paul, and W. H. Gerlich. 1990. Malignant transformation of immortalized transgenic hepatocytes after transfection with hepatitis B virus DNA. EMBO J. 9:1137-1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hu, Z., Z. Zhang, E. Doo, O. Coux, A. L. Goldberg, and T. J. Liang. 1999. Hepatitis B virus X protein is both a substrate and a potential inhibitor of the proteasome complex. J. Virol. 73:7231-7240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang, J., J. Kwong, E. C. Sun, and T. J. Liang. 1996. Proteasome complex as a potential cellular target of hepatitis B virus X protein. J. Virol. 70:5582-5591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim, C. M., K. Koike, I. Sato, T. Miyamura, and G. Jay. 1991. HBx gene of hepatitis B virus induces liver cancer in transgenic mice. Nature 351:317-320. [DOI] [PubMed] [Google Scholar]
- 19.Koike, K., K. Moriyama, and S. Iino. 1994. High-level expression of hepatitis B virus HBx gene and hepatocarcinogenesis in transgenic mice. Hepatology 19:810-819. [PubMed] [Google Scholar]
- 20.Koike, K. 1995. Hepatitis B virus HBx gene and hepatocarcinogenesis. Intervirology 38:134-142. [DOI] [PubMed] [Google Scholar]
- 21.Kubbutat, M. H., S. N. Jones, and K. H. Vousden. 1997. Regulation of p53 stability by Mdm2. Nature 387:299-303. [DOI] [PubMed] [Google Scholar]
- 22.Mandart, E., A. Kay, and F. Galibert. 1984. Nucleotide sequence of a cloned duck hepatitis B virus genome: comparison with woodchuck and human hepatitis B virus sequences. J. Virol. 49:782-792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Murakami, S. 1999. Hepatitis B virus X protein: structure, function and biology. Intervirology 42:81-99. [DOI] [PubMed] [Google Scholar]
- 24.Murakami, Y., S. Matsufuji, T. Kameji, S. Hayashi, K. Igarashi, T. Tamura, K. Tanaka, and A. Ichihara. 1992. Ornithine decarboxylase is degraded by the 26S proteasome without ubiquitination. Nature 360:597-599. [DOI] [PubMed] [Google Scholar]
- 25.Patel, A. H., D. F. Gaffney, J. H. Subak-Sharpe, and M. D. Stow. 1990. DNA sequence of the gene encoding a major secreted protein of vaccinia virus virus, strain Lister. J. Gen. Virol. 71:2013-2021. [DOI] [PubMed] [Google Scholar]
- 26.Reifenberg, K., H. Wilts, J. Lohler, P. Nusser, R. Hanano, L. G. Guidotti, F. V. Chisari, and H.-J. Schlicht. 1999. The hepatitis B virus X protein transactivates viral core gene expression in vivo. J. Virol. 73:10399-10405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Robek, M. D., S. F. Wieland, and F. V. Chisari. 2002. Inhibition of hepatitis B virus replication by interferon requires proteasome activity. J. Virol. 76:3570-3574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rousset, R., C. Desbois, F. Bantignies, and P. Jalinot. 1996. Effects on NF-κB1/p105 processing of the interaction between the HTLV-1 transactivator Tax and the proteasome. Nature 381:328-331. [DOI] [PubMed] [Google Scholar]
- 29.Schek, N., R. Bartenschlager, C. Kuhn, and H. Schaller. 1991. Phosphorylation and rapid turnover of hepatitis B virus X-protein expressed in HepG2 cells from a recombinant vaccinia virus virus. Oncogene 6:1735-1744. [PubMed] [Google Scholar]
- 30.Shafritz, D. A., and M. C. Kew. 1981. Identification of integrated hepatitis B virus DNA sequences in human hepatocellular carcinomas. Hepatology 1:1-8. [DOI] [PubMed] [Google Scholar]
- 31.Sirma, H., R. Weil, O. Rosmordue, S. Urban, A. Israel, D. Kremsdorf, and C. Brechot. 1998. Cytosol is the prime compartment of hepatitis B virus X protein where it colocalizes with the proteasome. Oncogene 16:2051-2063. [DOI] [PubMed] [Google Scholar]
- 32.Spandau, D. F., and C. Lee. 1988. Trans-activation of viral enhancers by the hepatitis B virus X protein. J. Virol. 62:427-434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Su, H., and J. K. Yee. 1992. Regulation of the hepatitis B virus gene expression by its two enhancers. Proc. Natl. Acad. Sci. USA 89:2708-2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Takada, S., Y. Gotoh, S. Hayashi, M. Yoshida, and K. Koike. 1990. Structural rearrangement of integrated hepatitis B virus DNA as well as cellular flanking DNA is present in chronically infected hepatic tissues. J. Virol. 64:822-828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Twu, J.-S., and R. H. Schloemer. 1987. Transcriptional trans-activating function of hepatitis B virus. J. Virol. 61:3448-3453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ullrich, S. J., Z. Z. Zeng, and G. Jay. 1994. Transgenic mouse models of human gastric and hepatic carcinomas. Semin. Cancer Biol. 5:61-68. [PubMed] [Google Scholar]
- 37.Weir, J. P. 2001. Regulation of herpes simplex virus gene expression. Gene 271:117-130. [DOI] [PubMed] [Google Scholar]
- 38.Wu, H. L., P. J. Chen, M. H. Lin, and D. S. Chen. 1991. Temporal aspects of major viral transcript expression in Hep G2 cells transfected with cloned hepatitis B virus DNA: with emphasis on the X transcript. Virology 185:644-651. [DOI] [PubMed] [Google Scholar]
- 39.Xu, Z., T. S. B. Yen, L. Wu, C. R. Madden, W. Tan, B. L. Slagle, and J. H. Ou. 2002. Enhancement of hepatitis B virus replication by its X protein in transgenic mice. J. Virol. 76:2579-2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yen, T. S. B. 1996. Hepadnaviral X protein: review of recent progress. J. Biomed. Sci. 3:20-30. [DOI] [PubMed] [Google Scholar]
- 41.Yen, T. S. B. 1993. Regulation of hepatitis B virus gene expression. Semin. Virol. 4:33-42. [Google Scholar]
- 42.Zoulim, F., J. Saputelli, and C. Seeger. 1994. Woodchuck hepatitis virus X protein is required for viral infection in vivo. J. Virol. 68:2026-2030. [DOI] [PMC free article] [PubMed] [Google Scholar]