

Abstract
Human immunodeficiency virus type 2 (HIV-2) infection is a serious problem in West Africa and Asia. However, there have been relatively few studies of HIV-2 reverse transcriptase (RT), a potential target for antiviral therapy. Detailed knowledge of HIV-2 RT activities is critical for development of specific high-throughput screening assays of potential inhibitors. Here, we have conducted a systematic evaluation of HIV-2 RT function, using assays that model specific steps in reverse transcription. Parallel studies were performed with HIV-1 RT. In general, under standard assay conditions, the polymerase and RNase H activities of the two enzymes were comparable. However, when the RT concentration was significantly reduced, HIV-2 RT was less active than the HIV-1 enzyme. HIV-2 RT was also impaired in its ability to catalyze secondary RNase H cleavage in assays that mimic tRNA primer removal during plus-strand transfer and degradation of genomic RNA fragments during minus-strand DNA synthesis. In addition, initiation of plus-strand DNA synthesis was much less efficient with HIV-2 RT than with HIV-1 RT. This may reflect architectural differences in the primer grip regions in the p66 (HIV-1) and p68 (HIV-2) palm subdomains of the two enzymes. The implications of our findings for antiviral therapy are discussed.
Human immunodeficiency virus type 2 (HIV-2), like HIV-1, is a retrovirus that causes AIDS. Although HIV-2 has only limited nucleotide sequence homology to HIV-1 (50 to 60% homology to gag and pol and even less homology to other HIV-1 genes) (26), it shows similar genomic organization, ultrastructural morphology, functional activities, and protein composition (7). The HIV-2 virus was originally isolated in West Africa, where it is still prevalent, but more recently, HIV-2 has spread to India (see reference 54 for a review). A large number of HIV-2-infected individuals are also found in Portugal and former Portuguese colonies, such as Guinea-Bissau and Brazil (54). In addition, there has been a large increase in the number of individuals in West Africa who are dually infected with HIV-1 and HIV-2 (54). HIV-2 is transmitted less efficiently than HIV-1, and progression to AIDS occurs more slowly (54), but the incidence of HIV-2 cases, and particularly HIV-1- HIV-2 dual infection, remains an important public health issue.
The virion-associated reverse transcriptase (RT) enzyme is a major target for anti-HIV therapy. RT plays a crucial role in virus replication: it catalyzes synthesis of a double-stranded DNA copy of the single-stranded RNA genome in a complex series of reactions known as reverse transcription (Fig. 1) (see below). HIV-1 RT has long been the focus of intensive investigation, and several X-ray crystal structures are available (13, 31, 33, 35, 58). Moreover, nucleoside RT inhibitors and nonnucleoside RT inhibitors (NNRTIs) that block HIV-1 RT activity are now being used routinely in AIDS treatment (http://www.amfar.org/td). Most NNRTIs do not inhibit HIV-2 RT activity (2, 42, 47, 62, 63), although there have been reports of new drugs that may be useful for this purpose (4, 41, 43, 55-57). A recent X-ray crystal structure of unliganded HIV-2 RT shows that structural differences between the HIV-1 and HIV-2 enzymes in the “NNRTI pocket” at conserved and nonconserved residues and conformational change resulting from substitution of Ile for Tyr at position 181 are significant factors that could account for the observed HIV-2 resistance to NNRTIs (55).
FIG. 1.

Schematic diagram of the events in reverse transcription. (Step 1) HIV reverse transcription is initiated by a cellular tRNA primer, tRNA3Lys, following annealing of the 3′ 18 nt of the tRNA to the 18-nt primer binding site (PBS) near the 5′ end of the genome. RT catalyzes synthesis of (−) SSDNA, which contains copies of the repeat (R) sequence and the unique 5′ genomic sequence (U5). (Step 2) As the primer is extended, the RNase H activity of RT degrades the genomic RNA sequences that have been reverse transcribed. (Step 3) (−) SSDNA is transferred to the 3′ end of viral RNA (minus-strand transfer). (Step 4) Elongation of minus-strand DNA and RNase H degradation continue. Plus-strand synthesis is initiated by the 15-nt PPT immediately upstream of the unique 3′ genomic sequence (U3). RT copies the u3, u5, and r regions in minus-strand DNA, as well as the 3′ 18 nt of the tRNA primer, thereby reconstituting the PBS. The product formed is termed (+) SSDNA. (Step 5) RNase H removal of the PPT and tRNA primers. (Step 6) Transfer of (+) SSDNA to the 3′ end of minus-strand DNA (plus-strand transfer) is followed by circularization of the two DNA strands and displacement synthesis. (Step 7) Minus- and plus-strand DNAs are elongated, resulting in a linear double-stranded DNA with a long terminal repeat at each end. Viral RNA is shown in red; minus-and plus-strand DNAs are shown in blue and green, respectively; and the tRNA primer is in orange. Minus-strand and plus-strand sequences are depicted in lower and upper case, respectively. The dashed red lines represent RNase H cleavage of genomic RNA.
In contrast to the proliferation of papers on HIV-1 RT, HIV-2 RT has received less attention. HIV-2 RT has ∼60% overall amino acid sequence homology to HIV-1 RT (26), but there are regions that are highly conserved in both enzymes. In fact, in an early study, an antiserum directed against a synthetic peptide containing 14 residues that are identical in HIV-1 and HIV-2 RTs was used to purify the HIV-2 enzyme (12). In other studies, it was shown that peptide antisera generated against regions in HIV-2 RT with at least 50% homology with HIV-1 RT not only recognized the HIV-2 enzyme but also cross-reacted with HIV-1 RT; the analogous HIV-1 sera were type specific (34).
HIV-2 RT activities have been investigated with enzyme preparations purified from virions (12) or following bacterial expression (15, 29, 39, 44). Like HIV-1 RT, the HIV-2 enzyme was shown to be a heterodimeric protein (12, 15, 39, 44) with a large (68-kDa) subunit and a small (54-kDa) subunit generated by cleavage of p68 at Met 484 (15). In early studies, performed primarily with homopolymeric substrates (16, 28, 29, 39, 44), it was established that HIV-2 RT behaves like a true retroviral RT, having both RNA- and DNA-dependent DNA polymerase activities, as well as RNase H activity.
There has been some discussion in the literature about the extent of quantitative differences between HIV-1 and HIV-2 RNase H activities. In a study by Fan et al. (16), it was observed that with a gag-based RNA-DNA hybrid for which RT has a high affinity, HIV-1 RT had only ∼2-fold-greater activity than HIV-2 RT. Hizi and colleagues reported that the RNase H activity of HIV-1 RT was 10-fold higher than that of HIV-2 RT, while differences in polymerase activities were less prominent (28, 29, 62). On the basis of results obtained with a substrate containing 5′ genomic RNA sequences and a series of HIV-1- HIV-2 chimeric enzymes, it was shown that the previously reported 10-fold difference in the RNase H activities of HIV-1 and HIV-2 RTs could be attributed to differences in the activities of the thumb subdomains of the small subunits (61) and, more specifically, to the identity of residue 294 (60).
Until now, there has not been a systematic evaluation of the functional activities of HIV-2 RT during specific steps that occur in reverse transcription (Fig. 1). In the absence of an X-ray crystal structure for the enzyme complexed to nucleic acid and given the urgent need to develop new drugs that can target HIV-2 RT function, such analysis is especially timely. Thus, the goal of the present work was to investigate the polymerase and RNase H activities of HIV-2 RT in reconstituted model systems and to conduct parallel studies with HIV-1 RT. This approach should aid in the development of additional targets for anti-HIV-2 therapy and new high-throughput screening assays.
Our results demonstrate that, in general, the polymerase and RNase H activities of the HIV-1 and HIV-2 enzymes give similar results in quantitative functional assays. However, under conditions where the RT concentration is limiting, HIV-1 RT is significantly more active than its HIV-2 counterpart. In addition, secondary RNase H cleavage measured in assays for 5′-directed RNase H activity and the tRNA removal step in plus-strand transfer are less efficient when catalyzed by HIV-2 rather than HIV-1 RT. Unexpectedly, we have also found that HIV-2 RT has great difficulty catalyzing the initiation of plus-strand DNA synthesis with the RNA polypurine tract (PPT) primer, and we propose that this may be due, at least in part, to architectural differences in the primer grip (33) regions in the p66 (HIV-1) and p68 (HIV-2) palm subdomains of the two enzymes.
MATERIALS AND METHODS
Materials.
T4 DNA polymerase was purchased from Roche (Indianapolis, Ind.). RNasin was obtained from Promega Biotech (Madison, Wis.). HIV-1 RT was purchased from Worthington Biochemical Corp. (Lakewood, N.J.). The radioactively labeled nucleotides [α-32P]dATP (3,000 Ci/mmol) and [γ-32P]ATP (3,000 Ci/mmol) were purchased from Amersham Biosciences Corp. (Piscataway, N.J.). RNA and DNA oligonucleotides were initially obtained from Oligos Etc., Inc. (Wilsonville, Oreg.). Subsequently, RNA oligonucleotides were purchased from Dharmacon, Inc., Lafayette, Colo., and DNA oligonucleotides were obtained from Integrated DNA Technologies, Coralville, Iowa.
RT enzymes.
Three versions of HIV-2 RT, all containing 559 residues in the p68 subunit and either 439, 469, or 483 residues in the p54 subunit, were prepared from HIV-2 ROD (GenBank accession number M15390), as previously described (38). For convenience, these enzymes will be referred to as HIV-2 RT 440, 470, and 484, in accordance with the numbering for HIV-1 RT (53). The small subunit of HIV-2 RT terminates at Met 484 (actually, Met 483) (15); HIV-1 p51 RT terminates at Phe 440 (6, 21). The three versions of HIV-2 RT were originally prepared to determine whether the number of C-terminal residues in the small subunit affects HIV-2 RT activity. In fact, all three HIV-2 enzymes had essentially equivalent activities in our assays (see below and data not shown). For this reason, when results for only one of the HIV-2 RTs are described in the text, we will refer to “HIV-2 RT” rather than to a specific version of the enzyme. However, in the figure legends, the HIV-2 RT(s) used is specified. Note that the data were obtained from at least three independent experiments. The results were reproducible, and representative data are shown in each case.
DNA polymerase assays.
Assays with RNA and DNA templates are described below.
(i) Assay for synthesis of HIV-1 minus-strand DNA.
The assay was carried out as described by Guo et al. (22), except that the RT concentration was reduced 10-fold. Briefly, 0.2 pmol of a 131-nt T7 RNA transcript, containing the entire R region and a portion (34 nucleotides [nt]) of the unique 5′ genomic sequence (U5) in HIV-1 NL4-3 (1), was heat annealed (65°C; 5 min) to 0.4 pmol of a 5′ 32P-labeled 20-nt DNA primer complementary to nt 565 to 584 in U5. Reaction buffer (50 mM Tris-HCl [pH 8.0], 75 mM KCl, and 1 mM dithiothreitol [DTT]) and 0.02 pmol of HIV-1 or HIV-2 RT, as specified, were added to the annealed donor-primer hybrid. Reactions (final volume, 20 μl) were initiated by the addition of MgCl2 (final concentration, 7 mM) and the four deoxyribonucleoside triphosphates (dNTPs) (each at a final concentration of 100 μM). For measuring the time course of DNA synthesis, the reactions were scaled up to a volume of 100 μl; 10-μl aliquots were removed after incubation at 37°C for the indicated times. EDTA (final concentration, 50 mM) was added to terminate the reactions, and the products were analyzed as described previously (22).
(ii) Assay for extension of a 15-nt DNA PPT primer.
The assay for extension of a 15-nt DNA PPT primer evaluates the ability of RT to synthesize plus-strand DNA from a DNA PPT primer. All of the HIV-1 PPT-containing oligonucleotides used in this assay, and others described below, have HIV-1 LAI (formerly LAV-1 or BRU) sequences (numbered according to the current version in GenBank, accession number K02013); the HIV-2 PPT-containing oligonucleotides have sequences derived from HIV-2 ROD (numbered according to GenBank accession number M15390).
The assay was performed as described by Powell et al. (51) with some modification. Ten picomoles of a 15-nt DNA version of the RNA PPT primer (RNA sequences are as follows: HIV-1, 5′-AAA AGA AAA GGG GGG-3′, nt 8662 to 8676; HIV-2, 5′-AAA AAC AAG GGG GGG-3′, nt 8925 to 8939) was annealed to 1 pmol of a 35-nt minus-strand DNA template (HIV-1 [JL186], complementary to nt 8662 to 8696; HIV-2 [JL257], complementary to nt 8925 to 8959) in 83 mM NaCl (total volume, 6 μl) by heating each oligonucleotide pair to 90°C for 5 min and then slowly cooling them to room temperature. Reaction buffer (50 mM Tris-HCl [pH 8.0], 25 mM KCl, 3 mM MgCl2, and 6 mM DTT), three dNTPs (dTTP, dGTP, and dCTP, each at a final concentration of 250 μM), cold dATP at a final concentration of 80 μM, [α-32P]dATP (10 μCi), and RT (as specified) were added to the annealed primer-template in a final volume of 15 μl. The final monocation concentration was 58 mM. The reaction mixtures were incubated at 37°C for 30 min, by which time maximal DNA synthesis was achieved (data not shown). Formamide STOP solution was added to terminate the reactions. The products were resolved in an 8% sequencing gel and visualized by autoradiography. The data were quantified by PhosphorImager analysis.
(iii) Assay for initiation of plus-strand DNA synthesis.
The ability to initiate plus-strand DNA synthesis from an RNA PPT (51, 52) was measured in an assay using 10 pmol of a 15-nt HIV-1 or HIV-2 RNA PPT primer (see above) annealed to 1 pmol of the complementary 35-nt minus-strand DNA templates (see above). Each reaction mixture (final volume, 15 μl) contained the components used in the assay for extension of the DNA PPT primer and 1 U of RNasin; in this case, 1 or 2 pmol of the HIV-1 or HIV-2 RT enzyme, respectively, was added. For time course experiments, the reaction mixtures were scaled up and aliquots were removed following incubation at 37°C for the indicated times. This assay measures extension and subsequent RNase H-mediated primer removal at the junction between the PPT and nascent plus-strand DNA. Since the initial 35-nt RNA-DNA extension product is not detectable (51, 52), only the 32P-labeled 20-nt DNA generated by RNase H cleavage is scored in the assay. The DNA product was analyzed as described under item ii above. No products were detected that would indicate premature termination of extension.
RNase H assays.
Three different RNase H assays were used, and they are described below.
(i) Assay of 5′-directed RNase H activity.
To measure 5′-directed RNase H activity, a 20-nt RNA primer (1 pmol), labeled at its 5′ end with 32P (25) and consisting of the 20-nt sequence immediately downstream of the HIV-1 LAI PPT (nt 8677 to 8696), was annealed to a 55-nt minus-strand DNA template (10 pmol) (complementary to nt 8657 to 8711) under the conditions described above for extension of the 15-nt DNA PPT primer. Since the primer was labeled, the ratio of primer to template was set at 1:10. Reaction buffer (50 mM Tris-HCl, pH 8.0, 25 mM KCl, 3 mM MgCl2, and 6 mM DTT), 1 U of RNasin, and 0.005 pmol of RT (HIV-1 or HIV-2 440, as specified) were added to the annealed primer-template in a final volume of 15 μl. For the time course study, reactions were scaled up from the unit 15-μl volume and were incubated at 37°C; aliquots were removed at the indicated times. RNA cleavage products were resolved by electrophoresis in a 12.5% sequencing gel and were quantified by PhosphorImager analysis.
(ii) Removal of an 18-nt RNA representing the 3′ end of the 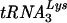 primer from a 32-nt HIV-1 minus-strand donor DNA.
primer from a 32-nt HIV-1 minus-strand donor DNA.
The assay for removal of the 18-nt RNA was performed as described by Wu et al. (71) except that minus-strand acceptor DNA and dNTPs were not added. The 18-nt RNA moiety attached to the 32-nt minus-strand DNA donor was labeled at its 5′ end with 32P (25). Aliquots were removed from the reaction mixtures at the indicated times, from 1 to 60 min. RNA cleavage products were resolved by electrophoresis in a 15% sequencing gel and were quantified by PhosphorImager analysis.
(iii) RNA PPT primer removal assay.
HIV-1 or HIV-2 RNA PPT primers were annealed to HIV-1 or HIV-2 minus-strand 35-nt DNA templates, respectively, and were then extended with T4 DNA polymerase in a reaction mixture containing buffer supplied by the manufacturer, three dNTPs (dTTP, dGTP, and dCTP, each at a final concentration of 250 μM), cold dATP at a final concentration of 80 μM, and [α-32P]dATP (10 μCi) (50); the reaction mixtures were incubated for 5 min at 37°C. One pmol of RT (HIV-1 or HIV-2, as specified) was then added to the 32P-labeled substrate. For time course studies, the reaction mixtures were scaled up from the unit 15-μl volume and were incubated at 37°C; aliquots were removed at the indicated times. The cleavage products were analyzed as detailed by Powell et al. (50) and were quantified by PhosphorImager analysis. The value for 100% cleavage is based on the amount of product that would result from total cleavage of the RNA-DNA substrate (50). In this assay, correct cleavage occurs at the junction between the PPT primer and nascent plus-strand DNA, resulting in the release of a 32P-labeled 20-nt plus-strand DNA fragment (50, 51). Note that T4 DNA polymerase does not exhibit RNase H activity under the conditions of this assay (52).
RESULTS
Steps in reverse transcription.
To obtain new functional information on HIV-2 RT and to compare its activities with the better-characterized HIV-1 enzyme, we exploited reconstituted systems that mimic specific steps in reverse transcription. Figure 1 is provided to illustrate the complex series of reactions that are involved in this process (5, 67); a detailed description of each step is given in the legend. Briefly, steps 1 to 7 include the following reactions: step 1, initiation of minus-strand synthesis by a cellular tRNA primer; step 2, completion of minus-strand strong-stop DNA [(−) SSDNA] synthesis and RNase H degradation of RNA sequences that have been reverse transcribed; step 3, minus-strand transfer; step 4, elongation of minus-strand DNA, further degradation of genomic RNA, and synthesis of (+) SSDNA by the PPT primer; step 5, RNase H removal of the tRNA and PPT primers; step 6, plus-strand transfer and strand displacement synthesis; and step 7, elongation of minus- and plus-strand DNAs to yield a full-length, linear, double-stranded DNA copy of the viral RNA genome.
RNA- and DNA-dependent DNA synthesis.
During the course of reverse transcription, RT must copy templates containing viral RNA or DNA sequences (Fig. 1). To assess the abilities of HIV-1 and HIV-2 RTs to catalyze such reactions, we performed two different assays that measure RNA- or DNA-dependent DNA synthesis. In the first of these assays (Fig. 2), we used an HIV-1 RNA template, containing 5′ genomic RNA sequences, to synthesize minus-strand DNA, including the first product of reverse transcription (Fig. 1), which in this system is a 131-nt (−) SSDNA (22). Total DNA synthesis was measured as a function of time. In addition to (−) SSDNA, the products consisted of pause products and self-priming products (SP DNAs) (Fig. 2A), which are minus-strand DNAs with plus-strand extensions (22). Self-priming is induced by the full-length complementary TAR stem-loop at the 3′ end of (−) SSDNA (3, 14, 22-24, 30, 37, 40) and also at sites within TAR DNA (22). SP DNAs characteristically increase with time of incubation and can be distinguished from pause products, which exhibit the opposite behavior (22).
FIG. 2.

Time course of RNA-dependent DNA synthesis catalyzed by HIV-1 and HIV-2 RTs. The RNA template contained nt 1 to 131 of HIV-1 genomic RNA (1). The reaction conditions, gel analysis, and quantification of the data by phosphorimaging are described in Materials and Methods and in Guo et al. (22). (A) Gel data. The time points are indicated above the lanes. The positions of (−) SSDNA, SP products (SP), and the primer are indicated to the left of the gel. (B) PhosphorImager analysis of gel data. Total DNA synthesized at each time point was determined by PhosphorImager analysis and was plotted as a function of time. Circles, HIV-1 RT; squares, HIV-2 RT.
The initial experiments indicated that under our usual conditions (equivalent concentrations of enzyme and primer-template), HIV-1 and HIV-2 RTs synthesized the same amount of total DNA (data not shown). However, when the enzyme concentration was reduced by 10-fold, visualization of the DNA products made in each reaction showed that HIV-1 RT had a greater amount of DNA polymerase activity than HIV-2 RT (Fig. 2A). It is interesting that with limiting enzyme concentration, long SP DNAs were not formed to any significant extent, while short SP DNAs were readily detected (Fig. 2A). As expected, since HIV-2 polymerase activity was lower than that of HIV-1, synthesis of SP DNAs was also reduced in the HIV-2 reaction. Further reductions in RT concentration (e.g., 100-fold) gave the same qualitative results, but the levels of activity at 30 min were very low (data not shown). PhosphorImager analysis of the gel data shown in Fig. 2A indicated that both the rate and extent of total DNA synthesis were greater with HIV-1 RT (∼2.5- to 2.7-fold and ∼2-fold, respectively) than with the HIV-2 enzyme (Fig. 2B). Taken together, these data indicate that primer extension during minus-strand DNA synthesis is less efficient with HIV-2 RT than with the HIV-1 enzyme.
It was next of interest to compare the two enzymes in an assay of DNA-dependent DNA polymerase activity (Fig. 1). We chose an assay that measures extension of a 15-nt DNA PPT primer annealed to a 35-nt minus-strand DNA template, since comparison of initiation of plus-strand DNA synthesis by DNA and RNA (see below) PPT primers can give useful information on the capabilities of RT (16a, 51, 52). The full-length product of this reaction is a 32P-labeled 35-nt plus-strand DNA (51, 52). The activities of HIV-1 and HIV-2 RTs were tested with both HIV-1 and HIV-2 primer-templates (lanes 1 to 6 and 7 to 12, respectively) as a function of enzyme concentration (Fig. 3). The 35-nt DNA was the major product formed in most of the reactions, although smaller pause products (32 to 34 nt) could also be detected. The product migrating more slowly than the 35-nt DNA was probably formed by blunt-end addition of a single dA to the 35-nt DNA; since this represents a nonspecific reaction (19, 48), the amount of 36-nt DNA was omitted from the calculation of total DNA products. For each set of reactions, i.e., with the HIV-1 or HIV-2 substrates, the values for total DNA synthesized at the highest amount of HIV-1 RT (1 pmol) were taken as 100%.
FIG. 3.

DNA-dependent DNA synthesis catalyzed by HIV-1 and HIV-2 RTs. A 15-nt HIV-1 or HIV-2 DNA PPT primer was extended by 20 nt in the presence of [α-32P]dATP using the appropriate minus-strand 35-nt DNA template, as described in Materials and Methods. HIV-1 primer-template (P-T), lanes 1 to 6; HIV-2 P-T, lanes 7 to 12. The amounts of enzyme added were as follows: 1 pmol, lanes 1, 2, 7, and 8; 0.1 pmol, lanes 3, 4, 9, and 10; and 0.01 pmol, lanes 5, 6, 11, and 12. The odd-numbered lanes represent reactions with HIV-1 RT, and the even-numbered lanes represent reactions with HIV-2 440 RT. The percentage of total products represented by 35-nt DNA and the percentage of the total amount of DNA synthesized relative to the control are given below each lane; 100% represents the amount of DNA synthesized by 1 pmol of HIV-1 RT with the HIV-1 substrate.
PhosphorImager analysis of the gel data in Fig. 3 showed that with either primer-template, ∼75 to 100% of the DNA products made by 1 and 0.1 pmol of HIV-1 (lanes 1, 3, 7, and 9) or HIV-2 (lanes 2, 4, 8, and 10) RT was present as the full-length 35-nt DNA. The total amounts of DNA synthesized were also approximately the same for both enzymes (90 to 100% of the control). In these reactions, the RT/primer-template ratios were 1 (lanes 1, 2, 7, and 8) or 0.1 (lanes 3, 4, 9, and 10). When 0.01 pmol of RT was used (RT/primer-template ratio, 0.01), pause products smaller than 35 nt were readily detected, regardless of which RT or primer-template was used. Approximately 35 to 40% of the total DNA made by HIV-2 RT was 35 nt in length (lanes 6 and 12), whereas 35-nt DNA constituted 55 to 60% of the products made by the HIV-1 enzyme (lanes 5 and 11). It is even more striking that the total amount of DNA synthesized by 0.01 pmol of HIV-1 RT was 3-fold greater than that synthesized by 0.01 pmol of HIV-2 RT with the HIV-1 substrate and was 10-fold higher than HIV-2 RT with the HIV-2 substrate (compare lane 5 with lane 6 and lane 11 with lane 12).
Taken together, the results indicate that at high concentrations, HIV-1 and HIV-2 RTs are able to catalyze RNA- and DNA-dependent DNA synthesis to approximately the same extent in two independent polymerase assay systems. However, by using limiting enzyme concentration to assay primer extension, differences in HIV-1 and HIV-2 DNA polymerase activities could be demonstrated (Fig. 2 and 3). The finding that overall DNA-dependent DNA synthesis catalyzed by 0.01 pmol of HIV-2 RT was especially inefficient when the HIV-2 PPT primer-template was used (Fig. 3, lane 12) may be related to the fact that the HIV-2 primer has seven dG residues at its 3′ end, in contrast to the six dG residues in the HIV-1 primer.
RNase H activities required for four steps in reverse transcription.
In addition to RNA- and DNA-dependent DNA polymerase activities, the RNase H activity of RT is also critical for the completion of viral DNA synthesis. There are four steps during reverse transcription in which RNase H activity is required (Fig. 1) (reviewed in reference 5): (i) cleavage of genomic, i.e., template RNA during synthesis of minus-strand DNA; (ii) 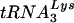 primer removal prior to plus-strand transfer; (iii) generation of the 15-nt RNA PPT primer from genomic RNA; and (iv) removal of the PPT primer from nascent plus-strand DNA. We have compared the activities of HIV-1 and HIV-2 RTs in assays that mimic reactions i, ii, and iv.
primer removal prior to plus-strand transfer; (iii) generation of the 15-nt RNA PPT primer from genomic RNA; and (iv) removal of the PPT primer from nascent plus-strand DNA. We have compared the activities of HIV-1 and HIV-2 RTs in assays that mimic reactions i, ii, and iv.
(i) 5′-Directed cleavage of large RNA fragments generated during synthesis of minus-strand DNA.
In the first group of experiments in this series, RNase H activity was measured in an assay that models polymerase-independent, 5′-directed, RNase H-catalyzed cleavage of template RNA (Fig. 1) (9-11, 45, 46, 59, 59a, 68-70). This reaction is required for efficient reverse transcription, since the initial RNase H cleavage products formed during synthesis of minus-strand DNA are too large to dissociate spontaneously from the DNA product (reviewed in reference 5). Thus, without prior cleavage of these large fragments, minus-strand transfer and subsequent elongation of minus-strand DNA cannot occur (5, 48, 66, 67).
Figure 4 shows the results of a time course experiment that was designed to measure 5′-directed RNase H activity with a substrate containing a recessed 32P-labeled 20-nt HIV-1 RNA annealed to a 55-nt complementary minus-strand DNA template. 32P-labeled cleavage products generated by HIV-1 or HIV-2 RT were visualized and quantified, as described in Materials and Methods. In initial experiments, we found that using an RT/primer-template ratio of 1 or 0.1 resulted in the same levels of activity of HIV-1 and HIV-2 RTs; with a ratio of 0.01, some differences in activity were apparent (data not shown). However, by using an RT/primer-template ratio of 0.005, we were able to detect even greater differences in the activities of the two enzymes.
FIG. 4.

Time course of 5′-directed RNase H cleavage activity. The 5′-directed RNase H cleavage activities of HIV-l and HIV-2 440 RTs were assayed as described in Materials and Methods. (A) Gel data. The time points are indicated above the lanes. Lane C is the zero-time-point (minus RT) control. The sizes of the RNA bands, shown on the left, were estimated by comparison with an RNA ladder generated by alkaline hydrolysis of the 20-nt RNA, run on the same gel as the samples. The 19-nt band in all samples is a contaminant and represents ∼3% of the total 20-nt RNA preparation. (B) PhosphorImager analysis of the gel data. Total cleavage was calculated by dividing the “volume” of all of the cleavage products in each lane by the total volume and then multiplying by 100. The values for total cleavage were plotted as a function of the time of incubation. Circles, HIV-1 RT; squares, HIV-2 RT. (C) Schematic diagram illustrating the 32P-labeled substrate. The 20-nt RNA primer is shown as an open rectangle; the 32P label at the 5′ end of the RNA is indicated by an asterisk. The 55-nt minus-strand DNA is shown as a hatched rectangle.
The gel data in Fig. 4A indicate that the RNase H activities of both RTs gave rise to the same cleavage products, but the rate and extent of cleavage were clearly higher in the HIV-1 RT reaction. The products ranged from 7 to 17 nt, representing multiple cleavage events (68-70). (Under our conditions, the primary cleavage product comigrated with a 17-nt, rather than an 18-nt, marker.) With HIV-1, there was a decrease in 15- to 17-nt products during incubation and an accumulation of products of 14 nt or smaller. In contrast, in the HIV-2 reaction, products of from 8 to 17 nt increased with time, suggesting that HIV-2 RT is impaired in its ability to catalyze secondary RNase H cleavage activity. (It is formally possible that each band in the HIV-2 reaction represents an independent primary cleavage event. However, extensive studies of the mechanism of 5′-directed RNase H activity demonstrating that a series of primary and secondary cleavages is involved [68-70] would appear to rule this out.) PhosphorImager analysis of the gel data (Fig. 4B) shows that for total cleavage, HIV-1 RT exhibited an ∼3- to 4-fold-greater rate of cleavage than HIV-2 RT. Moreover, total cleavage with HIV-1 RT reached a plateau value of nearly 100%, whereas the corresponding value for HIV-2 RT was 40 to 50%.
(ii) Primer removal during plus-strand DNA synthesis.
RNase H activity was also measured in an assay that models a key step in plus-strand DNA transfer, i.e., removal of the 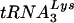 primer from the 5′ end of minus-strand DNA (Fig. 1) (references 65 and 71 and references therein; for earlier papers, also see the review in reference 5). This reaction is required for subsequent annealing of the 18-nt complementary primer binding site (PBS) sequences in (+) SSDNA and minus-strand acceptor DNA. In our system, an 18-nt RNA oligonucleotide, representing the 3′ 18 nt of the tRNA primer, is covalently attached to a 32-nt HIV-1 minus-strand DNA and labeled with 32P at its 5′ end; the chimeric RNA-DNA oligonucleotide is annealed to a 50-nt synthetic (+) SSDNA (Fig. 5A shows a schematic diagram).
primer from the 5′ end of minus-strand DNA (Fig. 1) (references 65 and 71 and references therein; for earlier papers, also see the review in reference 5). This reaction is required for subsequent annealing of the 18-nt complementary primer binding site (PBS) sequences in (+) SSDNA and minus-strand acceptor DNA. In our system, an 18-nt RNA oligonucleotide, representing the 3′ 18 nt of the tRNA primer, is covalently attached to a 32-nt HIV-1 minus-strand DNA and labeled with 32P at its 5′ end; the chimeric RNA-DNA oligonucleotide is annealed to a 50-nt synthetic (+) SSDNA (Fig. 5A shows a schematic diagram).
FIG. 5.

Time course of RNase H-catalyzed removal of an 18-nt RNA covalently attached to minus-strand DNA. Reaction mixtures containing the substrate (A) and the indicated RT enzymes were incubated at 37°C; aliquots were removed at the specified times. (A) Schematic diagram illustrating the substrate used for the assay. The 32-nt minus-strand DNA and the attached 18-nt RNA are shown as solid and open rectangles, respectively. The 32P label at the 5′ end of the 18-nt RNA moiety is indicated by an asterisk. The 50-nt (+) SSDNA is represented by a hatched rectangle. (B) Gel data. The time points are indicated above each lane. Lane C is the zero-time-point (minus RT) control. The sizes of the RNA bands were estimated as described in the legend to Fig. 4. The bands between the positions of the 50-nt chimeric DNA-RNA and the 17-nt product represent 2.8% contamination of the DNA-RNA preparation; no contaminating bands were observed in the region of the RNase H cleavage products. The nature of the contaminating bands is not known. (C and D) Analysis of gel data. The data in panel A were quantified by PhosphorImager analysis. (C) The values for total cleavage were determined as described in the legend to Fig. 4. The inset shows the data for the first 5 min of the time course study, using an expanded time scale. (D) A similar method was used to calculate the percent of the total volume represented by the final cleavage product (8 nt). Total cleavage and final cleavage were plotted as a function of the time of incubation. Circles, HIV-1 RT; inverted triangles, HIV-2 484 RT.
The experiment illustrated in Fig. 5 gives the time course of removal of the 18-nt RNA by the RNase H activities of HIV-1 and HIV-2 RTs during a 60-min incubation (Fig. 5B). In both cases, the primary cleavage product was a 17-nt RNA, which is formed by removal of the rA at the 3′ terminus of the 18-nt RNA; secondary cleavage products ranging from 8 to 16 nt were also observed, as has been documented previously (65, 71). Generation of the 8-nt RNA is especially significant, since its temporal appearance has been correlated with the onset of plus-strand DNA transfer (65, 71).
Inspection of the gel shown in Fig. 5B indicated that HIV-2 RNase H-catalyzed RNA removal was less efficient than cleavage by HIV-1 RT. This impression was confirmed when the total amount of cleavage products, as determined by PhosphorImager analysis, was plotted as a function of time (Fig. 5C). At early times (up to 5 min), HIV-1 RT had a higher rate of cleavage than HIV-2 RT (Fig. 5C, inset), but at later times, the difference in total cleavage products was less pronounced. A plot of the amount of the final 8-nt secondary cleavage product versus time gave more striking results (Fig. 5D). In this case, both the rate and extent of secondary cleavage were lower for HIV-2 RT, and at 60 min, the difference between the amounts of the HIV-1 and HIV-2 products was ∼3.2-fold. It is of interest to note that with HIV-1 RT, RNA products of from 13 to 17 nt decreased with time, while smaller products accumulated as the time of incubation was increased; this indicates that the larger RNAs were efficiently processed by secondary cleavages. With HIV-2 RT, the 15- to 17-nt products either reached a plateau value or decreased with time, whereas products of from 8 to 14 nt increased with time. The behavior of HIV-2 RT most likely reflects the lower rate and lesser extent of secondary RNase H cleavage and is in general accord with the results for assay of 5′-directed RNase H activity (Fig. 4).
(iii) Removal of the 15-nt RNA PPT primer from nascent plus-strand DNA.
To obtain a further comparison of the RNase H activities of the two HIV enzymes, we took advantage of an assay that mimics the RNA PPT primer removal step in plus-strand DNA synthesis (Fig. 1) (reference 5 and references therein; 50, 51, 59a). A schematic diagram illustrating the experimental protocol is given in Fig. 6C. Note that correct cleavage of the labeled substrate by the RNase H activities of HIV-1 or HIV-2 RTs occurs at the RNA-DNA junction, yielding the unlabeled primer and labeled 20-nt DNA (Fig. 6C, step 2).
FIG. 6.

Time course of RNase H-catalyzed RNA PPT primer removal. Aliquots were removed from reaction mixtures at the indicated times, and samples were run in 8% sequencing gels. The amount of 32P-labeled 20-nt product formed after PPT primer removal was quantified by PhosphorImager analysis. The percent cleavage was calculated by comparing the volume of the 20-nt band at each time point with the value for the volume of the 35-nt product synthesized with T4 DNA polymerase, set at 100%. (A) HIV-1 substrate. Circles, HIV-1 RT; squares, HIV-2 440 RT. (B) HIV-2 substrate. Circles, HIV-1 RT; squares, HIV-2 440 RT; triangles, HIV-2 470 RT; inverted triangles, HIV-2 484 RT. (C) Schematic diagram of the PPT primer removal assay. Step 1, extension by T4 DNA polymerase. The 15-nt RNA PPT and the plus-strand 20-nt DNA extension are represented by an open rectangle and a thick dashed line, respectively. The asterisk indicates that the 20-nt product is internally labeled with 32P. Step 2, PPT primer removal by RT. The DNA moiety attached to the PPT is shown as a solid rectangle. The vertical arrow indicates the site of cleavage, i.e., at the junction of the 3′ end of the RNA primer and the 5′ end of nascent plus-strand DNA. The 20-nt labeled DNA product and the unlabeled 15-nt primer, which are released, are also shown.
Figure 6 shows that with an HIV-1 primer-template, HIV-1 RT catalyzed cleavage of almost 80% of the HIV-1 substrate (Fig. 6A) and ∼70% of the HIV-2 substrate (Fig. 6B) in 30 min. Using an HIV-2 primer-template (Fig. 6B), the initial rates of cleavage by the three HIV-2 enzymes (440, 470, and 484) were similar. The plateau levels were almost the same, and in repeated experiments, none of the three enzymes appeared to be significantly more active than the other two (data not shown). These observations indicate that the three HIV-2 RTs, which differ only in the C-terminal position of the p54 subunit, had similar RNase H activities in this assay. Similar results were also obtained when the HIV-2 enzyme was assayed with the HIV-1 substrate (Fig. 6A). The highest rate of primer removal was achieved in the reaction with the HIV-1 enzyme and the HIV-1 substrate but was not significantly different from the cleavage rates in the other reactions. The extent of cleavage reached by HIV-1 RT with either substrate was ∼2-fold higher than that observed for the HIV-2 enzymes.
Collectively, the results of the three RNase H assays indicate that the HIV-1 and HIV-2 enzymes have similar cleavage activities, although HIV-2 RT was less active than HIV-1 RT (Fig. 4 to 6), especially at low enzyme concentrations. Interestingly, the assays for 5′-directed RNase H cleavage (Fig. 4) and tRNA removal (Fig. 5) also indicate that HIV-2 RT catalyzes secondary cleavages less efficiently than HIV-1 RT.
Initiation of plus-strand DNA synthesis.
During reverse transcription, HIV-1 plus-strand DNA synthesis is initiated by a 15-nt RNA PPT primer (Fig. 1) (see the review in reference 5). Once the primer is selected, it is extended by the polymerase activity of RT and is then rapidly removed by RNase H. Thus, an assay that measures PPT extension will also measure primer removal (52). We have already shown above (Fig. 6) that the PPT primer removal activities of HIV-1 and HIV-2 are not dramatically different. Thus, if we use an assay that models both PPT extension and removal (52) and find major differences in HIV-1 and HIV-2 RT activities, we can attribute these differences primarily to differences in the ability to extend the primer. Previously, it was found (16a, 18, 20, 51) that the ability of HIV-1 RT to extend the RNA PPT primer is one of the most stringent tests of RT activity. For example, certain HIV-1 RT mutants with single substitutions in primer grip residues can easily extend a DNA version of the PPT primer but completely lack the ability to extend the RNA PPT (18, 51).
The amount of HIV-1 or HIV-2 RT used in the RNA PPT removal assay was 1 pmol (Fig. 6). In the plus-strand initiation assay, 1 pmol of HIV-1 RT was also used and was sufficient to detect activity. However, the HIV-2 RTs were active only when 2 pmol was added. In addition, HIV-2 RT activity was sensitive to high salt concentrations, and the monocation and MgCl2 concentrations in the reaction mixture were therefore adjusted to 58 and 3 mM, respectively. It should be noted that both the HIV-1 and HIV-2 enzymes produced the expected 20-nt DNA product.
Figure 7 shows that HIV-1 RT was able to extend 100% of the PPT primer in 60 min with either HIV-1 (Fig. 7A) or HIV-2 (Fig. 7B) substrate. In contrast, with HIV-2 RT and the HIV-1 substrate, the rate at which the labeled 20-nt DNA product was generated, as well as the amount produced in 60 min (∼25 to 30% of the control), were severalfold lower than that seen with HIV-1 RT (Fig. 7A), even though twice as much HIV-2 RT was used. Interestingly, even with the homologous HIV-2 substrate (Fig. 7B), all three HIV-2 RTs exhibited low rates of activity and reached plateau levels of ∼25 to 35% of the control. Increasing the amount of HIV-2 RT in the reaction mixture to as high as 10 pmol did not increase initiation activity under our assay conditions (data not shown). However, when the monocation concentration was reduced to 5 mM (Fig. 7C), extension of the HIV-2 PPT by 1 pmol of HIV-2 RT reached a plateau value only twofold lower than that reached by 1 pmol of HIV-1 RT with an HIV-1 substrate. In contrast, similar levels of HIV-1 RT activity were observed at the two different salt concentrations (compare Fig. 7C with A).
FIG. 7.

Time course of initiation of plus-strand DNA synthesis. The assay for initiation of HIV-1 or HIV-2 plus-strand DNA synthesis was performed as described in Materials and Methods. Aliquots were removed at the indicated times, and samples were run in 8% sequencing gels. The amount of 32P-labeled 20-nt DNA formed was calculated by comparing the volume of the 20-nt band at each time point with the value for the volume of the 35-nt RNA-DNA extension product synthesized with T4 DNA polymerase, which was set at 100%. (A) HIV-1 substrate. Reactions were carried out with the following amounts of enzyme: 1 pmol of HIV-1 RT (circles) and 2 pmol of HIV-2 440 RT (squares). (B) HIV-2 substrate. Reactions were carried out with 1 pmol of HIV-1 (circles) or 2 pmol of the three HIV-2 RTs: 440 (squares), 470 (triangles), and 484 (inverted triangles). (C) HIV-1 and HIV-2 substrates. The reaction mixtures contained a final monocation concentration of 5 mM instead of 58 mM, and reactions were carried out with the HIV-1 substrate and 1 pmol of HIV-1 RT (circles) and with the HIV-2 substrate and 1 pmol of HIV-2 440 RT (squares). (D) Schematic diagram of the initiation assay. The procedure was the same as that used for the experiment in Fig. 6, except that in this assay, both primer extension and primer removal are catalyzed by RT. The representation of the nucleic acid reactants is the same as in Fig. 6C. The vertical arrow in step 2 indicates the site of RNase H cleavage.
These findings indicate that the levels at which RT initiated plus-strand DNA synthesis were the same regardless of whether the homologous or heterologous RNA PPT primer was extended. The results also demonstrate that the three HIV-2 RTs have similar levels of PPT priming activity. Interestingly, compared with HIV-1, the HIV-2 activity is more salt sensitive.
DISCUSSION
A major goal of this study was to perform an in-depth investigation of the enzymatic activities of HIV-2 RT during various steps in reverse transcription and to provide a comparison with the analogous activities of HIV-1 RT. In certain instances, the activity of each enzyme was assessed with both HIV-1 and HIV-2 substrates (Fig. 3, 6, and 7). To determine whether the difference in the C-terminal position of the smaller subunits of HIV-1 and HIV-2 RT (15) has functional significance, three HIV-2 RTs were expressed, each differing in the length of the small subunit, i.e., HIV-2 RTs 440, 470, and 484. Our data indicate that despite the fact that the authentic p54 terminates at residue 484 (15), there are no significant differences between the polymerase or RNase H activities of the three HIV-2 RT heterodimers in our assays (Fig. 6 and 7 and data not shown).
A critical finding of this work is that in general, the functional activities of the HIV-1 and HIV-2 RT enzymes are comparable, although it is possible to detect significant quantitative differences under certain assay conditions. In earlier work (16, 28, 29, 61), the DNA polymerase activity of HIV-2 RT was found to be similar to that of HIV-1 RT. The results for RNA-dependent synthesis of minus-strand DNA with an RT/primer-template ratio of 1 showed no difference in HIV-1 and HIV-2 RT activities, in agreement with these previous studies; however, when this ratio was reduced to 0.1, HIV-1 RT exhibited a higher rate and greater extent of DNA synthesis (almost 3-fold and ∼2-fold, respectively) than the HIV-2 enzyme (Fig. 2). In the case of DNA-dependent DNA polymerase activity primed by an HIV-1 or HIV-2 DNA version of the PPT, HIV-2 RT was also as efficient in extending the primer as HIV-1 RT at an RT/primer-template ratio of 1. However, when this ratio was reduced to 0.01, total DNA synthesis in HIV-2 RT-catalyzed reactions was significantly reduced, whereas the effect on HIV-1 RT reactions was less pronounced (Fig. 3). This indicates that intrinsic differences in the HIV-1 and HIV-2 DNA polymerase activities are more apparent when the enzyme concentration is lowered.
In previous in vitro studies of HIV-1 plus-strand initiation, it was demonstrated that residues 224 to 235 in the primer grip region in the p66 palm subdomain (33) are important participants in this process (18, 51). Thus, single-amino-acid substitutions in these residues abrogate the ability of RT to extend the RNA PPT primer, but surprisingly, extension of a DNA version of this primer is still detected (18, 51). It is interesting that in the present study we find that while HIV-2 RT can extend the DNA PPT as well as HIV-1 RT under standard assay conditions (Fig. 3) (see above), the two enzymes exhibit comparable abilities to extend the natural RNA PPT primer only when the Mg2+ and salt concentrations are drastically reduced (Fig. 7).
One possible cause for the discordant activities of HIV-1 and HIV-2 RT may be a difference in the architecture of the respective primer grip regions (33, 55). X-ray crystallographic studies of HIV-1 RT indicate that residues in β strands 12 and 13 are involved in forming the hydrophobic pocket needed to bind nevirapine and other NNRTIs (35, 64). Although the structures of the HIV-1 and HIV-2 binding sites show some overall similarity (55), HIV-2 RT is resistant to NNRTIs (2, 42, 47, 62, 63), presumably because it lacks some of the amino acids that induce formation of the requisite binding pocket (2, 42, 55, 63, 72). For example, residue 235 in the primer grip is His in HIV-1 RT and Trp in HIV-2 RT; this change results in the widening of the binding pocket in HIV-2 RT (55). In addition, Phe 227 in the HIV-1 primer grip is one of the critical residues lining the binding pocket, yet in HIV-2 RT, the corresponding residue is Tyr. Significantly, when Phe was substituted for Tyr in HIV-2 RT, the chimeric enzyme became almost as sensitive to TIBO (an NNRTI drug similar to nevirapine) as HIV-1 RT (72). These considerations lead us to conclude that structural differences in the HIV-1 and HIV-2 RT primer grip regions may contribute to the striking contrast in the abilities of the two enzymes to initiate plus-strand DNA synthesis with the RNA PPT primer and the greater salt sensitivity of HIV-2 RT.
Another factor to consider, particularly in light of the salt sensitivity of the HIV-2 enzyme, is that HIV-2 RT may have a lower affinity for binding to the RNA PPT primer-template than HIV-1 RT. Indeed, preliminary results from gel shift experiments indicate that binding of HIV-2 RT to an HIV-2 RNA PPT primer-template is less efficient than binding of HIV-1 RT to the corresponding HIV-1 primer-template (K. Post and J. G. Levin, unpublished observations). From the results of assays of PPT primer removal (Fig. 6) and plus-strand initiation (Fig. 7), it would appear that under the conditions used for these assays, potential differences in binding affinity and structure have a greater effect on the initiation reaction than on primer removal. Interestingly, a recent report demonstrates that the Km value for HIV-2 RT, as determined with a synthetic polynucleotide substrate [poly(rA) · poly(dT)] in an RNase H assay, is almost 10-fold greater than the corresponding value for HIV-1 RT; it was suggested that the identity of the residue at position 294 (Pro in HIV-1 and Gln in HIV-2) in the thumb subdomain of the small subunit may be responsible for this difference (60).
The issue of whether HIV-1 and HIV-2 have similar RNase H activities is also important, since several events in reverse transcription are absolutely dependent on RNase H cleavage (Fig. 1) (5). Here, we have analyzed RNase H function in three different model systems (Fig. 4 to 6). The cleavage patterns are qualitatively similar in HIV-1 and HIV-2 reactions, as was also reported for another RNase H assay (61).
In each of our model assays, we found relatively small quantitative differences between HIV-1 and HIV-2 RNase H activities under standard assay conditions, in agreement with Fan et al. (16). However, with lower enzyme concentrations, we observed that HIV-2 RT is compromised in its ability to catalyze total RNase H cleavage (Fig. 4 and 5), as reported in other studies (28, 29, 61, 62). In addition, secondary RNase H cleavage was less efficient under the conditions of the 5′-directed RNase H (Fig. 4) and tRNA removal (Fig. 5) assays. These results are consistent with the finding that HIV-2 RT is much less efficient than HIV-1 RT in an HIV-1 minus-strand transfer assay (61), which is dependent on primary and secondary RNase H cleavage events (5, 48, 66). Differences in binding affinity to the substrates in these assays (see above) could explain why HIV-2 RT activity is reduced relative to that of HIV-1 RT when the concentration of enzyme is decreased.
The finding that both the polymerase and RNase H activities are lower for HIV-2 RT than for HIV-1 RT under certain conditions raises the following question: do these differences have any impact on HIV-2 replication? It has been estimated that virions contain eight 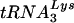 molecules for two copies of genomic RNA (32) and 50 to 100 copies of RT (8). From these calculations, it would appear that the reduced efficiency of HIV-2 RT seen under limiting conditions in vitro is unlikely to affect the extent of reverse transcription in vivo. This conclusion is supported by the observation that the levels of proviral DNA in HIV-1- and HIV-2-infected individuals are comparable (49).
molecules for two copies of genomic RNA (32) and 50 to 100 copies of RT (8). From these calculations, it would appear that the reduced efficiency of HIV-2 RT seen under limiting conditions in vitro is unlikely to affect the extent of reverse transcription in vivo. This conclusion is supported by the observation that the levels of proviral DNA in HIV-1- and HIV-2-infected individuals are comparable (49).
In the past, many drug-screening assays for RT inhibitors have been performed with homopolymeric substrates and may therefore not always accurately reflect in vivo activities. To increase the efficacy of drug discovery, it is clear that more specific in vitro assays are needed (e.g., see reference 17). The simple oligonucleotide assays used in this study are also attractive candidates for development of specific assays and could be incorporated into an overall strategy for drug-screening protocols. One example is the assay of plus-strand DNA initiation (Fig. 7) (51, 52). On the basis of the unusual structural features of the PPT (27, 36, 58), it has been suggested that the PPT or the RT complex with the RNA-DNA PPT-containing hybrid may be excellent targets for drug design (27, 58).
In summary, because HIV-2 infection is a major public health issue in parts of Africa and Asia, knowledge of the functional activities of HIV-2 RT is critical for developing effective drug screens, which have often been directed primarily at finding compounds that inhibit HIV-1 RT. The fact that most NNRTIs are not effective against HIV-2 RT or HIV-2 infection (2, 42, 47, 62, 63) and our finding that HIV-2 RT is less active than HIV-1 RT in the assay for plus-strand DNA initiation (Fig. 7) support the idea that structural differences between the HIV-1 and HIV-2 RTs (55) will have to be taken into account in the search for anti-HIV-2 agents. Thus, it is clear that strategies to combat HIV-2-induced AIDS will require more detailed analysis of the three-dimensional structure of HIV-2 RT and development of high-throughput assays based on specific steps in HIV replication, including reverse transcription.
Acknowledgments
We are grateful to Megan Dueck, Wilfredo Ayala-López, and Brenda Soto for outstanding assistance with many of these experiments. We also thank Jason Rausch for helpful comments.
This work was supported in part by funds from the National Institutes of Health Intramural AIDS Targeted Antiviral Program awarded to J.G.L.
REFERENCES
- 1.Adachi, A., H. E. Gendelman, S. Koenig, T. Folks, R. Willey, A. Rabson, and M. A. Martin. 1986. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J. Virol. 59:284-291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bacolla, A., C.-K. Shih, J. M. Rose, G. Piras, T. C. Warren, C. A. Grygon, R. H. Ingraham, R. C. Cousins, D. J. Greenwood, D. Richman, Y.-C. Cheng, and J. A. Griffin. 1993. Amino acid substitutions in HIV-1 reverse transcriptase with corresponding residues from HIV-2. Effect on kinetic constants and inhibition by non-nucleoside analogs. J. Biol. Chem. 268:16571-16577. [PubMed] [Google Scholar]
- 3.Brulé, F., G. Bec, G. Keith, S. F. J. Le Grice, B. P. Roques, B. Ehresmann, C. Ehresmann, and R. Marquet. 2000. In vitro evidence for the interaction of tRNA3Lys with U3 during the first strand transfer of HIV-1 reverse transcription. Nucleic Acids Res. 28:634-640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Buckheit, R. W., Jr., K. Watson, V. Fliakas-Boltz, J. Russell, T. L. Loftus, M. C. Osterling, J. A. Turpin, L. A. Pallansch, E. L. White, J.-W. Lee, S.-H. Lee, J.-W. Oh, H.-S. Kwon, S.-G. Chung, and E.-H. Cho. 2001. SJ-3366, a unique and highly potent nonnucleoside reverse transcriptase inhibitor of human immunodeficiency virus type 1 (HIV-1) that also inhibits HIV-2. Antimicrob. Agents Chemother. 45:393-400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Champoux, J. J. 1993. Roles of ribonuclease H in reverse transcription, p. 103-117. In A. M. Skalka and S. P. Goff (ed.), Reverse transcriptase. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 6.Chattopadhyay, D., D. B. Evans, M. R. Deibel, Jr., A. F. Vosters, F. M. Eckenrode, H. M. Einspahr, J. O. Hui, A. G. Tomasselli, H. A. Zurcher-Neely, R. L. Heinrikson, and S. K. Sharma. 1992. Purification and characterization of heterodimeric human immunodeficiency virus type 1 (HIV-1) reverse transcriptase produced by in vitro processing of p66 with recombinant HIV-1 protease. J. Biol. Chem. 267:14227-14232. [PubMed] [Google Scholar]
- 7.Clavel, F., D. Guétard, F. Brun-Vézinet, S. Chamaret, M.-A. Rey, M. O. Santos-Ferreira, A. G. Laurent, C. Dauguet, C. Katlama, C. Rouzioux, D. Klatzmann, J. L. Champalimaud, and L. Montagnier. 1986. Isolation of a new human retrovirus from West African patients with AIDS. Science 233:343-346. [DOI] [PubMed] [Google Scholar]
- 8.Coffin, J. M., S. H. Hughes, and H. E. Varmus. 1997. Retroviruses. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y. [PubMed]
- 9.DeStefano, J. J. 1995. The orientation of binding of human immunodeficiency virus reverse transcriptase on nucleic acid hybrids. Nucleic Acids Res. 23:3901-3908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.DeStefano, J. J., J. V. Cristofaro, S. Derebail, W. P. Bohlayer, and M. J. Fitzgerald-Heath. 2001. Physical mapping of HIV reverse transcriptase to the 5′ end of RNA primers. J. Biol. Chem. 276:32515-32521. [DOI] [PubMed] [Google Scholar]
- 11.DeStefano, J. J., L. M. Mallaber, P. J. Fay, and R. A. Bambara. 1993. Determinants of the RNase H cleavage specificity of human immunodeficiency virus reverse transcriptase. Nucleic Acids Res. 21:4330-4338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.DeVico, A. L., T. D. Copeland, F. di Marzo Veronese, S. Oroszlan, R. C. Gallo, and M. G. Sarngadharan. 1989. Purification and partial characterization of human immunodeficiency virus type 2 reverse transcriptase. AIDS Res. Hum. Retrovir. 5:51-60. [DOI] [PubMed] [Google Scholar]
- 13.Ding, J., K. Das, Y. Hsiou, S. G. Sarafianos, A. D. Clark, Jr., A. Jacobo-Molina, C. Tantillo, S. H. Hughes, and E. Arnold. 1998. Structure and functional implications of the polymerase active site region in a complex of HIV-1 RT with a double-stranded DNA template-primer and an antibody Fab fragment at 2.8 Å resolution. J. Mol. Biol. 284:1095-1111. [DOI] [PubMed] [Google Scholar]
- 14.Driscoll, M. D., and S. H. Hughes. 2000. Human immunodeficiency virus type 1 nucleocapsid protein can prevent self-priming of minus-strand strong stop DNA by promoting the annealing of short oligonucleotides to hairpin sequences. J. Virol. 74:8785-8792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fan, N., K. B. Rank, J. W. Leone, R. L. Heinrikson, C. A. Bannow, C. W. Smith, D. B. Evans, S. M. Poppe, W. G. Tarpley, D. J. Rothrock, A. G. Tomasselli, and S. K. Sharma. 1995. The differential processing of homodimers of reverse transcriptases from human immunodeficiency viruses type 1 and 2 is a consequence of the distinct specificities of the viral proteases. J. Biol. Chem. 270:13573-13579. [PubMed] [Google Scholar]
- 16.Fan, N., K. B. Rank, S. M. Poppe, W. G. Tarpley, and S. K. Sharma. 1996. Characterization of the p68/p58 heterodimer of human immunodeficiency virus type 2 reverse transcriptase. Biochemistry 35:1911-1917. [DOI] [PubMed] [Google Scholar]
- 16a.Fuentes, G. M., L. Rodríguez-Rodríguez, P. J. Fay, and R. A. Bambara. 1995. Use of an oligonucleotide containing the polypurine tract sequence as a primer by HIV-1 reverse transcriptase. J. Biol. Chem. 270:28169-28176. [DOI] [PubMed]
- 17.Gabbara, S., W. R. Davis, L. Hupe, D. Hupe, and J. A. Peliska. 1999. Inhibitors of DNA strand transfer reactions catalyzed by HIV-1 reverse transcriptase. Biochemistry 38:13070-13076. [DOI] [PubMed] [Google Scholar]
- 18.Ghosh, M., J. Williams, M. D. Powell, J. G. Levin, and S. F. J. Le Grice. 1997. Mutating a conserved motif of the HIV-1 reverse transcriptase palm subdomain alters primer utilization. Biochemistry 36:5758-5768. [DOI] [PubMed] [Google Scholar]
- 19.Golinelli, M.-P., and S. H. Hughes. 2002. Nontemplated base addition by HIV-1 RT can induce nonspecific strand transfer in vitro. Virology 294:122-134. [DOI] [PubMed] [Google Scholar]
- 20.Götte, M., M. Kameoka, N. McLellan, L. Cellai, and M. A. Wainberg. 2001. Analysis of efficiency and fidelity of HIV-1 (+)-strand DNA synthesis reveals a novel rate-limiting step during retroviral reverse transcription. J. Biol. Chem. 276:6711-6719. [DOI] [PubMed] [Google Scholar]
- 21.Graves, M. C., M. C. Meidel, Y.-C. E. Pan, M. Manneberg, H.-W. Lahm, and F. Grüninger-Leitch. 1990. Identification of a human immunodeficiency virus-1 protease cleavage site within the 66,000 dalton subunit of reverse transcriptase. Biochem. Biophys. Res. Commun. 168:30-36. [DOI] [PubMed] [Google Scholar]
- 22.Guo, J., L. E. Henderson, J. Bess, B. Kane, and J. G. Levin. 1997. Human immunodeficiency virus type 1 nucleocapsid protein promotes efficient strand transfer and specific viral DNA synthesis by inhibiting TAR-dependent self-priming from minus-strand strong-stop DNA. J. Virol. 71:5178-5188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guo, J., T. Wu, J. Anderson, B. F. Kane, D. G. Johnson, R. J. Gorelick, L. E. Henderson, and J. G. Levin. 2000. Zinc finger structures in the human immunodeficiency virus type 1 nucleocapsid protein facilitate efficient minus- and plus-strand transfer. J. Virol. 74:8980-8988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guo, J., T. Wu, B. F. Kane, D. G. Johnson, L. E. Henderson, R. J. Gorelick, and J. G. Levin. 2002. Subtle alterations of the native zinc finger structures have dramatic effects on the nucleic acid chaperone activity of human immunodeficiency virus type 1 nucleocapsid protein. J. Virol. 76:4370-4378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guo, J., W. Wu, Z. Y. Yuan, K. Post, R. J. Crouch, and J. G. Levin. 1995. Defects in primer-template binding, processive DNA synthesis, and RNase H activity associated with chimeric reverse transcriptases having the murine leukemia virus polymerase domain joined to Escherichia coli RNase H. Biochemistry 34:5018-5029. [DOI] [PubMed] [Google Scholar]
- 26.Guyader, M., M. Emerman, P. Sonigo, F. Clavel, L. Montagnier, and M. Alizon. 1987. Genome organization and transactivation of the human immunodeficiency virus type 2. Nature 326:662-669. [DOI] [PubMed] [Google Scholar]
- 27.Han, G. W., M. L. Kopka, D. Cascio, K. Grzeskowiak, and R. E. Dickerson. 1997. Structure of a DNA analog of the primer for HIV-1 RT second strand synthesis. J. Mol. Biol. 269:811-826. [DOI] [PubMed] [Google Scholar]
- 28.Hizi, A., R. Tal, and S. H. Hughes. 1991. Mutational analysis of the DNA polymerase and ribonuclease H activities of human immunodeficiency virus type 2 reverse transcriptase expressed in Escherichia coli. Virology 180:339-346. [DOI] [PubMed] [Google Scholar]
- 29.Hizi, A., R. Tal, M. Shaharabany, and S. Loya. 1991. Catalytic properties of the reverse transcriptases of human immunodeficiency viruses type 1 and type 2. J. Biol. Chem. 266:6230-6239. [PubMed] [Google Scholar]
- 30.Hong, M. K., E. J. Harbron, D. B. O'Connor, J. Guo, P. F. Barbara, J. G. Levin, and K. Musier-Forsyth. 2003. Nucleic acid conformational changes essential for HIV-1 nucleocapsid protein-mediated inhibition of self-priming in minus-strand transfer. J. Mol. Biol. 325:1-10. [DOI] [PubMed] [Google Scholar]
- 31.Huang, H., R. Chopra, G. L. Verdine, and S. C. Harrison. 1998. Structure of a covalently trapped catalytic complex of HIV-1 reverse transcriptase: implications for drug resistance. Science 282:1669-1675. [DOI] [PubMed] [Google Scholar]
- 32.Huang, Y., J. Mak, Q. Cao, Z. Li, M. A. Wainberg, and L. Kleiman. 1994. Incorporation of excess wild-type and mutant tRNA 3Lys into human immunodeficiency virus type 1. J. Virol. 68:7676-7683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jacobo-Molina, A., J. Ding, R. G. Nanni, A. D. Clark, Jr., X. Lu, C. Tantillo, R. L. Williams, G. Kamer, A. L. Ferris, P. Clark, A. Hizi, S. H. Hughes, and E. Arnold. 1993. Crystal structure of human immunodeficiency virus type 1 reverse transcriptase complexed with double-stranded DNA at 3.0 Å resolution shows bent DNA. Proc. Natl. Acad. Sci. USA 90:6320-6324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Klutch, M., A. M. Woerner, C. J. Marcus-Sekura, and J. G. Levin. 1998. Generation of HIV-1/HIV-2 cross-reactive peptide antisera by small sequence changes in HIV-1 reverse transcriptase and integrase immunizing peptides. J. Biomed. Sci. 5:192-202. [DOI] [PubMed] [Google Scholar]
- 35.Kohlstaedt, L. A., J. Wang, J. M. Friedman, P. A. Rice, and T. A. Steitz. 1992. Crystal structure at 3.5 Å resolution of HIV-1 reverse transcriptase complexed with an inhibitor. Science 256:1783-1790. [DOI] [PubMed] [Google Scholar]
- 36.Kvaratskhelia, M., S. R. Budihas, and S. F. J. Le Grice. 2002. Pre-existing distortions in nucleic acid structure aid polypurine tract selection by HIV-1 reverse transcriptase. J. Biol. Chem. 277:16689-16696. [DOI] [PubMed] [Google Scholar]
- 37.Lapadat-Tapolsky, M., C. Gabus, M. Rau, and J.-L. Darlix. 1997. Possible roles of HIV-1 nucleocapsid protein in the specificity of proviral DNA synthesis and in its variability. J. Mol. Biol. 268:250-260. [DOI] [PubMed] [Google Scholar]
- 38.Le Grice, S. F. J., C. E. Cameron, and S. J. Benkovic. 1995. Purification and characterization of human immunodeficiency virus type 1 reverse transcriptase. Methods Enzymol. 262:130-144. [DOI] [PubMed] [Google Scholar]
- 39.Le Grice, S. F. J., R. Zehnle, and J. Mous. 1988. A single 66-kilodalton polypeptide processed from the human immunodeficiency virus type 2 pol polyprotein in Escherichia coli displays reverse transcriptase activity. J. Virol. 62:2525-2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li, X., Y. Quan, E. J. Arts, Z. Li, B. D. Preston, H. de Rocquigny, B. P. Roques, J.-L. Darlix, L. Kleiman, M. A. Parniak, and M. A. Wainberg. 1996. Human immunodeficiency virus type 1 nucleocapsid protein (NCp7) directs specific initiation of minus-strand DNA synthesis primed by human tRNA3Lys in vitro: studies of viral RNA molecules mutated in regions that flank the primer binding site. J. Virol. 70:4996-5004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Loya, S., M. Bakhanashvili, Y. Kashman, and A. Hizi. 1995. Peyssonols A and B, two novel inhibitors of the reverse transcriptases of human immunodeficiency virus types 1 and 2. Arch. Biochem. Biophys. 316:789-796. [DOI] [PubMed] [Google Scholar]
- 42.Loya, S., M. Bakhanashvili, R. Tal, S. H. Hughes, P. L. Boyer, and A. Hizi. 1994. Enzymatic properties of two mutants of reverse transcriptase of human immunodeficiency virus type 1 (tyrosine 181 → isoleucine and tyrosine 188 → leucine), resistant to nonnucleoside inhibitors. AIDS Res. Hum. Retrovir. 10:939-946. [DOI] [PubMed] [Google Scholar]
- 43.Milton, J., M. J. Slater, A. J. Bird, D. Spinks, G. Scott, C. E. Price, S. Downing, D. V. S. Green, S. Madar, R. Bethell, and D. K. Stammers. 1998. Biaryl acids: novel non-nucleoside inhibitors of HIV reverse transcriptase types 1 and 2. Bioorg. Med. Chem. Lett. 8:2623-2628. [DOI] [PubMed] [Google Scholar]
- 44.Müller, B., T. Restle, H. Kühnel, and R. S. Goody. 1991. Expression of the heterodimeric form of human immunodeficiency virus type 2 reverse transcriptase in Escherichia coli and characterization of the enzyme. J. Biol. Chem. 266:14709-14713. [PubMed] [Google Scholar]
- 45.Palaniappan, C., G. M. Fuentes, L. Rodríguez-Rodríguez, P. J. Fay, and R. A. Bambara. 1996. Helix structure and ends of RNA/DNA hybrids direct the cleavage specificity of HIV-1 reverse transcriptase RNase H. J. Biol. Chem. 271:2063-2070. [PubMed] [Google Scholar]
- 46.Palaniappan, C., M. Wisniewski, P. S. Jacques, S. F. J. Le Grice, P. J. Fay, and R. A. Bambara. 1997. Mutations within the primer grip region of HIV-1 reverse transcriptase result in loss of RNase H function. J. Biol. Chem. 272:11157-11164. [DOI] [PubMed] [Google Scholar]
- 47.Pauwels, R., K. Andries, J. Desmyter, D. Schols, M. J. Kukla, H. J. Breslin, A. Raeymaeckers, J. Van Gelder, R. Woestenborghs, J. Heykants, K. Schellekens, M. A. C. Janssen, E. De Clercq, and P. A. J. Janssen. 1990. Potent and selective inhibition of HIV-1 replication in vitro by a novel series of TIBO derivatives. Nature 343:470-474. [DOI] [PubMed] [Google Scholar]
- 48.Peliska, J. A., and S. J. Benkovic. 1992. Mechanism of DNA strand transfer reactions catalyzed by HIV-1 reverse transcriptase. Science 258:1112-1118. [DOI] [PubMed] [Google Scholar]
- 49.Popper, S. J., A. D. Sarr, A. Guèye-Ndiaye, S. Mboup, M. E. Essex, and P. J. Kanki. 2000. Low plasma human immunodeficiency virus type 2 viral load is independent of proviral load: low virus production in vivo. J. Virol. 74:1554-1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Powell, M. D., W. A. Beard, K. Bebenek, K. J. Howard, S. F. J. Le Grice, T. A. Darden, T. A. Kunkel, S. H. Wilson, and J. G. Levin. 1999. Residues in the αH and αI helices of the HIV-1 reverse transcriptase thumb subdomain required for the specificity of RNase H-catalyzed removal of the polypurine tract primer. J. Biol. Chem. 274:19885-19893. [DOI] [PubMed] [Google Scholar]
- 51.Powell, M. D., M. Ghosh, P. S. Jacques, K. J. Howard, S. F. J. Le Grice, and J. G. Levin. 1997. Alanine-scanning mutations in the “primer grip” of p66 HIV-1 reverse transcriptase result in selective loss of RNA priming activity. J. Biol. Chem. 272:13262-13269. [DOI] [PubMed] [Google Scholar]
- 52.Powell, M. D., and J. G. Levin. 1996. Sequence and structural determinants required for priming of plus-strand DNA synthesis by the human immunodeficiency virus type 1 polypurine tract. J. Virol. 70:5288-5296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ratner, L., W. Haseltine, R. Patarca, K. J. Livak, B. Starcich, S. F. Josephs, E. R. Doran, J. A. Rafalski, E. A. Whitehorn, K. Baumeister, L. Ivanoff, S. R. Petteway, Jr., M. L. Pearson, J. A. Lautenberger, T. S. Papas, J. Ghrayeb, N. T. Chang, R. C. Gallo, and F. Wong-Staal. 1985. Complete nucleotide sequence of the AIDS virus, HTLV-III. Nature 313:277-284. [DOI] [PubMed] [Google Scholar]
- 54.Reeves, J. D., and R. W. Doms. 2002. Human immunodeficiency virus type 2. J. Gen. Virol. 83:1253-1265. [DOI] [PubMed] [Google Scholar]
- 55.Ren, J., L. E. Bird, P. P. Chamberlain, G. B. Stewart-Jones, D. I. Stuart, and D. K. Stammers. 2002. Structure of HIV-2 reverse transcriptase at 2.35-Å resolution and the mechanism of resistance to non-nucleoside inhibitors. Proc. Natl. Acad. Sci. USA 99:14410-14415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ren, J., J. Diprose, J. Warren, R. M. Esnouf, L. E. Bird, S. Ikemizu, M. Slater, J. Milton, J. Balzarini, D. I. Stuart, and D. K. Stammers. 2000. Phenylethylthiazolylthiourea (PETT) non-nucleoside inhibitors of HIV-1 and HIV-2 reverse transcriptases. Structural and biochemical analyses. J. Biol. Chem. 275:5633-5639. [DOI] [PubMed] [Google Scholar]
- 57.Rubinek, T., J. B. McMahon, and A. Hizi. 1994. Inhibition of reverse transcriptase of human immunodeficiency virus type 1 and chimeric enzymes of human immunodeficiency viruses types 1 and 2 by two novel non-nucleoside inhibitors. FEBS Lett. 350:299-303. [DOI] [PubMed] [Google Scholar]
- 58.Sarafianos, S. G., K. Das, C. Tantillo, A. D. Clark, Jr., J. Ding, J. M. Whitcomb, P. L. Boyer, S. H. Hughes, and E. Arnold. 2001. Crystal structure of HIV-1 reverse transcriptase in complex with a polypurine tract RNA:DNA. EMBO J. 20:1449-1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schultz, S. J., M. Zhang, C. D. Kelleher, and J. J. Champoux. 1999. Polypurine tract primer generation and utilization by Moloney murine leukemia virus reverse transcriptase. J. Biol. Chem. 274:34547-34555. [DOI] [PubMed] [Google Scholar]
- 59a.Schultz, S. J., M. Zhang, C. D. Kelleher, and J. J. Champoux. 2000. Analysis of plus-strand primer selection, removal, and reutilization by retroviral reverse transcriptases. J. Biol. Chem. 275:32299-32309. [DOI] [PubMed]
- 60.Sevilya, Z., S. Loya, N. Adir, and A. Hizi. 2003. The ribonuclease H activity of the reverse transcriptases of human immunodeficiency viruses type 1 and type 2 is modulated by residue 294 of the small subunit. Nucleic Acids Res. 31:1481-1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sevilya, Z., S. Loya, S. H. Hughes, and A. Hizi. 2001. The ribonuclease H activity of the reverse transcriptases of human immunodeficiency viruses type 1 and type 2 is affected by the thumb subdomain of the small protein subunits. J. Mol. Biol. 311:957-971. [DOI] [PubMed] [Google Scholar]
- 62.Shaharabany, M., and A. Hizi. 1992. The catalytic functions of chimeric reverse transcriptases of human immunodeficiency viruses type 1 and type 2. J. Biol. Chem. 267:3674-3678. [PubMed] [Google Scholar]
- 63.Shih, C.-K., J. M. Rose, G. L. Hansen, J. C. Wu, A. Bacolla, and J. A. Griffin. 1991. Chimeric human immunodeficiency virus type 1/type 2 reverse transcriptases display reversed sensitivity to nonnucleoside analog inhibitors. Proc. Natl. Acad. Sci. USA 88:9878-9882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Smerdon, S. J., J. Jäger, J. Wang, L. A. Kohlstaedt, A. J. Chirino, J. M. Friedman, P. A. Rice, and T. A. Steitz. 1994. Structure of the binding site for nonnucleoside inhibitors of the reverse transcriptase of human immunodeficiency virus type 1. Proc. Natl. Acad. Sci. USA 91:3911-3915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Smith, C. M., J. S. Smith, and M. J. Roth. 1999. RNase H requirements for the second strand transfer reaction of human immunodeficiency virus type 1 reverse transcription. J. Virol. 73:6573-6581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tanese, N., A. Telesnitsky, and S. P. Goff. 1991. Abortive reverse transcription by mutants of Moloney murine leukemia virus deficient in the reverse transcriptase-associated RNase H function. J. Virol. 65:4387-4397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Telesnitsky, A., and S. P. Goff. 1993. Strong-stop strand transfer during reverse transcription, p. 49-83. In A. M. Skalka and S. P. Goff (ed.), Reverse transcriptase. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 68.Wisniewski, M., M. Balakrishnan, C. Palaniappan, P. J. Fay, and R. A. Bambara. 2000. The sequential mechanism of HIV reverse transcriptase RNase H. J. Biol. Chem. 275:37664-37671. [DOI] [PubMed] [Google Scholar]
- 69.Wisniewski, M., M. Balakrishnan, C. Palaniappan, P. J. Fay, and R. A. Bambara. 2000. Unique progressive cleavage mechanism of HIV reverse transcriptase RNase H. Proc. Natl. Acad. Sci. USA 97:11978-11983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wisniewski, M., Y. Chen, M. Balakrishnan, C. Palaniappan, B. P. Roques, P. J. Fay, and R. A. Bambara. 2002. Substrate requirements for secondary cleavage by HIV-1 reverse transcriptase RNase H. J. Biol. Chem. 277:28400-28410. [DOI] [PubMed] [Google Scholar]
- 71.Wu, T., J. Guo, J. Bess, L. E. Henderson, and J. G. Levin. 1999. Molecular requirements for human immunodeficiency virus type 1 plus-strand transfer: analysis in reconstituted and endogenous reverse transcription systems. J. Virol. 73:4794-4805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yang, G., Q. Song, M. Charles, W. C. Drosopoulos, E. Arnold, and V. R. Prasad. 1996. Use of chimeric human immunodeficiency virus types 1 and 2 reverse transcriptases for structure-function analysis and for mapping susceptibility to nonnucleoside inhibitors. J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 11:326-333. [DOI] [PubMed] [Google Scholar]