

Abstract
Intranasal infection of mice with murine gammaherpesvirus 68 (MHV-68), a virus genetically related to the human pathogen Kaposi's sarcoma-associated herpesvirus, results in a persistent, latent infection in the spleen and other lymphoid organs. Here, we have determined the frequency of virus infection in splenic dendritic cells, macrophages, and several B-cell subpopulations, and we quantified cell type-dependent virus transcription patterns. The frequencies of virus genome positive cells were maximal at 14 days postinfection in all splenic cell populations analyzed. Marginal zone and germinal center B cells harbored the highest frequency of infection and the former population accounted for approximately half the total number of infected B cells. Analysis of virus transcription during the establishment of latency revealed that virus gene expression in B cells was restricted and dependent on the differentiation stage of the B cell. Notably, transcription of ORF73 was detected in germinal center B cells, a finding in agreement with the predicted latent genome maintenance function of ORF73 in dividing cells. At late times after infection, virus DNA could only be detected in newly formed and germinal center B cells, which suggests that B cells play a critical role in facilitating life-long latency.
Epstein-Barr virus (EBV) and Kaposi's sarcoma-associated herpesvirus (KSHV) are human gammaherpesviruses, belonging to the gamma-1 and gamma-2 subgroups, respectively, which establish latent infections in B lymphocytes. In the case of EBV, persistence is established in memory B cells after infection of naive B cells (1, 2, 27). Proliferation and associated expansion of latently infected cells culminates with the generation of a persistently infected memory B-cell pool (2). In contrast to EBV, comparatively little is known about the natural sequence of events during KSHV infection or the phenotype of B cells during long-term latent infection.
In order to elucidate the molecular mechanisms governing the establishment of latent infection in B cells by gamma-2-herpesviruses, we have used a gammaherpesvirus, designated murine herpesvirus 68 (MHV-68), since its pathogenesis can be readily investigated in the laboratory mouse (12, 33a, 36). Intranasal inoculation of mice with MHV-68 results in an acute self-limiting infection of lung epithelial cells, which is followed by a persistent, latent infection of lungs (41) and lymphoid organs (42). Within the spleen, the levels of latent virus are maximal around 14 days postinfection (p.i.), decline quickly thereafter, and remain stable for life (8, 42). Spread of infection to the spleen generates an antigen-nonspecific, T-cell-dependent B-cell activation (32, 38) and an infectious mononucleosis-like disease similar to that observed in adolescent EBV infection (13, 43).
Previous studies have shown that MHV-68 infection of the spleen results in the establishment of latency in B lymphocytes, macrophages, and dendritic cells (14, 42, 56). Within B cells latency is preferentially associated with germinal-center (GC) cells (14), and this finding is in agreement with previous reports showing abundant virus-encoded tRNA-like (vtRNA) transcripts in GCs (6, 34). The pivotal role that B cells play in latency is confirmed by the inability after intranasal inoculation to establish splenic latency in mice deficient in B cells (μMT) (46, 55) and their requirement for spreading of infection from the lung to the spleen (41). The combined observation of GC infection (14, 34), non-antigen-specific B-cell activation (38), and CD4-dependent increase in viral load after infection of mice with MHV-68 (32) may reflect an exploitation of the normal GC reaction by the virus.
Thus, the identification of viral genes expressed during the establishment of infection in GC B cells is central for an understanding of MHV-68 pathogenesis. During the establishment of latent infection in the spleen a restricted number of transcribed viral genes have been identified. These include M2, M3, K3, M8, and M9 (20, 30, 34, 44, 52). Of these putative latency-associated genes, only K3 and M2 have been formally shown to be necessary for the establishment of a normal latent load (21, 39). K3 downregulates major histocompatibility complex I surface expression via a proteosome-dependent mechanism (4, 18). No function has as yet been attributed to M2. With the exception of K3 (39), whether M2, M3, or any of the other putative latency-associated genes are expressed in GC B cells or in any of the other proposed latent sites, i.e., macrophages and dendritic cells, is still unknown.
In the present study, we have determined the frequency of infection in several cell types in the spleen, including total B cells, follicular (Fo) B cells, GC B cells, marginal-zone (MZ) B cells, newly formed (NF) B cells, macrophages, and dendritic cells, during the establishment of latency after intranasal challenge with MHV-68. For each cell type, cell-specific patterns of virus transcription were quantified at 14 days p.i. The virus open reading frames (ORFs) analyzed included unique genes and cellular homologues that are predicted to be involved in activities, such as immune evasion and latency.
MATERIALS AND METHODS
Cell lines, virus stocks, and infections.
NS0 and NIH 3T3 cell cultures were grown in Dulbecco modified Eagle medium containing 10% fetal calf serum (FCS). S11 cells (45) were cultured in RPMI medium plus 10% FCS. Baby hamster kidney cells (BHK-21) were cultured in Glasgow modified Eagle medium supplemented with 10% FCS and 10% tryptose phosphate. Virus working stocks were prepared by a low multiplicity of infection (MOI; 0.001 PFU/cell) of BHK-21 cells (33). Female BALB/c mice (Gulbenkian Institute of Science, Oeiras, Portugal), 6 to 8 weeks of age, were inoculated intranasally under the effect of light halothane anesthesia with 104 PFU of virus in 20 μl of phosphate-buffered saline (PBS). At different time points after infection, mice were killed by inhalation of CO2. Lungs or spleens were removed and kept at −80°C or immersed in PBS-2% FCS at 4°C for subsequent use, respectively.
Virus assays.
Infectious virus was plaque assayed on BHK-21 cells, and latent virus was assayed by explant coculture of fluorescence-activated cell-sorted (FACS) splenocytes with BHK-21 cells, incubated for 5 days, fixed with 10% formal saline, and counterstained with toluidine blue, and plaques were counted with a plate microscope (33). FACS-purified populations of splenocytes were also freeze-thawed and sonicated prior to plaque assay in order to determine the level of preformed infectious virus.
Purification of different splenocyte populations.
Single-cell suspensions of at least five pooled spleens were prepared per time point and passed trough a 100-μm (pore-size) filter to remove stromal debris. Cell suspensions were washed in PBS-2% FCS and red blood cells lysed in 154 mM ammonium chloride, 14 mM sodium hydrogen carbonate, and 1 mM EDTA (pH 7.3). For surface staining, single-cell suspensions were preincubated with 2.4G2 [anti-CD16/CD32 (FcγIII/II receptor), rat immunoglobulin G2b(κ) culture supernatant; Pharmingen] before staining with the following monoclonal antibodies (Pharmingen) and lectins (Vector Laboratories): anti-CD19, anti-CD11b, anti-CD11c, anti-CD21, anti-CD23, anti-B220 (CD45R), and peanut agglutinin (PNA). Using a MoFlo cytometer (Cytomation, Fort Collins, Colo.), the following enriched cell subpopulations were obtained: CD19+ for total B cells, B220+ CD21int CD23hi for Fo cells, B220+ CD21− CD23− for NF B cells, B220+ CD21hi CD23+ for MZ B cells, B220+ PNAhi for GC cells B cells, B220− CD11b+ CD11c− for macrophages, and B220− CD11c+ for dendritic cells. Sorted populations were analyzed in a FACScan flow cytometer, and the data were processed by using CellQuest software (Becton Dickinson Immunocytometry Systems, San Jose, Calif.). The purities of sorted populations were usually >95% and always >90%.
Real-time PCR.
Real-time PCR was performed by using a LightCycler from Roche Molecular Biochemicals (Mannheim, Germany) according to the manufacturer's instructions by using sequence-specific fluorescence detection oligonucleotide hybridization probes coupled to suitable fluorophores (see Table 1 for probe genome coordinates). Labeled probes were designed and supplied by TIB Biomol. PCR primers specific for several MHV-68 ORFs were used (see Table 1 for primer genome coordinates), and amplification was as follows: a melting step of 95°C for 10 min was followed by 45 cycles of 95°C for 10 s, 50 to 60°C (depending on the primer set utilized) for 10 s, and 72°C for 20 s, followed by a melting analysis step from 50 to 95°C at 0.1°C/s. In this system one oligonucleotide is directly coupled at the 3′ end with Fluorescin and is excited by an external light source provided by the LightCycler. It then passes on part of its excitation energy to the adjacent LC Red-640, which is directly conjugated to the 5′ end of another oligonucleotide. The excited LC Red-640 then emits measurable light. Because interaction of the two dyes can only occur when both oligonucleotides are hybridized to their target on a head-to-tail arrangement at a distance of one to five nucleotides, this system is highly specific.
TABLE 1.
Probe and primer genome coordinates
| Genea | Oligonucleotide coordinates (range)
|
|||
|---|---|---|---|---|
| Upper primer | FL probeb | LC probec | Lower primer | |
| M1 | 2596-2613 | 2657-2678 | 2629-2655 | 2902-2888 |
| M2d | 4212-4232 | 4518-4541 | 4544-4568 | 5840-5820 |
| M3 | 6167-6183 | 6336-6360 | 6308-6334 | 6642-6625 |
| M4 | 8745-8761 | 8811-8835 | 8784-8808 | 8942-8920 |
| ORF4 | 10333-10348 | 10566-10591 | 10593-10615 | 10616-10632 |
| ORF6 | 11246-11263 | 11334-11360 | 11363-11386 | 11397-11417 |
| K3 | 24832-24850 | 24973-24998 | 25000-25026 | 25049-25071 |
| ORF50 | 68514-68531 | 68836-68860 | 68862-68886 | 68956-68976 |
| M7 | 69754-69776 | 70087-70116 | 70118-70150 | 70220-70240 |
| M8e | 76065-76083 | 76139-76164 | 76166-76189 | 76241-76261 |
| M9f | 94055-94071 | 94105-94126 | 94081-94103 | 94250-94271 |
| ORF72 | 102626-102641 | 102818-102840 | 102790-102815 | 103049-103065 |
| M11 | 103418-103429 | 103833-103858 | 103861-103886 | 103908-103929 |
| ORF73 | 104026-104045 | 104256-104229 | 104228-104254 | 104425-104444 |
| ORF74 | 105360-105379 | 105418-105442 | 105392-105417 | 105933-105956 |
| Hprtg | 15263734-15263752 | 15264732-15264756 | 15264758-15264782 | 15264802-15264820 |
According to GenBank accession no. U97553.
Oligonucleotide labeled at the 3′ end with Fluorescin (Roche).
Oligonucleotide labeled at the 5′ end with LC-Red fluorophore and modified at the 3′ end by phosphorylation (Roche).
Spanning the intron from coordinates 4610 to 5814 (20).
According to Mackett et al. (25), M8 forms a 5′ exon in a spliced transcript with ORF57 to generate an immediate-early transcript homologue to the EBV-M transactivator.
M9 has been reclassified as ORF65 by its homology with its counterpart in HVS.
According to GenBank accession no. NW042621, spanning the 15263777 to 15263948 and 15264025 to 15264691 introns.
For every PCR performed in our study, homologous standard curves were determined and were always within the concentration range of the target sample. Of importance, the target sample had kinetics of amplification identical to those of the standard amplification reactions, i.e., the slope of the target sample at each cross point, over a large range, was the same as for the standard samples. Different primer sets had, however, slightly different amplification kinetics, but they were all capable of detecting one copy of plasmid template and reproducibly detected 10 copies.
Limiting-dilution analysis.
FACS-purified single-cell suspensions were serially diluted 1.5-, 2-, or 3-fold, and 8 to 12 replicates of each cell dilution were lysed overnight (0.45% Tween 20, 0.45% NP-40, 2 mM MgCl2, 50 mM KCl, 10 mM Tris [pH 8.3], 0.5 mg of proteinase K/ml) at 37°C. Proteinase K was then inactivated (5 min at 95°C), and the samples were analyzed by real-time PCR (as described above), with primer-probe sets specific for K3 in a final volume of 10 μl per PCR (2 mM MgCl2, 4 ng of each primer/μl, a 0.02 mM concentration of each internal probe, a 1× DNA mix [Roche], and 1 μl of cell lysate). Although different primer sets used in the present study had slightly different amplification kinetics, they all amplified viral genomes with equivalent sensitivity, i.e., S11 cells (harboring ca. 40 viral genomes per cell) were detected at a frequency of 1:1 × 105 in an uninfected control population. Therefore, any primer-probe combination could have been used for the detection of viral genomic DNA. The K3 genomic region was chosen because its primer set had rapid amplification kinetics. Our data were compatible with the single-hit Poisson model (SHPM) as tested by modeling the limiting dilution data according to a generalized linear log-log model fitting the SHPM and checking this model by an appropriate slope test as described by Bonnefoix et al. (5). A regression plot of input cell number against log fraction-negative samples was used to estimate the frequency of cells with viral genomes. Estimation of the cell subset frequency of MHV-68 infection consisted of computation by maximal-likelihood estimation as follows: let f be the estimate of the cell frequency; the maximum likelihood of f is the value of f that maximizes
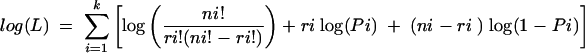 |
where log(L) is the natural logarithm of the likelihood function L and Pi is given by Pi = exp(−f xi) according to the SHPM. The variance of f was calculated as the negative reciprocal of the second derivative of log(L), var(f ) = −1/[d2 log(L)/df 2]. The 95% confidence interval (CI) for f was calculated as 95% CI (f) = f ± 1.96SE (f). Abbreviations are as follows: k = the number of groups of replicate PCRs, numbered i = 1, 2,… k; ni = the number of replicate reactions; ri = the number of observed negative PCRs; and mi = the observed fraction of negatives (mi = ri/ni).
Reverse transcription-PCR (RT-PCR) analysis of virus transcription.
RNA was isolated from 106 to 3 × 106 splenocytes purified by sorting from pools of five spleens and NIH 3T3 cells by using the RNeasy Minikit with the RNase-free DNase set protocol (Qiagen) according to the manufacturer's instructions. RNA was also extracted from lung tissue by using Trizol as specified by the manufacturer (Gibco-BRL). For cDNA synthesis, 2 μg of RNA was incubated at 70°C for 10 min with 500 μg of pd(T)12-18-5′PO4 sodium salt in a total volume of 23 μl. Samples were then reverse transcribed in a total reaction volume of 40 μl containing 0.5 mM concentrations of each of deoxynucleoside triphosphate, 1× first-strand buffer (Gibco-BRL), 1 U of RNase OUT RNase inhibitor (Gibco-BRL), and 400 U of Superscript II reverse transcriptase (Gibco-BRL). Reactions were performed for 50 min at 37°C, followed by 5 min at 90°C. Viral cDNAs were quantified by using real-time PCR, as described above. For every PCR, only cDNA samples corresponding to a minimum of 10, 000 copies of the housekeeping gene Hprt were utilized. Primer and probe genome coordinates are listed in Table 1. Tenfold serial dilutions of plasmid template spiked into total splenic cDNA equivalent to a minimum of 10,000 copies of Hprt were used to establish, for each gene, a linear relationship between the input template copy number, and the cycle number at which an arbitrary fluorescence threshold was crossed. This gave the relative quantity of each viral transcript in each sample. The signal in RT-negative controls, indicative of residual viral DNA, was subtracted from the total to give a cDNA-specific signal. In the present study, only RT-PCRs yielding 10 or more copies of viral transcript per RT-PCR were considered.
RESULTS
MHV-68 persistence in GC and NF B cells.
The frequency of virus genome-positive cells in different splenocyte populations during the establishment and maintenance of latent infection was determined by limiting dilution analyses and real-time PCR (Fig. 1 and 2 and Table 2). During the establishment of latency at 14 days p.i., virus DNA was detected in all populations of cells analyzed. For all cell populations, levels of virus genome positive cells were maximal at 14 days p.i., with MZ B cells and GC B cells harboring the highest frequency of infection. In MZ B cells and GC B cells, the estimated frequency of virus genome-positive cells (1 infected cell per 63 cells and 1 infected cell per 89 cells, respectively) was at least 2 to 3 orders of magnitude higher than in Fo B cells, dendritic cells, and macrophages. This result showed that B cells constitute the principal target for virus infection during the establishment of latency. Furthermore, within B cells, GC B cells accounted for the majority of infected B cells in the spleen.
FIG. 1.

Splenocyte populations analyzed in this study. After intranasal infection of BALB/c mice with 10,000 PFU. MHV-68, single-cell suspensions from pools of at least five spleens were stained with the following combination of conjugated antibodies and lectins: anti-CD19 (A); anti-B220 and PNA (B); anti-B220, -CD21, and -CD23 (C); and anti-B220, -CD11b, and -CD11c (D). Cells were then purified by using a MoFlo cytometer to obtain the following enriched cell subpopulations: CD19+ for total B cells, B220+ CD21− CD23− for NF B cells, B220+ CD21int CD23hi for Fo B cells, B220+ CD21hi CD23+ for MZ B cells, B220+ PNAhi for GC B cells, B220− CD11b+ CD11c− for macrophages, and B220− CD11c+ for dendritic cells. The purity of sorted populations was analyzed by FACS and was always >90% and usually >95%.
FIG.2.

Graphical representation of limiting-dilution data estimating the frequency of MHV-68 genome-positive cells in different splenocyte populations. Real-time PCR was performed on limiting dilutions of purified splenocytes and frequencies of infection were determined by limiting-dilution analysis. (A to C) Total B cells at 14, 21, and 153 days p.i., respectively; (D to H) GC B cells at 14, 21, 80, 122, and 253 days p.i., respectively; (I to K) Fo B cells at 14, 21, and 112 days p.i., respectively; (L to N) NF B cells at 14, 21, and 112 days p.i., respectively; (O to Q) MZ B cells at 14, 21, and 112 days p.i., respectively; (R to T) dendritic cells at 14, 21, and 70 days p.i., respectively; (U to X) macrophages at 14, 21, 70, and 147 days p.i., respectively. Data are plotted as the log of the fraction of negative PCRs as a function of cells per reaction (○). The prediction equation of the regression line (plain line) is −log(μi) = f xi, where f is the cell frequency estimated by the maximum-likelihood method according to the SHPM hypothesis. Upper and lower dotted lines are plotted by using upper and lower values of the 95% CI of f. When all PCRs were negative to extrapolate the maximum value of the cell frequency f in the assay, the log of (ni − 1/ni, where ni is the number of replicate reactions) is plotted as a function of the maximum number of cells analyzed (cross) and the prediction inequation is −log(μi) > f xi (shaded area).
TABLE 2.
Frequency of MHV-68 infection in splenocyte populationsa
| Cell subpopulation | Time (days) p.i. | Reciprocal frequencyb of viral DNA-positive cells (95% CI) | % Purityc | % Cellsd | Tot no. of cellse | No. of viral DNA-positive cellsf |
|---|---|---|---|---|---|---|
| Total B cells | 14 | 311 (117-1237) | 99 | 53.2 | 1.1 × 108 | 353,370 |
| 21 | 14,663 (8,598-49767) | 98 | 50.1 | 1.0 × 108 | 6,820 | |
| 153 | >741,979 | 99 | 51.6 | 1.0 × 108 | <135 | |
| GC B cells | 14 | 89 (57-208) | 96 | 5.5 | 1.1 × 107 | 123,596 |
| 21 | 880 (563-2,015) | 98 | 4.5 | 9.0 × 106 | 10,227 | |
| 80 | 4,054 (2,535-10,117) | 97 | 2.5 | 5.0 × 106 | 1,233 | |
| 122 | 28,495 (18,241-65,083) | 97 | 2.4 | 4.8 × 106 | 168 | |
| 235 | 24,551 (15,790-55,163) | 98 | 2.0 | 4.0 × 106 | 163 | |
| Fo B cells | 14 | 9,198 (5,593-25,854) | 99 | 26.5 | 5.2 × 107 | 5653 |
| 21 | 104,144 (67,169-231,685) | 99 | 27.0 | 5.4 × 107 | 519 | |
| 112 | >3,977,009 | 99 | 25.0 | 5.0 × 107 | <12 | |
| NF B cells | 14 | 783 (465-2,475) | 96 | 5.0 | 1.0 × 107 | 12,771 |
| 21 | 11,020 (6,741-30,176) | 95 | 6.0 | 1.2 × 107 | 1,089 | |
| 112 | 11,000 (6,839-28,097) | 98 | 5.5 | 1.2 × 107 | 1,090 | |
| MZ B cells | 14 | 63 (40-140) | 91 | 1.5 | 3.0 × 106 | 47,619 |
| 21 | 2,421 (1,507-6,154) | 93 | 1.0 | 2.0 × 106 | 826 | |
| 112 | >411,874 | 92 | 1.1 | 2.2 × 106 | <5 | |
| Dendritic cells | 14 | 4,054 (2,535-10,118) | 91 | 0.8 | 1.6 × 106 | 395 |
| 21 | >119,072 | 98 | 0.6 | 1.2 × 106 | <10 | |
| 70 | >671,792 | 92 | 0.7 | 1.4 × 106 | <2 | |
| Macrophages | 14 | 9,947 (6,276-23,960) | 95 | 7.0 | 1.4 × 107 | 1,407 |
| 21 | 10,037 (6,203-26,276) | 99 | 6.1 | 1.2 × 107 | 1,196 | |
| 70 | >1,117,137 | 97 | 6.6 | 1.3 × 107 | <12 | |
| 147 | >1,117,137 | 98 | 6.5 | 1.3 × 107 | <12 |
Data were obtained from pools of at least five spleens.
Frequencies of infection were calculated by limiting-dilution analysis with the 95% CI as shown.
That is, the purity of sorted cells as determined by FACS analysis.
That is, the percentage of each population of total spleen was determined by FACS analysis.
The total numbers of cells were estimated from the percentage of the total spleen, based on an estimate of 2 × 108 cells/spleen.
The number of latently infected cells was based on the frequency of latency within each cell type and the estimated total number of cells.
After 14 days p.i. the frequency of virus genome-positive cells decreased sharply in all cell populations analyzed. In GC B cells the frequency of infection fell to 1 infected cell per 880 cells at 21 days p.i. to reach stable but low levels at late times after infection, i.e., on average 1 infected cell per 26,000 cells at 122 and 235 days p.i. In NF B cells stable levels of virus persistence were also reached but at a higher frequency than GC B cells of 1 infected cell per 11,000 cells. In contrast, to GC and NF B cells, no virus DNA could be detected in dendritic cells beyond 14 days p.i. and in Fo B cells, MZ B cells, total B cells, and macrophages beyond 21 days p.i.
Absence of productive infection during the establishment of infection in spleen.
In order to determine the nature of the infection within different splenic cell populations, plaque assays and infectious center assays were performed to detect the presence of preformed infectious virus or latent virus, respectively (Table 3). The time point chosen was 14 days p.i., which corresponded to the time after infection when the frequencies of infection were at maximal levels. None of the populations analyzed, contained detectable preformed infectious virus. In contrast, the presence of latent virus could be readily shown in all populations. Total B cells and macrophages harbored the highest levels of reactivation-competent latent virus. By comparing the frequencies of viral DNA-positive cells with levels of infectious centers, it was possible to estimate the efficiency of reactivation within a cell population. By performing this analysis it was immediately apparent that the efficiency of virus reactivation within macrophages and dendritic cells was 2 to 3 orders of magnitude higher compared to total B cells and GC B cells, respectively. When the same analysis was performed between total B cells and GC B cells, it became apparent that, although GC B cells accounted for majority of the total number of infected B cells, they contributed <5% of infectious centers observed for total B cells.
TABLE 3.
Quantification of reactivation-competent virus and preformed infectious virus in splenic populations from pools of five spleens at 14 days p.i.
| Cell type | Virus (PFU) as determined by:
|
Reactivation efficiencyc | |
|---|---|---|---|
| PAa | ICAb | ||
| Total spleen cells | <44 | 5,000 | NDd |
| Total B cells | <100 | 3,300 | 0.0093 |
| GC B cells | <13 | 110 | 0.0009 |
| Dendritic cells | <5 | 80 | 0.2025 |
| Macrophages | <14 | 980 | 0.6965 |
PA, plaque assay. Preformed infectious virus was not detected, and the values indicate the limit of detection, taking into account the maximal number of cells used in the assay and based on a estimate of 2 × 108 cells/spleen and on the percentage of each population of total spleen as determined by FACS analysis.
The infectious center assay (ICA) gives the number of infectious centers per total spleen based on a estimate of 2 × 108 cells/spleen and on the percentage of each population of total spleen as determined by FACS analysis.
The reactivation efficiency was estimated from the ratio of the number of infectious centers to the number of viral DNA+ cells.
ND, not done.
Analysis of virus transcription during lytic infection.
The virus ORFs analyzed are listed in Table 1 and include unique genes and cellular homologues that are predicted to encode particular biological functions, e.g., immune evasion, latency, and disease. To control for patterns of lytic transcription, we have included in our analysis ORF50, an immediate-early gene, which has been shown to play a central role both in initiation of lytic replication and reactivation from latency (25, 57, 58), and ORF6 and M7, which encode for an early single-stranded DNA-binding protein (51) and a late structural glycoprotein (40), respectively. The relative quantity of each viral transcript was normalized to the housekeeping gene Hprt.
Virus transcription was first analyzed in RNA extracted from NIH 3T3 cells infected with MHV-68 at an MOI of 5 PFU/cell and harvested at 4, 8, and 12 h p.i. (Fig. 3). All ORFs tested were transcribed during lytic replication in NIH 3T3 cells. The most abundant transcripts were those corresponding to M3, M9, and ORF6. Consistent with being immediate-early genes, transcripts to ORF50, M8/57, and ORF73 were readily detectable at 4 h p.i. Likewise, M7 transcripts could only be detected from 8 h p.i. in agreement with the known late kinetics of this gene (30).
FIG. 3.

MHV-68 lytic cycle transcription. RNA extracted from NIH 3T3 cells infected with MHV-68 at an MOI of 5 PFU per cell and harvested at 4, 8, and 12 h p.i. (A) and from lungs obtained from three individual mice at 14 days p.i. (B) was reverse transcribed and analyzed by real-time PCR. The data are presented as the number of viral transcripts normalized for one copy of the housekeeping gene Hprt. For panel B, the geometric mean is presented, and the error bars represent the standard deviation of the mean.
We next analyzed virus transcription during lytic infection in lungs after intranasal infection. To this end, RNA was extracted from lung tissue from three indivual mice at 4 days p.i. As for infection in NIH 3T3 cells, all virus ORFs analyzed were transcribed during lytic infection in lung tissue, with M3, M9, and ORF6 transcripts being the most abundant.
MHV-68 transcription during the establishment of latent infection is cell type dependent.
In order to determine cell type-specific patterns of virus transcription in vivo during the establishment of latency in the spleen at 14 days p.i., two independent animal infections were established, and RNA was extracted from FACS-purified populations of different splenocyte populations from pools of five spleens. For each RNA sample, two independent cDNAs were derived, and virus transcripts were quantified by real-time PCR. Given that distinct splenocyte populations presented different frequencies of infection, data were expressed as the number of transcripts per one copy of Hprt per infected cell (Fig. 4).
FIG. 4.

MHV-68 transcription during the establishment of latency is cell type specific. RNA extracted from FACS-purified macrophages with a purity of 97.7% and no contamination from dendritic cells (A), dendritic cells with a purity of 96.4% and a contamination fraction of 0.13% of macrophages (B), Fo B cells with a purity of 99.3% and a contamination fraction of 0.08% MZ B cells and 0.13% NF B cells (C), GC B cells with a purity of 98.5% and contamination fraction of 1.25% B220− cells (D), NF B cells with a purity of 95.7% and a contamination fraction of 0.06% MZ B cells and 0.79% NF B cells (E), and MZ B cells with a purity of 93.3% and a contamination fraction of 1.89% Fo B cells and 0.23% NF (F) was subjected to RT and quantified by real-time PCR. Four cDNAs were analyzed for each cell population, corresponding to RNA extractions from two independent pools of five spleens obtained from two separate animal infections that were subjected to two independent cDNA syntheses. Data are presented as the geometric mean of the number of viral transcripts per infected cell normalized for one copy of the housekeeping gene Hprt. The number of transcripts per infected cell was determined, taking into account the frequency of infected cells in each cell population. The error bars represent the standard deviation of the mean.
Virus transcription was first quantified in macrophages and dendritic cells. With the exception of M2, M7, ORF4, ORF73, and ORF74 in macrophages and ORF4 in dendritic cells, all of the ORFs analyzed were transcribed in these two cell types. The fact that transcripts corresponding to ORF6, ORF50, and M7, the latter only in dendritic cells, were detected was consistent with a pattern of lytic transcription although, overall, the levels of transcripts were very low. However, in macrophages no transcripts to the immediate-early gene ORF73 could be detected, which would argue against a pattern of lytic infection.
In contrast to macrophages and dendritic cells, in all of the different B-cell subpopulalions analyzed no transcripts corresponding to the lytic cycle genes ORF50 and M7 could be detected (Fig. 4). This result is consistent with an overall transcription pattern of latent infection. The exception was ORF6, which is predicted to be a dedicated lytic cycle gene and was transcribed in NF, MZ, and GC B cells. Significantly, ORF73 specific transcripts were detected exclusively in GC and MZ B cells. It is also worthwhile noting that from all of the ORFs considered in the present study, transcripts corresponding to ORF4 were not detected in any of the populations analyzed.
One issue that arises from the RT-PCR analysis is whether the data obtained for Fo, NF, and MZ B cells are representative of each subset or are a consequence of a degree of contamination between these populations. This is particularly relevant in the case of Fo B cells in which at 14 days p.i. the frequency of infection was 2 orders of magnitude lower than in MZ B cells. Thus, the purities of each sorted population and respective contaminant fractions were determined by FACS analysis (refer to Fig. 4 legend). In the case of Fo B cells, the purity was 99.3% with the contamination fractions of 0.08% MZ cells and 0.13% NF cells. Taking into account the frequencies of infection within each population, we estimated that there is a contamination of ca. 1 infected MZ or NF cell per 10 or 100 infected Fo cells, respectively. Comparison of the transcription profiles between these B-cell populations shows, however, that this level of contamination is not detectable by the RT-PCR methodology used. Whereas transcripts corresponding to ORF73 and M11 in MZ cells and M11 in NF cells were readily detectable, they were below the limit of detection in Fo cells.
We have also determined that ca. 50% of the B220+ PNAhi population fell within the gates defining the Fo B-cell population (B220+ CD21int CD23hi; data not shown). However, transcription for ORF73 could be readily detected in GC B cells whereas no ORF73 transcripts could be detected in Fo B cells. Thus, the pattern of transcription observed for GC B cells could not be attributed only to cells of Fo phenotype. Moreover, the GC transcription pattern observed either occurs in a subset of GC cells that does not overlap with the follicular population as defined by our gates, or else the overlap between the two is insufficient for ORF73 transcripts to be detectable.
DISCUSSION
In the present study we have analyzed the cellular sites of latent infection in different splenocyte subpopulations and respective cell-specific patterns of virus transcription after intranasal infection of BALB/c mice with MHV-68. Although the virus established infection in B cells, dendritic cells, and macrophages, by late after infection we observed latency in GC and NF B cells only. The frequencies of virus genome-positive cells were maximal at 14 days p.i. in all splenic populations analyzed, a time at which no preformed infectious virus could be detected. In contrast, reactivation-competent latent virus could be readily demonstrated, indicating that, overall, the majority of infected cells within each cell population analyzed were latently infected. MZ and GC B cells harbored the highest frequency of virus genome-positive cells, and the latter population accounted for approximately half of the total number of infected B cells in the spleen. This result is consistent with the fact that we had earlier shown an extensive amplification of latently infected cells within GCs during the establishment of latency in the spleen (6, 34). Thus, our results confirm those of Flano et al. (14), who demonstrated that, during the establishment of latency, infection in the spleen is associated with GC B cells, dendritic cells, and macrophages. In contrast to our results, however, Flano et al. (14) reported that dendritic cells harbored the highest frequency of latent virus. This apparent discrepancy may be explained by the fact that these authors measured the levels of reactivation-competent latent virus, whereas in the present study we determined the frequencies of virus genome-positive cells. Recently, Flano et al. (15) reported that during the establishment of latency, one in eight GC B cells harbored MHV-68 DNA. This frequency, although 10-fold higher than that one reported in the present study, is consistent with GC B cells being the main target of viral infection during the establishment phase of viral latency.
By comparing the frequencies of virus genome-positive cells with the levels of reactivation-competent latent virus, we have shown that macrophages and dendritic cells reactivate virus with an efficiency 2 to 3 orders of magnitude higher than B cells, namely, GC B cells. Thus, these data indicate that the efficiency of virus reactivation from latency is cell type dependent in a conventional explant coculture assay. It is important to note that at 14 days p.i. ORF50 transcription was detected in macrophages and dendritic cells but not in any of the B-cell subsets analyzed. Given the role played by this gene product in the induction of lytic cycle replication (25, 57, 58), it is reasonable to suggest that the higher reactivation efficiency observed in macrophages and dendritic cells could be attributed to expression of ORF50. On the other hand, expression of ORF50 was not sufficient to induce levels of productive replication detectable by our virus plaque assay. Indeed, in addition to ORF50, active transcription of ORF6 in macrophages and ORF6 and M7 in dendritic cells (which encode for an early single-stranded DNA-binding protein and a late structural glycoprotein, respectively) was consistent with an overall pattern of productive lytic replication. Thus, the different patterns of lytic transcription may reflect immune-mediated elimination of infected cells. The presence of ORF6 but the absence of M7 transcripts is consistent with the early elimination of macrophages. Dendritic cells would be more resistant to immune clearance, hence, the presence of M7 transcripts. Importantly, all lytically infected cells would be eliminated, hence, the absence of detectable preformed infectious virus. Alternatively, the absence of infectious virus may indicate that only a minority of cells within these populations is undergoing productive infection, which is beyond the limit of detection of our plaque assay. The pattern of virus transcription is, however, complex since in a context of lytic cycle, transcripts for ORF73, an immediate-early gene, were not detected in macrophages and ORF4 transcripts were not detected in either macrophages or dendritic cells.
In contrast to dendritic cells and macrophages, virus transcription in B cells was characterized by the absence of detectable transcripts to ORF50 and M7, indicating that, on average, the majority of the infected cells in the Fo, NF, MZ, and GC B-cell subsets were latently infected. This interpretation does not exclude the possibility that a small proportion of B cells are undergoing lytic infection.
Analyses of total RNA (20, 30, 39, 44, 52) and in situ hybridization studies (34) in spleens of mice infected with MHV-68 have demonstrated that, during the establishment of latency in the spleen, a restricted number of viral ORFs, including M2, M3, K3, M8, and M9, are transcribed. In the present study we confirmed and extended these results by quantifying virus transcription, at 14 days p.i. when the frequencies of infection were higher, in macrophages, dendritic cells, and several B-cell subsets, including Fo, NF, MZ, and GC cells.
Interestingly, the most abundant transcripts in macrophages and dendritic cells were those corresponding to M3. Thus, macrophages and dendritic cells are more likely to be a source of M3 in vivo than GC B cells. M3 encodes a secreted protein (49), which binds chemokines blocking chemokine signaling (22, 29, 47). The role of M3 in vivo during the establishment of latency remains controversial (7, 48). However, in one study recombinant viruses with disrupted M3 genes showed reduced levels of latent virus in lymphoid tissue, a phenotype that can could be partially reversed by CD8+-T-cell depletion (7). Therefore, it is possible that lytic infection of macrophages and dendritic cells may protect latent infection in B cells.
Virus transcription in B cells was clearly restricted to fewer genes and, interestingly, depended upon the differentiation stage of the B cell. ORF73 transcripts were detected in MZ and GC B cells but not in Fo B cells or NF B cells. This finding is consistent with the demonstration that the ORF73 gene product in KSHV (3, 11) and herpesvirus saimiri (HVS) (24, 35) has functions essential for virus episome maintenance in dividing cells. Therefore, we predict that ORF73 of MHV-68, like its counterparts in KSHV and HVS, encodes functions necessary for maintenance of virus infection in latently infected proliferating GC B cells. The significance of selective transcription of ORF73 in MZ B cells is not obvious, since they do not appear to be clonally expanded (9). In the case of M11, transcripts were detected in NF and MZ B cells but not in Fo or CG B cells. The sequence of M11 shows low homology to cellular bcl-2 (51) but, like its cellular homologue, has been shown to encode antiapoptotic functions (31, 53). Although our failure to detect M11 transcripts in both Fo and GC B cells may simply reflect the sensitivity limit of the technique we have used, M11 transcripts could be readily detected in both macrophages and dendritic cells. Significantly, in vivo studies of mice infected with a recombinant virus containing a disrupted M11 gene have shown that this gene is dispensable for efficient acute replication and the establishment of latency but is required for efficient ex vivo reactivation (17). Thus, our results confirm previous studies reporting M11 transcription during the establishment of latency in spleen (31, 52) and extend these observations by assigning transcription to macrophages, dendritic cells, and NF and MZ B cells.
Viral ORFs that were transcribed in all B-cell subsets analyzed include M1, M2, M3, M4, K3, M8, M9, and 72. Given that B cells accounted for the overwhelming majority of the virus genome-positive cells in spleen, it is predicted that these genes will encode functions necessary for the efficient establishment of latency. This has been shown for M2 (21), M3 (7), and K3 (39), although for M3 an independent study reported little if any effect in latency establishment (48). On the other hand, studies with M1-deficient recombinant viruses showed an enhanced reactivation phenotype from latency, perhaps suggesting that this ORF functions to suppress virus reactivation (10). Similar studies with ORF72 have shown that its gene product is required for efficient reactivation from latency (19, 50). It is possible that expression of ORF72 in B cells is critical for reactivation of latent genomes, although at a less efficient rate than those observed in macrophages and dendritic cells. The functions that M4, M8, and M9 may have during the establishment of latency are not immediately apparent, and their gene products await further characterization. M8 has been shown to form a 5′ exon in a spliced transcript with ORF57 to generate an immediate early transcript homologue to the EBV-M transactivator (26) and M9 has been reclassified as ORF65 on the basis of its similarity with its HVS counterpart (28). No function has as yet been attributed to M4.
Of all of the virus ORFs analyzed in the present study, ORF4 was the only ORF to which no transcripts could be detected in any of the splenic populations analyzed, although transcription could be readily detected in infection of NIH 3T3 cells or lung tissue. This observation is consistent with a recent study showing that ORF4 is dispensable for the establishment of normal levels of latent infection in the spleen (23).
A key observation in the present study was that beyond day 21 p.i., virus genomes could only be detected in NF and GC B cells, although, at very low frequencies. This result is consistent with the observed detection of small foci of vtRNA-positive cells within follicles as late as 90 days p.i. (34) and with the recent demonstration that maintenance of latent infection is preferentially associated with GC and memory B cells (15). It is possible that long-term latency is established in activated GC B cells. Alternatively, infection of GC B cells at late times p.i. could represent sites of latent expansion that originated either from reactivation of long-term latently infected resting memory B cells or from de novo infection of naive B cells that initiate a GC reaction. The latter hypothesis is consistent with the observation made in the present study that NF B cells are infected at late times p.i. NF B cells constitute a subset of naive B cells that are recent emigrants from the bone marrow that colonize lymphoid follicles in the spleen (9). A proportion of these cells acquire the ability to recirculate and give rise to mature naive recirculating Fo B cells. Thus, upon infection, NF B cells could become activated to give rise to latently infected GC B cells, which in turn would differentiate into latently infected long-lived memory B cells. Of significance is the recent demonstration by Flano et al. (15) of MHV-68 latent infection in GC and resting memory B cells long after infection. De novo infection of NF B cells would, however, require a source of productive lytic infection in the spleen. Both the present study and others (14, 34, 54, 55) have failed to detect productive lytic infection in the spleen at late times after infection. However, it is possible that there is low-grade productive lytic infection in the spleen and that our inability to detect it may simply reflect the limit of detection of the assays used. In support of this is the continued presence throughout infection of activated CD4+ and CD8+ T cells specific for virus lytic cycle proteins (16, 37). Alternatively, NF B cells could become infected in the bone marrow, which has been shown to be a site of long-term infection (8).
The data presented in the present study argues for a role of NF and GC B cells during long-term maintenance of MHV-68 latent infection. The biological significance of the broader tropism during the establishment of latent infection, including infection of macrophages, dendritic cells, and MZ and Fo B cells, is not immediately apparent. They could simply reflect bystander infection or represent a strategy of immune modulation employed by the virus. Thus, the identification in the present study of the viral genes transcribed during the establishment of infection in these different cell types, combined with future phenotypic characterization of recombinant viruses with these genes disrupted, should contribute to an understanding of the role played by these different cell types during the establishment of infection.
In conclusion, although gamma-2 herpesviruses do not have the same set of latency associated genes as members of the gamma-1 subgroup and therefore the precise mechanisms for achieving latency in B cells may differ, the overall strategy of targeting B cells for long-term maintenance of latent infection appears to be common.
Acknowledgments
This work was supported by a project grant from the Ministry of Science and Technology of Portugal (PRAXIS/10265/98) to J.P.S. and by a Wellcome Trust Biomedical Research Collaborative Grant (054458/Z/98/MEP/LEC/CRD) to S.E. and J.P.S.
We thank Jorge Carneiro for statistical help, Philip Stevenson for critical review of the manuscript, and Dominique Ostler and Alexandra Teixeira for technical help.
REFERENCES
- 1.Babcock, G. J., L. L. Decker, M. Volk, and D. A. Thorley-Lawson. 1998. EBV persistence in memory B cells in vivo. Immunity 9:395-404. [DOI] [PubMed] [Google Scholar]
- 2.Babcock, G. J., D. Hochberg, and A. D. Thorley-Lawson. 2000. The expression pattern of Epstein-Barr virus latent genes in vivo is dependent upon the differentiation stage of the infected B cell. Immunity 13:497-506. [DOI] [PubMed] [Google Scholar]
- 3.Ballestas, M. E., P. A. Chatis, and K. M. Kaye. 1999. Efficient persistence of extrachromosomal KSHV DNA mediated by latency-associated nuclear antigen. Science 284:641-644. [DOI] [PubMed] [Google Scholar]
- 4.Boname, J. M., and P. G. Stevenson. 2001. MHC class I ubiquitination by a viral PHD/LAP finger protein. Immunity 15:627-636. [DOI] [PubMed] [Google Scholar]
- 5.Bonnefoix, T., P. Bonnefoix, M. Callanan, P. Verdiel, and J. J. Sotto. 2001. Graphical representation of a generalized linear model-based statistical test estimating the fit of the single-hit Poisson model to limiting dilution assays. J. Immunol. 167:5725-5730. [DOI] [PubMed] [Google Scholar]
- 6.Bowden, R. J., J. P. Simas, A. J. Davis, and S. Efstathiou. 1997. Murine gammaherpesvirus 68 encodes tRNA-like sequences which are expressed during latency. J. Gen. Virol. 78:1675-1687. [DOI] [PubMed] [Google Scholar]
- 7.Bridgeman, A., P. G. Stevenson, J. P. Simas, and S. Efstathiou. 2001. A secreted chemokine binding protein encoded by murine gammaherpesvirus-68 is necessary for the establishment of a normal latent load. J. Exp. Med. 194:301-312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cardin, R. D., J. W. Brooks, S. R. Sarawar, and P. C. Doherty. 1996. Progressive loss of CD8+ T cell-mediated control of a gamma-herpesvirus in the absence of CD4+ T cells. J. Exp. Med. 184:863-871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cariappa, A., and S. Pillai. 2002. Antigen-dependent B-cell development. Curr. Opin. Immunol. 14:241-249. [DOI] [PubMed] [Google Scholar]
- 10.Clambey, E. T., H. W. T. Virgin, and S. H. Speck. 2000. Disruption of the murine gammaherpesvirus 68 M1 open reading frame leads to enhanced reactivation from latency. J. Virol. 74:1973-1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cotter, M. A., II, and E. S. Robertson. 1999. The latency-associated nuclear antigen tethers the Kaposi's sarcoma-associated herpesvirus genome to host chromosomes in body cavity-based lymphoma cells. Virology 264:254-264. [DOI] [PubMed] [Google Scholar]
- 12.Doherty, P. C., J. P. Christensen, G. T. Belz, P. G. Stevenson, and M. Y. Sangster. 2001. Dissecting the host response to a gamma-herpesvirus. Philos. Trans. R. Soc. Lond. B Biol. Sci. 356:581-593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Doherty, P. C., D. J. Topham, R. A. Tripp, R. D. Cardin, J. W. Brooks, and P. G. Stevenson. 1997. Effector CD4+ and CD8+ T-cell mechanisms in the control of respiratory virus infections. Immunol. Rev. 159:105-117. [DOI] [PubMed] [Google Scholar]
- 14.Flano, E., S. M. Husain, J. T. Sample, D. L. Woodland, and M. A. Blackman. 2000. Latent murine gamma-herpesvirus infection is established in activated B cells, dendritic cells, and macrophages. J. Immunol. 165:1074-1081. [DOI] [PubMed] [Google Scholar]
- 15.Flano, E., I. J. Kim, D. L. Woodland, and M. A. Blackman. 2002. Gammaherpesvirus latency is preferentially maintained in splenic germinal center and memory B cells. J. Exp. Med. 196:1363-1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Flano, E., D. L. Woodland, M. A. Blackman, and P. C. Doherty. 2001. Analysis of virus-specific CD4+ T cells during long-term gammaherpesvirus infection. J. Virol. 75:7744-7748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gangappa, S., L. F. van Dyk, T. J. Jewett, S. H. Speck, and H. W. T. Virgin. 2002. Identification of the in vivo role of a viral bcl-2. J. Exp. Med. 195:931-940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hewitt, E. W., L. Duncan, D. Mufti, J. Baker, P. G. Stevenson, and P. J. Lehner. 2002. Ubiquitylation of MHC class I by the K3 viral protein signals internalization and TSG101-dependent degradation. EMBO J. 21:2418-2429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hoge, A. T., S. B. Hendrickson, and W. H. Burns. 2000. Murine gammaherpesvirus 68 cyclin D homologue is required for efficient reactivation from latency. J. Virol. 74:7016-7023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Husain, S. M., E. J. Usherwood, H. Dyson, C. Coleclough, M. A. Coppola, D. L. Woodland, M. A. Blackman, J. P. Stewart, and J. T. Sample. 1999. Murine gammaherpesvirus M2 gene is latency-associated and its protein a target for CD8+ T lymphocytes. Proc. Natl. Acad. Sci. USA 96:7508-7513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jacoby, M. A., H. W. T. Virgin, and S. H. Speck. 2002. Disruption of the M2 gene of murine gammaherpesvirus 68 alters splenic latency following intranasal, but not intraperitoneal, inoculation. J. Virol. 76:1790-1801. [DOI] [PMC free article] [PubMed]
- 22.Jensen, K. K., S. C. Chen, R. W. Hipkin, M. T. Wiekowski, M. A. Schwarz, C. C. Chou, J. P. Simas, A. Alcami, and S. A. Lira. 2003. Disruption of CCL21-induced chemotaxis in vitro and in vivo by M3, a chemokine-binding protein encoded by murine gammaherpesvirus 68. J. Virol. 77:624-630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kapadia, S. B., B. Levine, S. H. Speck, and H. W. T. Virgin. 2002. Critical role of complement and viral evasion of complement in acute, persistent, and latent gammaherpesvirus infection. Immunity 17:143-155. [DOI] [PubMed] [Google Scholar]
- 24.Kung, S. H., and P. G. Medveczky. 1996. Identification of a herpesvirus Saimiri cis-acting DNA fragment that permits stable replication of episomes in transformed T cells. J. Virol. 70:1738-1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu, S., I. V. Pavlova, H. W. T. Virgin, and S. H. Speck. 2000. Characterization of gammaherpesvirus 68 gene 50 transcription. J. Virol. 74:2029-2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mackett, M., J. P. Stewart, V. P. S. De, M. Chee, S. Efstathiou, A. A. Nash, and J. R. Arrand. 1997. Genetic content and preliminary transcriptional analysis of a representative region of murine gammaherpesvirus 68. J. Gen. Virol. 78:1425-1433. [DOI] [PubMed] [Google Scholar]
- 27.Miyashita, E. M., B. Yang, G. J. Babcock, and D. A. Thorley-Lawson. 1997. Identification of the site of Epstein-Barr virus persistence in vivo as a resting B cell. J. Virol. 71:4882-4891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nash, A. A., B. M. Dutia, J. P. Stewart, and A. J. Davison. 2001. Natural history of murine gamma-herpesvirus infection. Philos. Trans. R. Soc. Lond. B Biol. Sci. 356:569-579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Parry, C. M., J. P. Simas, V. P. Smith, C. A. Stewart, A. C. Minson, S. Efstathiou, and A. Alcami. 2000. A broad spectrum secreted chemokine binding protein encoded by a herpesvirus. J. Exp. Med. 191:573-578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rochford, R., M. L. Lutzke, R. S. Alfinito, A. Clavo, and R. D. Cardin. 2001. Kinetics of murine gammaherpesvirus 68 gene expression following infection of murine cells in culture and in mice. J. Virol. 75:4955-4963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Roy, D. J., B. C. Ebrahimi, B. M. Dutia, A. A. Nash, and J. P. Stewart. 2000. Murine gammaherpesvirus M11 gene product inhibits apoptosis and is expressed during virus persistence. Arch. Virol. 145:2411-2420. [DOI] [PubMed] [Google Scholar]
- 32.Sangster, M. Y., D. J. Topham, S. D'Costa, R. D. Cardin, T. N. Marion, L. K. Myers, and P. C. Doherty. 2000. Analysis of the virus-specific and nonspecific B-cell response to a persistent B-lymphotropic gammaherpesvirus. J. Immunol. 164:1820-1828. [DOI] [PubMed] [Google Scholar]
- 33.Simas, J. P., R. J. Bowden, V. Paige, and S. Efstathiou. 1998. Four tRNA-like sequences and a serpin homologue encoded by murine gammaherpesvirus 68 are dispensable for lytic replication in vitro and latency in vivo. J. Gen. Virol. 79:149-153. [DOI] [PubMed] [Google Scholar]
- 33a.Simas, J. P., and S. Efstathiou. 1998. Murine gamma herpesvirus 68: a model for the study of gammaherpesvirus pathogenesis. Curr. Opin. Microbiol. 2:403-409. [DOI] [PubMed] [Google Scholar]
- 34.Simas, J. P., D. Swann, R. Bowden, and S. Efstathiou. 1999. Analysis of murine gammaherpesvirus-68 transcription during lytic and latent infection. J. Gen. Virol. 80:75-82. [DOI] [PubMed] [Google Scholar]
- 35.Smith, P. G., P. L. Coletta, A. F. Markham, and A. Whitehouse. 2001. In vivo episomal maintenance of a herpesvirus saimiri-based gene delivery vector. Gene Ther. 8:1762-1769. [DOI] [PubMed] [Google Scholar]
- 36.Speck, S. H., and H. W. Virgin. 1999. Host and viral genetics of chronic infection: a mouse model of gamma-herpesvirus pathogenesis. Curr. Opin. Microbiol. 2:403-409. [DOI] [PubMed] [Google Scholar]
- 37.Stevenson, P. G., G. T. Belz, J. D. Altman, and P. C. Doherty. 1999. Changing patterns of dominance in the CD8+ T-cell response during acute and persistent murine gammaherpesvirus infection. Eur. J. Immunol. 29:1059-1067. [DOI] [PubMed] [Google Scholar]
- 38.Stevenson, P. G., and P. C. Doherty. 1999. Non-antigen-specific B-cell activation following murine gammaherpesvirus infection is CD4 independent in vitro but CD4 dependent in vivo. J. Virol. 73:1075-1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stevenson, P. G., J. S. May, X. G. Smith, S. Marques, H. Adler, U. H. Koszinowski, J. P. Simas, and S. Efstathiou. 2002. K3-mediated evasion of CD8+ T cells aids amplification of a latent gammaherpesvirus. Nat. Immunol. 3:733-740. [DOI] [PubMed] [Google Scholar]
- 40.Stewart, J. P., N. J. Janjua, S. D. Pepper, G. Bennion, M. Mackett, T. Allen, A. A. Nash, and J. R. Arrand. 1996. Identification and characterization of murine gammaherpesvirus 68 gp150: a virion membrane glycoprotein. J. Virol. 70:3528-3535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stewart, J. P., E. J. Usherwood, A. Ross, H. Dyson, and T. Nash. 1998. Lung epithelial cells are a major site of murine gammaherpesvirus persistence. J. Exp. Med. 187:1941-1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sunil-Chandra, N. P., S. Efstathiou, and A. A. Nash. 1992. Murine gammaherpesvirus 68 establishes a latent infection in mouse B lymphocytes in vivo. J. Gen. Virol. 73:3275-3279. [DOI] [PubMed] [Google Scholar]
- 43.Tripp, R. A., A. M. Hamilton-Easton, R. D. Cardin, P. Nguyen, F. G. Behm, D. L. Woodland, P. C. Doherty, and M. A. Blackman. 1997. Pathogenesis of an infectious mononucleosis-like disease induced by a murine gamma-herpesvirus: role for a viral superantigen? J. Exp. Med. 185:1641-1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Usherwood, E. J., D. J. Roy, K. Ward, S. L. Surman, B. M. Dutia, M. A. Blackman, J. P. Stewart, and D. L. Woodland. 2000. Control of gammaherpesvirus latency by latent antigen-specific CD8+ T cells. J. Exp. Med. 192:943-952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Usherwood, E. J., J. P. Stewart, and A. A. Nash. 1996. Characterization of tumor cell lines derived from murine gammaherpesvirus-68-infected mice. J. Virol. 70:6516-6518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Usherwood, E. J., J. P. Stewart, K. Robertson, D. J. Allen, and A. A. Nash. 1996. Absence of splenic latency in murine gammaherpesvirus 68-infected B-cell-deficient mice. J. Gen. Virol. 77:2819-2825. [DOI] [PubMed] [Google Scholar]
- 47.van Berkel, V., J. Barrett, H. L. Tiffany, D. H. Fremont, P. M. Murphy, G. McFadden, S. H. Speck, and H. I. Virgin. 2000. Identification of a gammaherpesvirus selective chemokine binding protein that inhibits chemokine action. J. Virol. 74:6741-6747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.van Berkel, V., B. Levine, S. B. Kapadia, J. E. Goldman, S. H. Speck, and H. W. T. Virgin. 2002. Critical role for a high-affinity chemokine-binding protein in gamma-herpesvirus-induced lethal meningitis. J. Clin. Investig. 109:905-914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.van Berkel, V., K. Preiter, H. W. T. Virgin, and S. H. Speck. 1999. Identification and initial characterization of the murine gammaherpesvirus 68 gene M3, encoding an abundantly secreted protein. J. Virol. 73:4524-4529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.van Dyk, L. F., H. W. T. Virgin, and S. H. Speck. 2000. The murine gammaherpesvirus 68 v-cyclin is a critical regulator of reactivation from latency. J. Virol. 74:7451-7461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Virgin, H. W. T., P. Latreille, P. Wamsley, K. Hallsworth, K. E. Weck, A. J. Dal Canto, and S. H. Speck. 1997. Complete sequence and genomic analysis of murine gammaherpesvirus 68. J. Virol. 71:5894-5904. [DOI] [PMC free article] [PubMed]
- 52.Virgin, H. W. T., R. M. Presti, X. Y. Li, C. Liu, and S. H. Speck. 1999. Three distinct regions of the murine gammaherpesvirus 68 genome are transcriptionally active in latently infected mice. J. Virol. 73:2321-2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang, G. H., T. L. Garvey, and J. I. Cohen. 1999. The murine gammaherpesvirus-68 M11 protein inhibits Fas- and TNF-induced apoptosis. J. Gen. Virol. 80:2737-2740. [DOI] [PubMed] [Google Scholar]
- 54.Weck, K. E., M. L. Barkon, L. I. Yoo, S. H. Speck, and H. I. Virgin. 1996. Mature B cells are required for acute splenic infection, but not for establishment of latency, by murine gammaherpesvirus 68. J. Virol. 70:6775-6780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Weck, K. E., S. S. Kim, H. I. Virgin, and S. H. Speck. 1999. B cells regulate murine gammaherpesvirus 68 latency. J. Virol. 73:4651-4661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Weck, K. E., S. S. Kim, H. I. Virgin, and S. H. Speck. 1999. Macrophages are the major reservoir of latent murine gammaherpesvirus 68 in peritoneal cells. J. Virol. 73:3273-3283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wu, T. T., L. Tong, T. Rickabaugh, S. Speck, and R. Sun. 2001. Function of Rta is essential for lytic replication of murine gammaherpesvirus 68. J. Virol. 75:9262-9273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wu, T. T., E. J. Usherwood, J. P. Stewart, A. A. Nash, and R. Sun. 2000. Rta of murine gammaherpesvirus 68 reactivates the complete lytic cycle from latency. J. Virol. 74:3659-3667. [DOI] [PMC free article] [PubMed] [Google Scholar]