

Abstract
In order to predict the binding regions within the complex formed by Toll-like receptor 5 (TLR-5) and flagellin, a complementary hydropathy between the two proteins was sought. A region common to the flagellins of Salmonella enterica serovar Typhimurium, Pseudomonas aeruginosa, and Listeria monocytogenes was shown to be hydropathically complementary to the 552-to-561 fragment of TLR-5, whose sequence is EILDISRNQL. The hydrophobicity profile of this region is shared with flagellins of 377 bacterial species out of a total of 723 publicly available sequences. A conformational analysis of the predicted binding site of TLR-5, whose structure is still unknown, was carried out with a methodology already applied to similar problems. To sample the conformations available to the peptide chain, a plot of the number of conformations per unit energy interval (density of states) versus energy was built. Following a theoretical argument, conformations belonging to maxima in this plot were selected. The most stable structure obtained in this search, an α-helical conformation, was shown to form the electrostatic interactions Glu552-Gln89, Asp555-Arg92, and Arg558-Glu93 with the predicted binding site of the flagellin of S. enterica serovar Typhimurium, formed by the 88-to-97 chain fragment (LQRVRELAVQ), which is likewise α helical.
Binding of bacterial flagellin to Toll-like receptor 5 (TLR-5) has been shown (13) to elicit recognition and response by the innate immune system. The sequences of the flagellins of Salmonella enterica serovar Typhimurium (11), Pseudomonas aeruginosa (31), and Listeria monocytogenes (27), for which TLR-5 activity has been demonstrated (13), have similar regions that may be the binding sites of TLR-5. The same sequence identity, limited to regions that include those considered important in this work, is extended to hundreds of known (http://us.expasy.org/sprot/sprot-top.html) flagellin sequences but not to all of them. Presumably, for this subset of flagellins, a common structure of the TLR-5-flagellin complex may be found.
To accomplish such a study, we made use of a strategy that, in this laboratory (6, 21, 28, 34) and elsewhere (14), has proved its value for the prediction of sequences of putative peptide ligands and the location of receptor binding sites. A hypothesis (15) that enables prediction of the structure of a peptide ligand was also used.
The complementary-hydropathy principle (3, 4, 5, 25) postulates the existence of stabilizing interactions between two peptide chains in whose sequences there is a correspondence between hydrophobic (hydrophilic) residues in one chain and hydrophilic (hydrophobic) residues in the other chain. According to this principle, two complementary peptide chains may form a complex, even though a pairwise binding of all complementary amino acid residues is not necessary.
There are now 69 reports of the successful exploitation of complementary hydropathy for the identification of interacting proteins (14), as opposed to only 3 negative reports (1, 9, 12), 1 of which (5) has already been refuted. Furthermore, the definition through this strategy of the binding site of collagenase (28) was subsequently confirmed (19) by site-directed mutagenesis. Also, the predicted site in the cellular prion molecule to which a putative ligand receptor would bind (21) was confirmed after the complete characterization of this molecule (34) by peptide inhibition of binding between the two molecules, as well as by removal (34) of the cellular prion binding site by sire-directed mutagenesis.
In the following discussion, it is shown how a search for complementary hydropathy between the sequences of TLR-5 and flagellin enables the prediction of binding sites. There still remains the problem, however, that although the structure of the flagellin of S. enterica serovar Typhimurium (26) has been determined by X-ray diffraction analysis, the structure of TLR-5 has not been experimentally determined.
To determine the structure of the TLR-5 fragment predicted to bind flagellin, we made use of a hypothesis that had already been applied to similar problems, such as α-MSH (18) and substrates (17) of oligopeptidase EC 24.15. According to this hypothesis, the structures adopted by peptide chains belong to conformational families that remain the same in different chemical environments. A transition to a new chemical environment causes a change in the probabilities of occurrence of the various conformational families so that, for instance, a conformational family that has a very low probability in an aqueous solution may have a very high probability in a lipid solution and vice versa.
According to the same hypothesis, a peptide chain and a protein fragment with the same sequence also occupy the same conformational families, although with different probabilities. Thus, various conformational families occupied by the predicted TLR-5 binding site may be tested against flagellin.
MATERIALS AND METHODS
To find the hydropathically complementary regions that are the predicted binding sites of TLR-5 and flagellin, the sequence of TLR-5 (7) was scanned against the sequences of the flagellins of S. enterica serovar Typhimurium (11), P. aeruginosa (31), and L. monocytogenes (27) with the in-house software HYDROLOG. These sequences were also submitted to a comparison, performed with the CLUSTALW (30) software, with 723 flagellin sequences downloaded from the SwissProt database (http://us.expasy.org/sprot/sprot-top.html). A conformational search (16) of the TLR-5 binding site was then carried out with five main chain rotamers (A [φ = −57, ψ = −47], B [φ = −139, ψ = 135], G [φ = −60, ψ = −30], D [φ = −90, ψ = 0], and E [φ = 70, ψ = −60], where φ and ψ [reference 18 and references quoted therein] are main chain dihedral angles) as described in the Appendix. For the side chain conformations, we made use of the gauche minus, trans, and gauche plus rotamers of the classification of Ponder and Richards (23) (numerals 1, 2, and 3 indicate, respectively, the gauche minus, trans, and gauche plus rotamers of χ1). Internal energies were calculated with the ECEPP/2 (35) force field. A search for complementary features on the surfaces of the binding site of TLR-5 and the S. enterica serovar Typhimurium flagellin, whose structure (26) was downloaded from the Protein Data Bank (2) website, was then carried out with translations and rotations of the two structures until a maximum score of hydrogen bonds and salt bridges was found. This search was done by hand with the software WEBLAB (provided by Accelrys) and WHAT IF (32). After that, the complex was docked with the HEX (24) software by using the parameters listed on page 11 of the manual (http://www.biochem.abdn.ac.uk/hex). Structures of the TLR-5-flagellin complexes of L. monocytogenes and P. aeruginosa were obtained with WHAT IF by making point mutations and structural optimizations in the TLR-5-flagellin complex of S. enterica serovar Typhimurium.
RESULTS AND DISCUSSION
Table 1 lists hydropathically complementary fragments belonging to TLR-5 and the flagellins of three bacterial species. Although, as shown in Table 1, many instances of complementary hydropathy between TLR-5 and the flagellins were found, there is only one TLR-5 region that, in all three cases, meets the criteria C ≥ Cm and L = 10 (C [defined in the equation in the Appendix], Cm [= 0.75], and L are, respectively, the actual and minimum complementary hydropathies and the fragment length). It was therefore concluded that this chain fragment is the binding site of TLR-5.
TABLE 1.
Regions of the TLR-5 sequence for which a complementary hydropathy with the flagellins of three bacterial species was founda
| TLR-5 | Flagellinb
|
||
|---|---|---|---|
| I | II | III | |
| LLERLQLLEL | ERLASGKRIN | ||
| LRILDLGQSQ | NLQRVRELAV | ||
| RILDLGQSQI | LQRVRELAVQ | LQRIRDLALQ | |
| IEVLNRDAFQ | RVRELAVQSA | RIRDLALQSA | |
| FNQIFTICED | NILQRIRDLA | ||
| NQIFTICEDE | ILQRMRQLAV | ILQRIRDLAL | |
| QIFTICEDEL | LQRVRELAVQ | LQRIRDLALQ | |
| RGVRLETLDL | LINIDLFNAK | ||
| VDLQRNHIGI | KIDAALAQVD | ||
| KLVHLPHIHF | VKNVQVANAD | ||
| IHFTANFLEL | EINNNLQRVR | ||
| NFLELSENRL | LQRVRELAVQ | LQRIRDLALQ | |
| LYFLLRVPQL | EINNNLQRVR | ||
| LPANLEILDI | TIDIDLKQIN | ||
| LEILDISRNQ | NLQRVRELAV | ||
| EILDISRNQL | LQRVRELAVQ | LQRMRQLAVQ | LQRIRDLALQ |
| EILDISRNQL | LQKIDAALAQ | ||
| ILDISRNQLL | QRVRELAVQS | QRIRDLALQS | |
| LRVLDITHNE | NLQRVRELAV | ||
| RVLDITHNEF | LQRVRELAVQ | LQRIRDLALQ | |
| LFLVITLVVI | KTQEYLRKNN | ||
| LVVIKFRGIC | NYNNIKLLDQ | ||
A complementary region common to the three flagellins is shown in bold.
I, S. enterica serovar Typhimurium; II, L. monocytogenes; III, P. aeruginosa.
In Fig. 1, the resemblance between the hydrophobicity profiles of the predicted binding sites of the three flagellins and their complementary hydropathies to the TLR-5 binding site are shown.
FIG. 1.

Hydrophobicity profiles (H, hydrophobicity measured by the Kyte and Doolittle scale [20]; aa, amino acid) of the peptides listed in Table 1. Solid line, TLR-5 binding site; dotted line, peptide I; dashed line, peptide II. The hydrophobicity profiles of peptides I and III are almost indistinguishable.
More specifically, the predicted binding site of TLR-5 is the 552-to-561 chain fragment, whose sequence is EILDISRNQL. A comparison with 723 flagellin sequences downloaded from the SwissProt (http://us.expasy.org/sprot/sprot-top.html) database showed that 377 flagellin sequences share with the flagellins of Table 1 binding regions having a sequence identity equal to or greater than 60% and hydrophobicity profiles similar to those of the predicted flagellin binding sites. This finding is in accordance with the previous finding (22) that hydropathy profiles tend to be conserved despite a lower sequence identity level. For these flagellins, a common TLR-5-flagellin complex may exist.
However, an additional difficulty is posed by the nonavailability of an experimentally determined structure of TLR-5. Although the structures of TLR-1 and TLR-2 are known (33), there is only partial homology among the sequences of TLR-1, TLR-2, and TLR-5 and this sequence homology does not encompass the predicted binding site. Therefore, to carry out the intended study, we counted only on the sequences of flagellin binding sites, the structure of S. enterica serovar Typhimurium flagellin, and the sequence of the TLR-5 binding site.
To cope with the absence of a TLR-5 structure, we did an exhaustive search of the conformations of its binding site that generated 14,414 different structures. The assumption that this high number of structures may be classified in a limited (15, 18) number of conformational families facilitated the, otherwise almost impossible, task of determining the active structure.
A set of conformational families available to the binding regions of TLR-5 was thus generated as described in the Appendix. Structures belonging to some of these conformational families are represented in Fig. 2.
FIG. 2.

Density-of-states (δ) versus energy (E, in kilocalories per mole) plot obtained for the 552-to-561 (EILDISRNQL) fragment of TLR-5. The energy was rescaled so that the minimum energy corresponds to zero. Thirteen density-of-states maxima are labeled. Some of the conformations found within these maxima are GGGAAAAAAD3112122121 (I), GGGAAAAAAD3112122111, GGGAAAAAGD3112122111 (II), GGGAAAAAAA3112122111 (III), GGGAAAAABA3112122131 (IV), GDDAAAAAGD1311122121 (V), GDDAAAAAAD1111121111 (VI), DDDGGAAAAG3311121121 (VII), GDDDAAAAAG2113211111 (VIII), DDDAAAAABD1311122131 (IX), GDDDDGAAAD1213111111 (X), DDDAAAAAGD1111231221 (XI), EGABBBBGAA1112221111 (XII), and BBDBGBDABG1311123131 (XIII), where the letters and numerals indicate, respectively, main chain and side chain rotamers, as defined in Materials and Methods. Structures found within three maxima are depicted. Stick representations of the polypeptide chain and ribbon representations of the secondary structure are shown.
In the density-of-states versus energy plot shown in Fig. 2 (see the Appendix for details), there are 350 density-of-states maxima. It has been argued (15, 16) that each maximum contains the most stable conformations in the corresponding energy range. Although the number of maxima approaches the number of conformational families, more than one conformational family may be found within the same density-of-states maximum, so that the determination of conformational families is not a straightforward procedure.
In the present study, however, the condition that TLR-5 forms a stable complex with flagellin considerably restricted the search. The most obvious choice, the ground level conformation (lists of rotamers that define chain conformations are shown in the legend to Fig. 2) that in Fig. 2 corresponds to the maximum labeled I in the density-of-states versus energy plot and to conformation I, was considered first.
Docking (see Materials and Methods) of this α-helical conformation to the, likewise α-helical, predicted binding site of flagellin showed that, as shown in Fig. 3, three electrostatic interactions may form. More specifically, these are the TLR-5-flagellin interactions Glu552-Gln89, Asp555-Arg92, and Arg558-Glu93. Model building (data not shown) showed that the Arg558-Glu (or other residue in position) 93 interaction is maintained even when Glu93 is replaced with glutamine in the L. monocytogenes flagellin and by aspartate in the P. aeruginosa flagellin.
FIG. 3.

Complex formed between the binding sites of flagellin and TLR-5 predicted by the complementary-hydropathy principle. (A) Flagellin complexed to the binding site of TLR-5. Stick representations of the 87-to-97 and 552-to-561 chain fragments, respectively, are shown. Ribbon representations of the flagellin structure and the TLR-5 binding site are also shown. (B) Stick representation of the binding sites of flagellin (gray) and TLR-5 (black). Hydrogen-bonded side chains are represented by dotted lines.
The same complementary positioning between side chains of TLR-5 and flagellin would occur, in principle, in other TLR-5 conformations whose 552-to-558 fragment has the same conformation of the previously described structure (GGGAAAA) and whose Glu552, Asp555, and Arg558 rotamers are also the same (respectively, 3, 2, and 2). Five such conformations were found within the density-of-states maxima labeled I, II, III, and IV in Fig. 2.
On the other hand, TLR-5 conformations distorted in relation to the above-shown conformation would not have the required positioning of side chains. Inspection of the conformations found within the density-of-states maxima labeled V to XIII in Fig. 2 showed that, in every case, the three described electrostatic interactions cannot form because the side chain positioning has been disrupted, as a comparison between conformations I and VIII and I and XII, depicted in Fig. 2, would show. These findings were confirmed by docking (see Materials and Methods) conformations VIII and XII (depicted in Fig. 2) to flagellin.
The binding site of flagellin found in the present study is located within constant domain D1, which is relatively conserved (8) among different species. Two regions required for TLR-5 activation were experimentally (8) identified in this domain. One region, on the N-terminal side, includes amino acids 57 to 190 and therefore includes a fragment sequentially homologous to the predicted binding site at positions 88 to 97. The other region is on the C-terminal side. It has been shown (8), with the aid of linker insertion mutagenesis experiments, that within this region, the short stretch from 485 to 489 (GAIQN) is necessary for interleukin-8 release. A search for complementary hydropathy with the parameters Cm ≥ 0.75 and L = 5 identified the 484-to-488 chain fragment (LGAIQ) in the sequence (29) of enteroaggregative Escherichia coli, which is hydropathically complementary to the 323-to-327 (KINKI) TLR-5 fragment. These fragments were not included in this molecular modeling study because the 484-to-489 flagellin fragment is not part of the experimentally determined (26) structure and because a search of short (L = 5), hydropathically complementary peptide fragments is too unrestricted and is likely to yield false positives.
The present study is part of the broader research field of protein-protein interactions. It has been shown (24) that surface complementarity, i.e., optimal positioning of interacting side chains, is an effective predictor of complexes formed by proteins. This criterion is in accordance with our calculations that showed that the side chains of Gln89, Arg92, and Glu93 on the surface of flagellin are optimally positioned to interact, respectively, with the side chains of Glu552, Asp555, and Arg558 on the surface of the predicted TLR-5 binding site.
Although the proposed TLR-5 and flagellin binding sites are hydropathically complementary, the predicted complex is stabilized by electrostatic interactions between hydrophilic side chains. In this study, we used an in silico search for complementary peptides in interacting proteins, combined with conformational analysis calculations, to predict the binding sites and the structure of the complex.
APPENDIX
Search for complementary hydropathy. The theoretical concepts behind the complementary-hydropathy principle have been well described previously (3, 4, 14, 25). Herein, we describe the method developed to find complementary ligand and receptor regions.
Amino acid residue hydrophobicities are evaluated with the Kyte and Doolittle (20) scale. The degree of complementary hydropathy between two peptide fragments of length L is calculated (10) with the equation 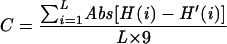
where H(i) and H′(i) are the Kyte and Doolittle indices of amino acid i in each sequence and Abs is absolute value. In the present study, the sequences of TLR-5 and flagellin were examined.
The receptor sequence is scanned against the ligand sequence in order to find regions corresponding to a high degree of complementary hydropathy, as calculated by the equation above. Important parameters in this search are the window length (L) and the minimum complementarity (Cm). Each i…i + L fragment in the receptor sequence is searched against fragments j…j + L in the ligand and those fragments for which C ≥ Cm are tabulated.
Determination of the conformational families of a peptide ligand. When there is substantial evidence that a certain peptide chain or a region of a longer chain is the binding site or peptide ligand that we are looking for but the corresponding structure is not known, we can generate a reasonably complete set of conformational families and test the binding of each one.
To carry out the search for conformational families, a plot of the density of states (δ, which is the number of conformations per unit energy interval) versus energy is obtained. It has been argued (15, 16) that minima in this plot correspond to kinetic barriers in the potential energy landscape and maxima correspond to energy minima that, in turn, correspond to conformational families. Therefore, the number of conformational families available to a peptide chain is approached by the number of maxima in a density-of-states versus energy plot.
The density-of-states versus energy plot shown in Fig. 2 includes 14,414 conformations of the 552-to-561 region of TLR-5. These conformations were generated with a matrix algorithm (16) and the adoption of a set (see Materials and Methods) of five main chain and three side chain conformations. The internal energy of each conformation was calculated with the ECEPP/2 (35) force field.
REFERENCES
- 1.Beattie, J., and D. J. Flint. 1992. Critical evaluation of a theory of molecular recognition using human insulin-like-growth-factor-I fragment 21 to 40 and its complementary peptide. Biochem. J. 283:473-478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Berman, J., Z. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, I. N. Shindyalov, and P. E. Bourne. 2000. The Protein Data Bank. Nucleic Acids Res. 28:235-242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blalock, J., and E. M. Smith. 1984. Hydropathic anti-complementarity of amino acids based on the genetic code. Biochem. Biophys. Res. Commun. 121:203-207. [DOI] [PubMed] [Google Scholar]
- 4.Brentani, R. R. 1988. Biological implications of complementary hydropathy of amino acids. J. Theor. Biol. 135:495-499. [DOI] [PubMed] [Google Scholar]
- 5.Brentani, R. R. 1990. Complementary hydropathy and the evolution of interacting polypeptides. J. Mol. Evol. 31:239-243. [Google Scholar]
- 6.Brentani, R. R., S. F. Ribeiro, P. Potocnjak, R. Pasqualini, J. D. Lopes, and C. R. Nakaie. 1988. Characterization of the cellular receptor for fibronectin through a hydropathic complementarity approach. Proc. Natl. Acad. Sci. USA 85:364-367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chaudhary, P. M., C. Ferguson, V. Nguyen, O. Nguyen, H. F. Massa, M. Eby, A. Jasmin, B. J. Trask, L. Hood, and P. S. Nelson. 1998. Cloning and characterization of two Toll/interleukin-1 receptor-like genes, TIL3 and TIL4: evidence for a multi-gene receptor family in humans. Blood 91:4020-4027. [PubMed] [Google Scholar]
- 8.Donnely, M. A., and T. S. Steiner. 2002. Two nonadjacent regions in enteroaggregative Escherichia coli flagellin are required for activation of Toll-liker receptor 5. J. Biol. Chem. 277:40456-40461. [DOI] [PubMed] [Google Scholar]
- 9.Eaton, H. L., R. E. Austin, S. W. Fesik, and S. F. Martin. 1989. NMR study of the possible interaction in solution of angiotensin II with a peptide encoded by angiotensin II complementary RNA. Proc. Natl. Acad. Sci. USA 86:9767-9769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fassina, G., S. S. Thorgeirson, and J. G. Omichinski. 1989. Sequence directed design of recognition peptides, p. 431-438. In B. Wittman Liebold (ed.) Methods in protein sequence and analysis. Springer, Berlin, Germany.
- 11.Gillen, K. L., and K. T. Hughes. 1991. Molecular characterization of flgM, a gene encoding a negative regulator of flagellin synthesis in Salmonella typhimurium. J. Bacteriol. 173:6453-6459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goldstein, A., and D. L. Brutlag. 1989. Is there a relationship between DNA sequences encoding peptide ligands and their receptors? Proc. Natl. Acad. Sci. USA 86:42-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hayashi, F., K. D. Smith, A. Ozinsky, T. R. Hawn, E. C. Yi, D. R. Goodlett, J. K. Eng, S. Akira, D. M. Underhill, and A. Aderem. 2001. The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5. Nature 410:1099-1103. [DOI] [PubMed] [Google Scholar]
- 14.Heal, J. R., G. W. Roberts, J. G. Raynes, A. Bhakoo, and A. D. Miller. 2002. Specific interactions between sense and complementary peptides: the basis for the proteomic code. ChemBioChem 3:136-151. [DOI] [PubMed] [Google Scholar]
- 15.Jacchieri, S. G. 1997. Conformational analysis of polypeptide chains with the aid of density of states calculations. Int. J. Quant. Chem. 65:1115-1124. [Google Scholar]
- 16.Jacchieri, S. G. 2001. Stepwise assembling of polypeptide chain energy distributions. Comput. Chem. 25:145-159. [DOI] [PubMed] [Google Scholar]
- 17.Jacchieri, S. G., M. D. Gomes, L. Juliano, and A. C. Camargo. 1998. A comparative conformational analysis of thimet oligopeptidase (EC 3.4.24.15) substrates. J. Pept. Res. 51:452-459. [DOI] [PubMed] [Google Scholar]
- 18.Jacchieri, S. G., and A. S. Ito. 1995. Characterization of structural transitions from aqueous solution to a lipid phase for α-MSH. Int. J. Quant. Chem. 53:335-341. [Google Scholar]
- 19.Knauper, V., A. J. Docherty, B. Smith, H. Tschesche, and G. Murphy. 1997. Analysis of the contribution of the hinge region of human neutrophil collagenase (HNC, MMP-8) to stability and collagenolytic activity by alanine scanning mutagenesis. FEBS Lett. 405:60-64. [DOI] [PubMed] [Google Scholar]
- 20.Kyte, J., and R. F. Doolittle. 1982. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 157:105-132. [DOI] [PubMed] [Google Scholar]
- 21.Martins, V. R., E. Graner, J. Garcia-Abreu, S. J. de Souza, A. F. Mercadante, S. S. Veiga, S. M. Zanata, V. M. Neto, and R. R. Brentani. 1997. Complementary hydropathy identifies a cellular prion protein receptor. Nat. Med. 3:1376-1382. [DOI] [PubMed] [Google Scholar]
- 22.Ohide, A., H. Ando, T. Yanagisawa, and A. Urano. 1996. Hydropathy profiles of predicted thyrotropin-releasing hormone precursors are highly conserved despite low similarity of primary structures. J. Neuroendocrinol. 8:695-701. [PubMed] [Google Scholar]
- 23.Ponder, J. W., and F. M. Richards. 1987. Tertiary templates for proteins. Use of packing criteria in the enumeration of allowed sequences for different structural classes. J. Mol. Biol. 193:775-791. [DOI] [PubMed] [Google Scholar]
- 24.Ritchie, D. W., and G. J. Kemp. 2000. Protein docking using spherical polar Fourier correlations. Proteins 39:178-194. [PubMed] [Google Scholar]
- 25.Root-Bernstein, R. S., and D. D. Holsworth. 1998. Antisense peptides: a critical mini-review. J. Theor. Biol. 190:107-119. [DOI] [PubMed] [Google Scholar]
- 26.Samatey, F. A., K. Imada, S. Nagashima, F. Vonderviszt, T. Kumasaka, M. Yamamoto, and K. Namba. 2001. Structure of the bacterial flagellar protofilament and implications for a switch for supercoiling. Nature 410:321-322. [DOI] [PubMed] [Google Scholar]
- 27.Sanchez-Campillo, M., S. Dramsi, J. M. Gomez-Gomez, E. Michel, P. Dehoux, P. Cossart, F. Baquero, and J. C. Perez-Diaz. 1995. Modulation of DNA topology by flaR, a new gene from Listeria monocytogenes. Mol. Microbiol. 18:801-811. [DOI] [PubMed] [Google Scholar]
- 28.Souza, S. J., and R. Brentani. 1992. Collagen binding site in collagenase can be determined using the concept of sense-antisense peptide interactions. J. Biol. Chem. 267:13763-13767. [PubMed] [Google Scholar]
- 29.Steiner, T. S., J. P. Nataro, C. E. Poteet-Smith, J. A. Smith, and R. L. Guerrant. 2000. Enteroaggregative Escherichia coli expresses a novel flagellin that causes IL-8 release from intestinal epithelial cells. J. Clin. Investig. 105:1769-1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thompson, J. D., D. G. Higgins, and T. J. Gibson. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, positions-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22:4673-4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Totten, P. A., and S. Lory. 1990. Characterization of the type a flagellin gene from Pseudomonas aeruginosa PAK. J. Bacteriol. 172:7188-7199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vriend, G. 1990. WHAT IF: a molecular modeling and drug design program. J. Mol. Graph. 8:52-56. [DOI] [PubMed] [Google Scholar]
- 33.Xu, Y., X. Tao, B. Shen, T. Horng, R. Medzhitov, J. L. Manley, and L. Tong. 2000. Structural basis for signal transduction by the Toll/interleukin-1 receptor domains. Nature 408:111-115. [DOI] [PubMed] [Google Scholar]
- 34.Zanata, S. M., M. H. Lopes, A. F. Mercadante, G. N. Hajj, L. B. Chiarini, R. Nomizo, A. R. Freitas, A. L. Cabral, K. S. Lee, M. A. Juliano, E. de Oliveira, S. G. Jachieri, A. Burlingame, L. Huang, R. Linden, R. R. Brentani, and V. R. Martins. 2002. Stress-inducible protein 1 is a cell surface ligand for cellular prion that triggers neuroprotection. EMBO. J. 21:3307-3316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zimmerman, S. S., M. S. Pottle, G. Nemethy, and H. Scheraga. 1977. Conformational analysis of the 20 naturally occurring amino acid residues using ECEPP. Macromolecules 10:1-9. [DOI] [PubMed] [Google Scholar]