

Abstract
Accumulating evidence suggests that regulatory T cells play a crucial role in preventing autoimmunity. Recently, a naturally occurring CD4+CD25+ T-cell subset that is anergic and also suppressive has been shown to suppress autoimmunity in several animal models. We used proteoglycan-induced arthritis (PGIA) as a study model to investigate the role of the CD4+CD25+ regulatory T cells in autoimmune arthritis. There was no significant change in the percentage of CD4+CD25+ T cells during the immunization period when proteoglycan- or ovalbumin-immunized BALB/c and C57BL/6 mice were compared. An adoptive transfer study showed that the CD4+CD25+ T cells did not protect severe combined immunodeficient mice from arthritis when they were cotransferred with splenocytes from arthritic animals. Similarly, depletion of the CD4+CD25+ T cells did not enhance the onset of the disease or disease severity in severe combined immunodeficient mice. Moreover, CD28-deficient mice, which have very few CD4+CD25+ T cells, were highly resistant to PGIA. These findings indicate that the CD4+CD25+ regulatory T cells may not play a critical role in controlling PGIA.
Keywords: arthritis, autoimmunity, peripheral tolerance, regulatory T cells
Introduction
Autoreactive T cells are primarily eliminated in the thymus by negative selection (central tolerance). Some of the autoreactive T cells, however, may escape from negative selection and are released into the periphery. These self-reactive T cells are exquisitely regulated, and their activation can result in autoimmune diseases. There is accumulating evidence that, in addition to activation-induced cell death or anergy, T-cell-mediated dominant control of self-reactive T cells represents one mechanism for maintaining immunological tolerance [1-3]. Studies conducted in a number of experimental models have demonstrated the existence of regulatory T-cell subsets that prevent activation of autoreactive T cells. Recently, a subset of CD4+ T cells was identified that is present on 5–10% of CD4+ T cells in normal naïve mice and expresses CD25 (the α-chain of IL-2 receptor) [4,5]. Functional analysis of murine CD4+CD25+ T cells showed that those cells, which constitutively express cytotoxic T-lymphocyte antigen (CTLA)-4 [6-8], fail to proliferate or secret cytokines in response to polyclonal or antigen-specific stimulation, but inhibit the activation of conventional responsive T cells [1-3,8,9]. The suppressive activity of the CD4+CD25+ T cells depends on signaling via the negative regulator of T-cell activation CTLA-4 [7] and requires a cell-cell interaction that possibly involves cell surface bound transforming growth factor (TGF)-β1 [1,10]. It has been shown that B7/CD28 costimulation is essential for the development and homeostasis of the CD4+CD25+ regulatory T cells [6], which play critical roles not only in preventing autoimmunity but also in controlling tumor immunity and transplantation tolerance [2,11].
Proteoglycan-induced arthritis (PGIA) is a novel autoimmune murine model that is induced by systemic immunization of BALB/c mice with cartilage proteoglycans [12,13]. The development of PGIA is based on the cross-reactive immune response between immunizing human and mouse (self) cartilage proteoglycans in genetically susceptible BALB/c mice [12,13]. Several lines of evidence indicate T-cell involvement in the pathogenesis of PGIA. First, CD4+ T cells selectively proliferate in response to proteoglycan antigens [14]. Second, prevention of arthritis can be achieved by in vivo treatment with anti-CD4 monoclonal antibodies [15]. Third, arthritis can be transferred to naïve BALB/c or severe combined immunodeficient (SCID) mice using T and B cells from arthritic animals [16-18]. Fourth, a proteoglycan-specific T-cell hybridoma (T-helper-1 type) can induce arthritis in BALB/c mice [19]. Finally, CD4+ T cells from arthritic animals are resistant to activation-induced cell death [20]. These data suggest a breakdown of peripheral tolerance and accumulation of autoreactive T cells in the periphery.
In order to determine whether CD4+CD25+ regulatory T cells play a role in the development of PGIA, we monitored the CD4+CD25+-expressing regulatory T cells in mice with PGIA during an entire immunization period. We also transferred purified CD4+CD25+ T cells from naïve BALB/c mice together with spleen cells from arthritic animals, or alternatively transferred CD4+CD25+-depleted spleen cells from arthritic animals into SCID mice, and then monitored disease development in SCID mice. In addition, we examined disease incidence in CD28-deficient mice, which have deficiency in CD4+CD25+ T cells. Our data suggest that the CD4+CD25+ regulatory T cells might not be essential for controlling the development of PGIA.
Materials and methods
Antigen, mice and immunization
High-density cartilage proteoglycan (aggrecan) was purified from human cartilage, by CsCl gradient centrifugation, and depleted of glycosaminoglycan side chains as described previously [21]. Female BALB/c mice (National Cancer Institute, Friedrich, MD, USA, or The Jackson Laboratory, Bar Harbor, ME, USA) and CD28-deficient mice (The Jackson Laboratory) were immunized intraperitoneally with cartilage proteoglycan (100 μg protein) in complete Freund's adjuvant on day 0, and boosted with proteoglycan in incomplete Freund's adjuvant on days 21 and 42. Female SCID mice on a BALB/c background (NCI/NCrC.B-17-scid/scid), aged 8–12 weeks or young retired breeders, were purchased from the National Cancer Institute and maintained under germ-free conditions.
Assessment of arthritis
A standard scoring system, based on swelling and redness of each paw, was used for the assessment of the severity of disease. The first clinical symptom of swelling was recorded as the time of onset of arthritis. Joint swelling was scored (ranging from 0 to 4 in each paw) and expressed as acute cumulative arthritis score, resulting in a possible maximum severity score of 16. Typically, in the primary form of PGIA, BALB/c mice developed swelling and redness in one or more limbs 7–14 days after the third injection with proteoglycan and adjuvant. In the transfer system, recipient SCID mice developed a more uniform disease from day 10 after transfer, with involvement of nearly all peripheral joints.
Flow cytometry analysis
The CD4+CD25+-expressing T cells were identified by staining spleen cells with fluorescein isothiocyanate (FITC)-labeled anti-CD4 and biotin-labeled anti-CD25 followed by CyChrome-labeled streptavidin (BD PharMingen, San Diego, CA, USA), and analyzed on a flow cytometer (Beckton Dickinson, San Jose, CA, USA). For analysis of intracellular CTLA-4, spleen cells were first stained with the above fluorescence-labeled antibodies, fixed and rendered permeable, and stained with R-phycoerythrin (PE)-labeled anti-CTLA-4 using a Cytofix/Cytoperm kit (BD PharMingen).
Purification of CD4+CD25+ and CD4+CD25- T cells and cell transfer in SCID mice
The CD4+CD25+ T cells were purified using a protocol described previously [6]. Briefly, CD4+ T cells were isolated from spleens of 10- to 12-week-old, naïve, BALB/c mice by negative selection using CD4-enrichment columns (R&D System, Minneapolis, MN, USA). Purified CD4+ T cells were incubated with PE-labeled anti-CD25, and the stained cells were incubated with anti-PE microbeads (Miltenyi Biotech Inc., Auburn, CA, USA). The CD25+ cells were selected on an LS+ column (Miltenyi Biotech Inc.). The purity of the CD4+CD25+ T cells was approximately 90%. The CD4+CD25- T cells, which did not bind to magnetic beads, were collected from the flow through the washing steps (purity >98%). A total of 5 × 105 CD4+CD25+ or CD4+CD25- T cells were injected intraperitoneally, along with 1 × 107 spleen cells from arthritic animals that were depleted of CD25+ cells and 100 μg proteoglycan, into SCID mice.
Depletion of CD4+CD25+ T cells and cell transfer
Spleen cells from arthritic animals were depleted of CD25+ cells using negative selection as described above (0.5 μg PE-labeled anti-CD25 per 1 × 106 cells). The depletion of CD4+CD25+ cells was detected by flow cytometry. Typically, less than 0.3% of the depleted cells expressed CD25. a total of 1 × 107 CD25+ and CD25- spleen cells, together with 100 μg proteoglycan, were injected intraperitoneally into SCID mice.
In vitro suppression assay
CD4+CD25+ T cells (1 × 104) isolated from naïve BALB/c mice were mixed with CD4+CD25- T cells (5 × 104) from arthritic mice in the presence of irradiated syngenic splenocytes as antigen-presenting cells (7.5 × 104) with 1 μg/ml soluble anti-CD3 for 3 days or 50 μg/ml proteoglycan for 5 days, and T-cell proliferation was determined by [3H]thymidine incorporation.
Measurements of antigen-specific T-cell responses, antibodies, and cytokine production
Antigen-specific T-cell responses were measured in quadriplicate samples of spleen cells (3 × 105 cells/well) cultured in the presence of 50 μg proteoglycan protein/ml. T-cell proliferation was assessed on day 5 by [3H]thymidine incorporation [14,22]. Antigen (proteoglycan)-specific interferon-γ, IL-10, and IL-4 production by T cells was determined in media collected on day 4 using capture enzyme-linked immunosorbent assays (ELISAs) from BioSource International, Inc. (Camarillo, CA, USA). For induction of TGF-β1, spleen cells were cultured in serum-free medium X-Vivo-20 (BioWhittaker, Inc., Walkersville, MD, USA) in the presence of proteoglycan. Total TGF-β1 was measured after acidification to activate latent TGF-β, followed by neutralization using an ELISA kit from Promega (Madison, WI, USA).
Maxisorp immunoplates (Nunc, Rochester, NY, USA) were coated with human or mouse cartilage proteoglycans (0.1 μg protein/100 μl per well) for ELISA, and free binding capacity of the wells was blocked by 1% fat-free milk in phosphate-buffered saline [23-25]. Sera were applied at increasing dilutions and isotypes of proteoglycan-specific antibodies were determined using peroxidase-conjugated rat antimouse IgG1 or IgG2a secondary antibodies (Zymed, South San Francisco, CA, USA) as previously described [24,26]. Serum antibody levels were calculated according to mouse IgG1 and IgG2a standards. Mouse IgG1 and IgG2a standards were purified by sepharose-coupled protein G from irrelevant (non-proteoglycan-specific) monoclonal antibody-containing ascites fluids and coated directly onto the microplate's surface.
Statistical analysis
Statistical analysis was performed using SPSS v7.5 (SPSS, Chicago, IL, USA). The Mann–Whitney and Wilcoxon tests were used for intergroup comparisons. P < 0.05 was considered statistically significant.
Results
Lack of correlation between development of arthritis and expression of CD4+CD25+ regulatory T cells in PGIA
To elucidate the role of these regulatory T cells in autoimmune arthritis, we first followed up the CD4+CD25+-expressing regulatory T cells in mice with PGIA during the entire course of immunization. To exclude the possibility that different CD4+CD25+-expressing T cells may be a strain- and/or antigen-dependent phenomenon, we immunized BALB/c mice (PGIA-susceptible strain) and C57BL/6 (arthritis-resistant strain) with human cartilage proteoglycan or ovalbumin. As shown in Fig. 1, BALB/c mice immunized with human cartilage proteoglycan started to develop arthritis after the third injection and the incidence of arthritis reached 100% at day 72 following the third immunization, whereas BALB/c mice immunized with ovalbumin or C57BL/6 mice immunized with proteoglycan were completely resistant to PGIA. The CD4+CD25+-expressing T cells detected 14 days after each immunization from each experimental group were similar during the entire course of immunization (Fig. 2a). This observation suggests that the CD4+CD25+ regulatory T cells might not be involved in the development of PGIA. Although it has been shown that the CD4+CD25+ regulatory T cells can suppress the development of autoimmunity, a recent report [27] demonstrated that depletion of the CD4+CD25+ T cells is necessary but not sufficient for induction of autoimmune gastritis.
Figure 1.
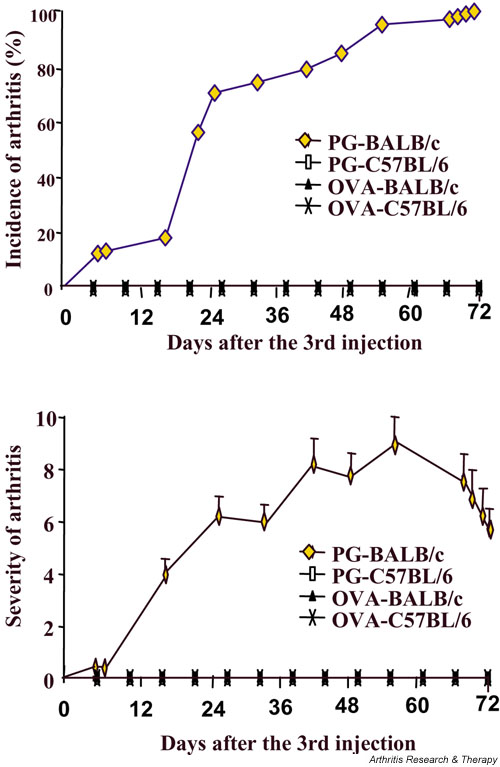
Incidence and severity of arthritis in BALB/c mice. BALB/c and C57BL/6 mice (15 mice/group) were immunized with proteoglycan or ovalbumin in complete Freund's adjuvant (day 0), and boosted with proteoglycan or ovalbumin in incomplete Freund's adjuvant at days 21 and 42. The incidence and severity of arthritis was determined.
Figure 2.
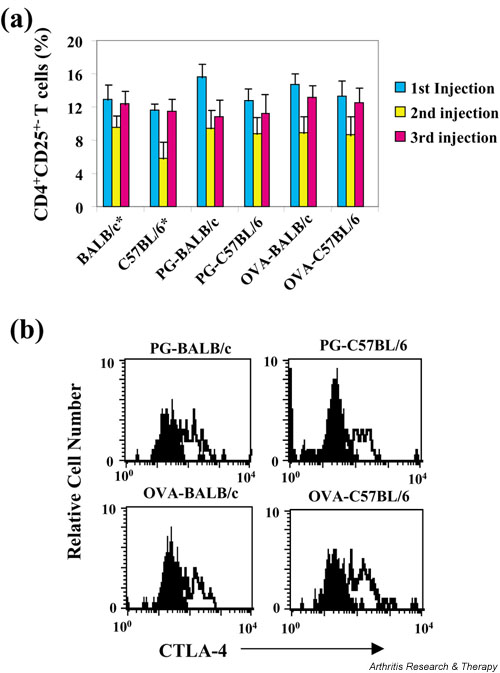
The number of CD4+CD25+-expressing regulatory T cells from BALB/c and C57BL/6 mice during immunization. (a) BALB/c and C57BL/6 mice, immunized with proteoglycan (PG) or ovalbumin (OVA), were killed at 14 days after each immunization, and levels of expression of the CD4+CD25+ T cells were determined by flow cytometry. *The CD4+CD25+ T cells from naïve BALB/c and C57BL/6 mice were also determined. (b) Cytotoxic T-lymphocyte antigen (CTLA)-4 expression on the CD4+CD25+ T cells from PG- or OVA-immunized animals (after the 3rd injection) was determined.
It has been shown that the CD4+CD25+ regulatory T cells express CTLA-4 [6-8], which is a negative regulator of T-cell activation. The suppressive activity of the CD4+CD25+ regulatory T cells may in part be ascribed to signaling through CTLA-4. CTLA-4 may transduce an activating signal to CD4+CD25+ regulatory T cells [2]. Although we did not detect any significant decrease in numbers of the CD4+CD25+ regulatory T cells between proteoglycan-immunized BALB/c and C57BL/6 mice, this might be due to reduced expression of CTLA-4 in the CD4+CD25+ regulatory T cells from proteoglycan-immunized BALB/c mice. To test this possibility, we then detected CTLA-4 expression on the CD4+CD25+ regulatory T cells using intracellular staining. There was no significant difference between levels of CTLA-4 expression in the CD4+CD25+ regulatory T cells from each group (Fig. 2b). This finding excludes the possibility that reduced expression of CTLA-4 in the CD4+CD25+ regulatory T cells from BALB/c mice with PGIA may mediate the induction of the disease.
CD4+CD25+ regulatory T cells may not control autoimmune arthritis
To further investigate whether the CD4+CD25+ T cells play a role in PGIA, we performed adoptive transfer experiments using SCID mice as recipients. Purified CD4+CD25+ regulatory T cells (3–20 × 105) from naïve BALB/c mice, together with 107 spleen cells from arthritic mice that were depleted of CD25+ T cells and 100 μg proteoglycan, were injected intraperitoneally into SCID mice [18], and the incidence and severity of disease were monitored. Note that we used different ratios of the CD4+CD25+ regulatory T cells to the effector cells, and we did not observe any suppression of arthritis development. As a control, the same number of CD4+CD25- T cells, together with arthritogenic spleen cells that were depleted of CD25+ cells, and proteoglycan, were also adoptively transferred into SCID mice. The transfers of CD4+CD25+ T cells did not have any impact on incidence and severity of the disease in SCID mice as compared with transfer of CD4+CD25- T cells (Fig. 3a), suggesting that the CD4+CD25+ regulatory T cells did not protect SCID mice from arthritis. To further verify these results, a depletion experiment was conducted. Spleen cells from arthritic animals, either depleted of CD25+ cells or without depletion, were injected into SCID mice together with proteoglycan. The incidence and severity of arthritis in SCID mice receiving spleen cells containing CD25+ cells or spleen cells depleted of CD25+ cells were identical (Fig. 3b). Proteoglycan-specific T-cell proliferation and production of IL-4, interferon-γ, IL-10, TGF-β1 and antiproteoglycan autoantibodies were comparable in both transfer groups (Fig. 3c). The inability of the CD4+CD25+ regulatory T cells to suppress PGIA was not due to loss of their suppression activity, because in vitro the CD4+CD25+ T cells could effectively inhibit the proliferation of the CD4+CD25- T cells (Fig. 3d, left panel). Interestingly, proteoglycan-induced proliferation of CD4+CD25- T cells could not be inhibited by the CD4+CD25+ T cells (Fig. 3d, right panel), which is consistent with a recent report that the effective function of the CD4+CD25+ regulatory T cells is T-cell receptor (TCR) specific [28]. Collectively, our data suggest that the CD4+CD25+ regulatory T cells may not be involved in the development of PGIA.
Figure 3.
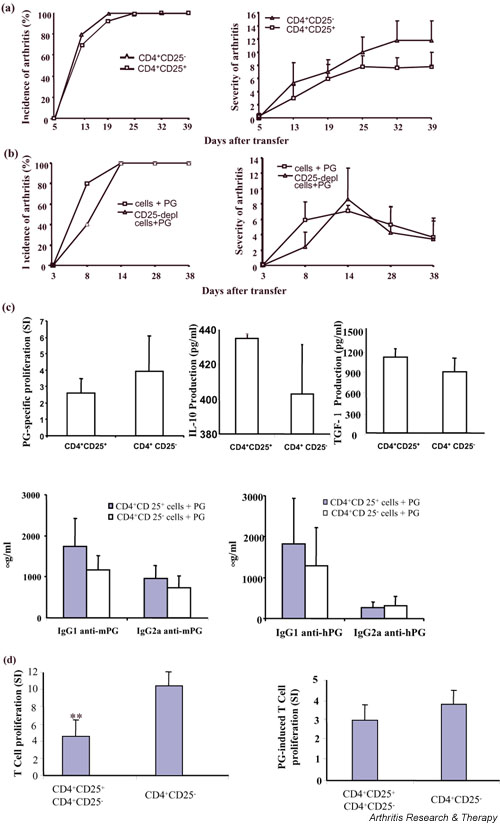
The incidence and severity of arthritis in severe combined immunodeficient (SCID) mice. (a) SCID mice (six to eight mice/group) were intraperitoneally injected with 5 × 105 purified CD4+CD25+ or CD4+CD25- T cells from naïve BALB/c mice, together with 1 × 107 CD25-depleted spleen cells from arthritic animals and 100 μg proteoglycan (PG). The incidence and severity of arthritis was determined. One representative experiment of two is shown. (b) A total of 1 × 107 spleen cells or spleen cells depleted of CD25-expressing cells from arthritic animals, together with 100 μg PG, were transferred intraperitoneally into SCID mice. Disease incidence and severity were determined. (c) Spleen cells from SCID mice that received cell transfer of the CD4+CD25+ or CD4+CD25- T cells and CD25-depleted spleen cells from arthritic mice were cultured in 96-well plate for 5 days in the presence of 25 μg/ml PG, and PG-specific T-cell proliferation was determined using [3H]thymidine incorporation, and expressed as stimulation index (SI; a ratio of incorporated [3H]thymidine [counts per minute] in antigen-stimulated cultures relative to counts per minute in nonstimulated cultures). PG-specific interferon-γ, IL-10 and IL-4 production by T cells was determined in media collected at day 4 using capture enzyme llinked immunosorbent assays (ELISAs). For induction of transforming growth factor (TGF)-β1, spleen cells were cultured in serum-free medium X-Vivo-20 (BioWhittaker, Inc.) in the presence of PG. Total TGF-β1 was measured after acidification to activate latent TGF-β, followed by neutralization using an ELISA kit from Promega. Serum antibody levels were determined by ELISA using mouse IgG1 and IgG2a as standards. (d) Purified splenic CD4+CD25+ T cells from naïve BALB/c mice were cocultured with CD4+CD25- T cells from arthritic mice in the presence of irradiated splenocytes and anti-CD3 for 72 hours or PG for 5 days at 37°C, and T-cell proliferation was determined by [3H]thymidine incorporation. **P < 0.005.
CD28-deficient mice have severely reduced CD4+CD25+ regulatory T cells but are resistant to PGIA
It has been shown that CD28–B7 interaction is essential for the homeostasis of the CD4+CD25+ regulatory T cells that control autoimmune diabetes [6]. Consistent with that report, the expression of CD4+CD25+ regulatory T cells was found to be significantly reduced as compared with that in wild-type BALB/c mice (Fig. 4a). To investigate whether a deficiency in the CD4+CD25+ regulatory T cells may lead to an increase in the development of PGIA, we immunized wild-type and CD28-deficient BALB/c mice with proteoglycan, and disease incidence was monitored. In contrast to the report in autoimmune diabetes [6], CD28-deficient BALB/c mice were highly resistant to induction of arthritis, albeit at a low percentage of the CD4+CD25+ regulatory T cells observed in these mice (Fig. 4b). Note that T cells from proteoglycan-immunized CD28-deficient mice proliferate in response to proteoglycan stimulation, albeit at a rate lower than that in wild-type T cells (Fig. 4c), suggesting that CD28-deficient T cells can be effectively primed.
Figure 4.
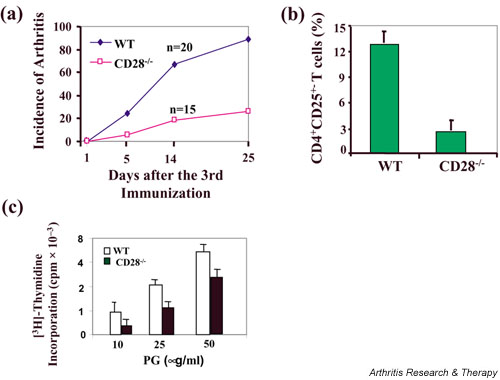
Resistance to proteoglycan-induced arthritis (PGIA) in CD28-deficient mice. (a) The expression of CD4+CD25+ T cells in naïve wild-type (WT) and CD28-deficient BALB/c mice was determined. (b) Wild-type and CD28-deficient BALB/c mice were immunized with proteoglycan (PG) in complete Freund's adjuvant, and boosted in incomplete Freund's adjuvant at days 21 and day 42. The mice developed arthritis after the third immunization. The incidence of arthritis in WT and CD28-deficient BALB/c mice was monitored. (c) CD4+ T cells (1 × 106/ml) from PG-immunized WT and CD28-deficient mice were cultured for 5 days with antigen-presenting cells (2500 rad-irradiated syngenic splenocytes, 2 × 106/ml) in the presence of proteoglycan (10, 25 and 50 μg/ml). The cell proliferation was determined by [3H]thymidine incorporation.
Discussion
A controlled balance between initiation and downregulation of immune responses (peripheral tolerance) is important for maintaining immune homeostasis, whereas dysfunctional immune regulation may lead to chronic inflammation or autoimmunity [29]. The CD4+CD25+ regulatory T cells have been shown to suppress autoimmunity in several animal models [2]. To evaluate the role of the CD4+CD25+ regulatory T cells in PGIA, three approaches were used. First, the numbers of the CD4+CD25+ cells in spleens of susceptible versus resistant or ovalbumin immunized mice were assessed by flow cytometry at day 14 following immunization. No significant change in the numbers of the CD4+CD25+cells was observed among experimental groups tested (Fig. 2a). Moreover, the levels of expression of CTLA-4 in the CD4+CD25+ cells was similar between groups. These data suggest that the CD4+CD25+ cells may not be involved in the development of PGIA. Although the immune system had been perturbed by immunization, CD4+ T cells isolated 14 days after immunization of each group did not show significant increase in CD25 expression as compared with that in naïve mice, suggesting that T cells activated by immunization had become resting memory T cells.
To further evaluate the role of the CD4+CD25+ cells in PGIA, an adoptive transfer system was employed. Different ratios of activated effector cells (CD25-depleted spleen cells) to CD4+CD25+ cells were tested in our cotransfer experiments. No suppression of development of PGIA in SCID mice was observed. Although one could argue that the ratio of effector cells to regulatory cells is not sufficiently great to inhibit disease, we tested a wide range of ratios of effector cells to regulatory cells (5–30 : 1) in our transfer system, and similar results were observed. We did not observe any inhibition at the ratios used in the present study. However, we cannot completely exclude the possibility that 1–4 : 1 ratios may be required for suppression of PGIA. In view of the published literature, this is unlikely because most studies of this type used ratios greater than 20 : 1 [6]. This finding is further supported by the evidence that adoptive transfer of CD25-depleted splenocytes from arthritic mice does not exacerbate disease in SCID mice. The CD4+CD25+ T cells are functionally normal because the CD4+CD25+ cells can significantly inhibit the proliferation of the CD4+CD25- cells induced by TCR stimulation (Fig. 3d, left panel). No alteration in IL-10 and TGF-β production could be detected in two transfer groups (Fig. 3c). Taken together, our data suggest that the CD4+CD25+ regulatory T cells may not regulate the development of PGIA.
We found that CD28-deficient mice, which have a low frequency of CD4+CD25+ T cells, are resistant to PGIA. Although one may argue that, in the absence of CD28, T-cell priming by proteoglycan immunization may be defective. However, CD28-deficient T cells from proteoglycan-immunized animals can proliferate in response to proteoglycan stimulation in vitro, albeit at a rate lower than that in wild-type T cells, which is consistent with the report by Oliveira-dos-Santos et al. [30]. It has been demonstrated that, in the absence of CD28 engagement, T cells require very high TCR occupancy and prolonged stimulation, whereas CD28 costimulation allows T cells to respond to lower degrees of TCR occupancy [30,31]. In support of this observation, CD28-/- mice can still mount immune responses, which vary in magnitude and efficiency depending on the antigen or infectious agent [30,32]. Our results and those reported by others [30] suggest that CD28 may regulate the threshold for T-cell activation. In support of this notion, immunization of CD28-deficient mice with high concentrations of myelin basic protein induces experimental autoimmune encephalomyelitis at similar prevalence and severity as in wild-type mice [30]. Our data suggest that the CD4+CD25+ regulatory T cells might be important for controlling spontaneous models of autoimmunity but not for induced models of autoimmunity. In induced models of autoimmunity, the CD28–B7 interaction has been shown to regulate disease susceptibility by rendering autoreactive T cells anergic, or alternatively by upregulating the threshold for autoreactive T-cell activation [30]. Furthermore, we previously showed that impaired Fas-mediated activation-induced cell death (AICD) of autoreactive T-helper-1 cells may be responsible for the development of PGIA [20]. Therefore, AICD and/or T-cell anergy, but not the CD4+CD25+ regulatory T cells, may be responsible for deletion or inactivation of autoreactive T cells in autoimmune arthritis.
Conclusion
Our results showed that the percentage of the CD4+CD25+ regulatory T cells does not change during the development of PGIA. The CD4+CD25+ regulatory T cells do not protect SCID mice from arthritis when they are cotransferred with spleen cells from arthritic mice and proteoglycan into SCID mice. Transfer of spleen cells from arthritic mice, depleted of the CD4+CD25+ regulatory T cells, into SCID mice does not accelerate the disease. Furthermore, CD28-deficient mice, which are deficient for CD4+CD25+ T cells, are resistant to PGIA. Our data suggest that the CD4+CD25+ regulatory T cells may not be essential for controlling experimentally induced autoimmune arthritis.
Competing interests
None declared.
Abbreviations
AICD = activation-induced cell death; CTLA = cytotoxic T-lymphocyte antigen; ELISA = enzyme-linked immunosorbent assay; FITC = fluorescein isothiocyanate; IL = interleukin; PE = R-phycoerythrin; PG = proteoglycan; PGIA, proteoglycan-induced arthritis; SCID = severe combined immunodeficient; TCR = T-cell receptor; TGF = transforming growth factor; WT = wild-type.
Acknowledgments
Acknowledgements
The authors thank Ms S Velins for assistance in the preparation of this manuscript. This work was supported in part by grants from the National Institutes of Health (AR45652 to KM and AF; AR47412 and AR49047 to JZ) and a grant from Arthritis National Research Foundation to JZ.
References
- Ozkaynak E, Gao W, Shemmeri N, Wang C, Gutierrez-Ramos JC, Amaral J, Qin S, Rottman JB, Coyle AJ, Hancock WW. Importance of ICOS-B7RP-1 costimulation in acute and chronic allograft rejection. Nat Immunol. 2001;2:591–596. doi: 10.1038/89731. [DOI] [PubMed] [Google Scholar]
- Anderton SM, Wraith DC. Selection and fine-tuning of the autoimmune T-cell repertore. Nat Rev Immunol. 2002;2:487–498. doi: 10.1038/nri842. [DOI] [PubMed] [Google Scholar]
- Ohashi PS. T-cell signaling and autoimmunity: molecular mechanisms of disease. Nat Rev Immunol. 2002;2:427–438. doi: 10.1038/nri822. [DOI] [PubMed] [Google Scholar]
- Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol. 1995;155:1151–1164. [PubMed] [Google Scholar]
- Shevach EM, McHugh RS, Piccirillo CA, Thornton AM. Control of T-cell activation by CD4+ CD25+ suppressor T cells. Immunol Rev. 2001;182:58–67. doi: 10.1034/j.1600-065X.2001.1820104.x. [DOI] [PubMed] [Google Scholar]
- Salomon B, Lenschow DJ, Rhee L, Ashourian N, Singh B, Sharpe A, Bluestone JA. B7/CD28 costimulation is essential for the homeostasis of the CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity. 2000;12:431–440. doi: 10.1016/s1074-7613(00)80195-8. [DOI] [PubMed] [Google Scholar]
- Read S, Malmström V, Powrie F. Cytotoxic T lymphocyte-associated antigen 4 plays an essential role in the function of CD25+CD4+ regulatory cells that control intestinal inflammation. J Exp Med. 2000;192:295–302. doi: 10.1084/jem.192.2.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T, Tagami T, Yamazaki S, Uede T, Shimizu J, Sakaguchi N, Mak TW, Sakaguchi S. Immunologic self-tolerance maintained by CD25+CD4+ regulatory T cells constitutively expressing cytotoxic T lymphocyte-associated antigen 4. J Exp Med. 2000;192:303–309. doi: 10.1084/jem.192.2.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T, Kuniyasu T, Toda M, Sakaguchi N, Itoh M, Iwata M, Shimizu J, Sakaguchi S. Immunologic self-tolerance maintained by CD25+CD4+ naturally anergic and suppressive T cells: induction of autoimmune disease by breaking their anergic/suppressive state. Int Immunol. 1998;10:1969–1980. doi: 10.1093/intimm/10.12.1969. [DOI] [PubMed] [Google Scholar]
- Nakamura K, Kitani A, Strober W. Cell contact-dependent immunosuppression by CD4+CD25+ regulatory T cells is mediated by cell surface-bound transforming growth factorβ. J Exp Med. 2001;194:629–644. doi: 10.1084/jem.194.5.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Read S, Powrie F. CD4+ regulatory T cells. Curr Opin Immunol. 2001;13:644–649. doi: 10.1016/S0952-7915(01)00273-4. [DOI] [PubMed] [Google Scholar]
- Ellis JS, Chain BM, Cooke A, Ibrahim MA, Katz DR. Adjuvant composition determines the induction of type II collagen-induced arthritis. Scand J Immunol. 1992;36:49–56. doi: 10.1111/j.1365-3083.1992.tb02939.x. [DOI] [PubMed] [Google Scholar]
- Buchmann K, Ostergaard L, Glamann J. Affinity purification of antigen-specific serum immunoglobulin from the European eel (Anguilla anguilla). Scand J Immunol. 1992;36:89–97. doi: 10.1111/j.1365-3083.1992.tb02944.x. [DOI] [PubMed] [Google Scholar]
- Buzás E, Mikecz K, Brennan FR, Glant TT. Mediators of autopathogenic effector cells in proteoglycan-induced arthritic and clinically asymptomatic BALB/c mice. Cell Immunol. 1994;158:292–304. doi: 10.1006/cimm.1994.1277. [DOI] [PubMed] [Google Scholar]
- Banerjee S, Webber C, Poole AR. The induction of arthritis in mice by the cartilage proteoglycan aggrecan: roles of CD4+ and CD8+ T cells. Cell Immunol. 1992;144:347–357. doi: 10.1016/0008-8749(92)90250-s. [DOI] [PubMed] [Google Scholar]
- Mikecz K, Glant TT, Buzás E, Poole AR. Proteoglycan-induced polyarthritis and spondylitis adoptively transferred to naive (nonimmunized) BALB/c mice. Arthritis Rheum. 1990;33:866–876. doi: 10.1002/art.1780330614. [DOI] [PubMed] [Google Scholar]
- Mikecz K, Glant TT. Migration and homing of lymphocytes to lymphoid and synovial tissues in proteoglycan-induced murine arthritis. Arthritis Rheum. 1994;37:1395–1403. doi: 10.1002/art.1780370919. [DOI] [PubMed] [Google Scholar]
- Bardos T, Mikecz K, Finnegan A, Zhang J, Glant TT. T and B cell recovery in arthritis adoptively transferred to SCID mice: antigen-specific activation is required for restoration of autopathogenic CD4+ Th1 cells in a syngeneic system. J Immunol. 2002;168:6013–6021. doi: 10.4049/jimmunol.168.12.6013. [DOI] [PubMed] [Google Scholar]
- Buzás EI, Brennan FR, Mikecz K, Garzó M, Negroiu G, Holló K, Cs-Szabo G, Pintye E, Glant TT. A proteoglycan (aggrecan)-specific T cell hybridoma induces arthritis in BALB/c mice. J Immunol. 1995;155:2679–2687. [PubMed] [Google Scholar]
- Zhang J, Bárdos T, Mikecz K, Finnegan A, Glant TT. Impaired Fas signaling pathway is involved in defective T cell apoptosis in autoimmune arthritis. J Immunol. 2001;166:4981–4986. doi: 10.4049/jimmunol.166.8.4981. [DOI] [PubMed] [Google Scholar]
- Glant TT, Cs-Szabó G, Nagase H, Jacobs JJ, Mikecz K. Progressive polyarthritis induced in BALB/c mice by aggrecan from human osteoarthritic cartilage. Arthritis Rheum. 1998;41:1007–1018. doi: 10.1002/1529-0131(199806)41:6<1007::AID-ART7>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Glant TT, Buzás EI, Finnegan A, Negroiu G, Cs-Szabó G, Mikecz K. Critical role of glycosaminoglycan side chains of cartilage proteoglycan (aggrecan) in antigen recognition and presentation. J Immunol. 1998;160:3812–3819. [PubMed] [Google Scholar]
- Otto JM, Cs-Szabó G, Gallagher J, Velins S, Mikecz K, Buzás EI, Enders JT, Li Y, Olsen BR, Glant TT. Identification of multiple loci linked to inflammation and autoantibody production by a genome scan of a murine model of rheumatoid arthritis. Arthritis Rheum. 1999;42:2524–2531. doi: 10.1002/1529-0131(199912)42:12<2524::AID-ANR4>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Otto JM, Chandrasekaran R, Vermes C, Mikecz K, Finnegan A, Rickert SE, Enders JT, Glant TT. A genome scan using a novel genetic cross identifies new susceptibility loci and traits in a mouse model of rheumatoid arthritis. J Immunol. 2000;165:5278–5286. doi: 10.4049/jimmunol.165.9.5278. [DOI] [PubMed] [Google Scholar]
- Glant TT, Bárdos T, Vermes C, Chandrasekaran R, Valdéz JC, Otto JM, Gerard D, Velins S, Lovász G, Zhang J, Mikecz K. Variations in susceptibility to proteoglycan-induced arthritis and spondylitis among C3H substrains of mice. Evidence of genetically acquired resistance to autoimmune disease. Arthritis Rheum. 2001;44:682–692. doi: 10.1002/1529-0131(200103)44:3<682::AID-ANR118>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Holló K, Glant TT, Garzó M, Finnegan A, Mikecz K, Buzás EI. Complex pattern of Th1 and Th2 activation with a preferential increase of autoreactive Th1 cells in BALB/c mice with proteoglycan (aggrecan)-induced arthritis. Clin Exp Immunol. 2000;120:167–173. doi: 10.1046/j.1365-2249.2000.01174.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHugh RS, Shevach EM. Cutting edge: depletion of CD4+CD25+ regulatory T cells is necessary, but not sufficient, for induction of organ-specific autoimmune disease. J Immunol. 2002;168:5979–5983. doi: 10.4049/jimmunol.168.12.5979. [DOI] [PubMed] [Google Scholar]
- Hori S, Haury M, Coutinho A, Demengeot J. Specificity requirements for selection and effector functions of CD25+4+ regulatory T cells in anti-myelin basic protein T cell receptor transgenic mice. Proc Natl Acad Sci USA. 2002;99:8213–8218. doi: 10.1073/pnas.122224799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Garra A, Steinman L, Gijbels K. CD4+ T-cell subsets in autoimmunity. Curr Opin Immunol. 1997;9:872–883. doi: 10.1016/S0952-7915(97)80192-6. [DOI] [PubMed] [Google Scholar]
- Oliveira-dos-Santos AJ, Ho A, Tada Y, Lafaille JJ, Tonegawa S, Mak TW, Penninger JM. CD28 costimulation is crucial for the development of spontaneous autoimmune encephalomyelitis. J Immunol. 1999;162:4490–4495. [PubMed] [Google Scholar]
- Viola A, Lanzavecchia A. T cell activation determined by T cell receptor number and tunable thresholds. Science. 1996;273:104–106. doi: 10.1126/science.273.5271.104. [DOI] [PubMed] [Google Scholar]
- Kane LP, Lin J, Weiss A. It's all Rel-ative: NF-kappaB and CD28 costimulation of T-cell activation. Trends Immunol. 2002;23:413–420. doi: 10.1016/S1471-4906(02)02264-0. [DOI] [PubMed] [Google Scholar]