

Abstract
Increasing attention has been directed towards identifying non-T-cell mechanisms as potential therapeutic targets in rheumatoid arthritis. Synovial fibroblast (SF) activation, a hallmark of rheumatoid arthritis, results in inappropriate production of chemokines and matrix components, which in turn lead to bone and cartilage destruction. We have demonstrated that SFs have an autonomous pathogenic role in the development of the disease, by showing that they have the capacity to migrate throughout the body and cause pathology specifically to the joints. In order to decipher the pathogenic mechanisms that govern SF activation and pathogenic potential, we used the two most prominent methods of differential gene expression analysis, differential display and DNA microarrays, in a search for deregulated cellular pathways in the arthritogenic SF. Functional clustering of differentially expressed genes, validated by dedicated in vitro functional assays, implicated a number of cellular pathways in SF activation. Among them, diminished adhesion to the extracellullar matrix was shown to correlate with increased proliferation and migration to this matrix. Our findings support an aggressive role for the SF in the development of the disease and reinforce the perspective of a transformed-like character of the SF.
Keywords: fibroblast, gene expression, migration, rheumatoid arthritis, tumor necrosis factor
Introduction
The etiology and pathogenesis of rheumatoid arthritis (RA), as well as of other inflammatory arthritides and chronic disorders, remain poorly understood [1,2]. By now, it is widely accepted that the development of the disease requires an orchestrated series of both autoimmune and inflammatory processes, as well as a complex interplay between different cell types.
Cytokines play an essential role in the regulation of the immune system and they have been implicated in inflammatory processes as well as in the pathogenesis of many diseases [3]. Tumor necrosis factor (TNF), a pleiotropic cytokine, is produced in response to infection or immunological injury and effects multiple responses that extend well beyond its well-characterized proinflammatory properties, to include diverse signals for cellular differentiation, proliferation, and death [4,5]. Elevated levels of TNF are found in the synovial fluid of RA patients [6,7], and synovial cells are triggered to proliferate by rTNF in vitro [8]. Transgenic studies provided in vivo evidence that deregulation of TNF production per se triggers the development of immunopathologies, including chronic destructive arthritis [9,10]. The minimal, if any, role of the adaptive immunity in the development of arthritis in these models has been confirmed in studies showing that the course of the disease in these transgenic mice is not affected by the absence of mature T and B cells [5,10]. The demonstration of the importance of TNF in synovial inflammation and disease progression has led to the successful therapeutic use of anti-TNF agents in RA [11], yet the precise molecular and cellular mechanisms of TNF function in disease have remained vague.
Increasing attention has been directed towards identifying non-T-cell mechanisms as potential therapeutic targets in RA. There is little disagreement that macrophages and fibroblasts, the majority of cells in both the normal and the hyperplastic synovium, which line diarthoidal joints, should play an essential part by providing the cytokine networks and destructive processes for the initiation and maintenance of disease [12-14]. Synovial fibroblasts (SFs), or fibroblast-like type B synoviocytes (FLS), are mesenchymal, nonvascular, nonepithelial, CD45-negative cells that display heterogeneous tissue localization (intimal and subintimal) [15]. Their physiological function is to provide nutrients for the cartilage and proteoglycans that lubricate the articular surfaces. They also express a variety of surface adhesion receptors that, presumably, help anchor them to the extracellular matrix (ECM) and regulate the flux of cells that pass into the synovial fluid space. In RA and under the influence of inflammatory cytokines, small-molecular-weight mediators, as well as from the interaction with other cell types and the extracellullar matrix, intimal SFs become activated and hyperplastic [16], while releasing a number of effector signals. These include proinflammatory and anti-inflammatory factors, chemoattractants, and factors that promote angiogenesis, matrix degradation and tissue remodeling, bone formation, and osteoclastogenesis [17].
Isolated human RA SFs were able to induce arthritis upon transfer to the knee of healthy SCID mice (mice with severe combined immunodeficiency) even in the absence of a functioning immune system. Similarly, in the present study, immortalized SFs, from an immune-independent animal model of RA [9,5], were shown to be able to induce an SF-specific, T/B-cell independent, TNF-dependent, arthritis-like disease to healthy mice upon transfer to the knee joint. Moreover, we employed two of the most prominent methods of differential gene expression analysis, differential display reverse transcriptase polymerase chain reaction (DD-RT-PCR) and DNA microarrays, in a search of pathways involved in SF activation and disease pathogenesis. Predicted deregulated functions were then validated in vitro.
Materials and methods
Animals
All mice were bred and maintained on a mixed CBA × C57BL/6 genetic background and kept at the animal facilities of the Biomedical Sciences Research Center 'Alexander Fleming' or the Hellenic Pasteur Institute under specific pathogen-free conditions, in compliance with the Declaration of Helsinki principles.
Cell isolation and culture
SFs were isolated from 6- to 8-week-old mice essentially as described previously [18]. Fibroblasts were selected by continuous culturing for at least 21 days and a minimum of 4 passages. Cells were grown at 37°C, 5% CO2 in complete Dulbecco's modified Eagle's medium (DMEM) (Gibco/Invitrogen, Paisley, UK) supplemented with 10% fetal calf serum (FCS) and 100 Units/ml of penicillin/streptomycin. Conditionally immortalized cells were grown similarly at the permissive conditions (33°C, 10 Units/ml of murine recombinant interferon gamma). For the generation of clones, SF populations were counted and diluted to 0.5 cells per well in a 96-well plate. To ensure clonicity, growth (which was observed in 30% of the plated wells, a statistical prerequisite for clonicity under these conditions) was monitored microscopically every day.
hTNF ELISA and measurement of TNF bioactivity
The enzyme-linked immunosorbent assay (ELISA) for hTNF (human tumor necrosis factor) was kindly provided by Dr Wim Buurman (University of Limburg, the Netherlands) and performed as described earlier [19]. TNF bioactivity was measured in tissue-culture supernatants by standard L929 cytotoxicity assay [20]. One unit of TNF bioactivity was taken as the amount of activity for LD50 (median lethal dose). Values are reported as units of TNF bioactivity/106 cells.
Transfer and blockade of disease
Single-cell suspensions of SF clones (2.5 × 106 cells per 20 μl) in phosphate-buffered saline (PBS) were injected into the right knee joint of adult RAG-1-deficient mice (mice deficient in recombination activating gene). Injection was from an anterolateral position using a Hamilton syringe with a 30G × 1/2 gauge needle (Becton Dickinson, Madrid, Spain). After the mice had been humanely killed, joints were fixed, embedded in paraffin wax, and assessed for histopathology, as previously described [9,10]. Sections were examined for histological signs of arthritis and classified accordingly, as previously described [9,10]. Disease induction occurred from 2 to 8 weeks after injection, with maximal incidence at around 4 weeks after injection. In order to block the transferred disease, mice were treated (2 weeks after transfer) with weekly intraperitoneal injections of anti-hTNF antibody (CB0006 5 μg/g) kindly provided by Celltech Ltd (Slough, UK).
Detection of tsTAg transgene by PCR
Tissue was removed by dissection, digested overnight with 20 μg/ml proteinase K (Sigma, L'Isle d'Abeau, France) in 50 mM Tris, 100 mM NaCl, 100 mM EDTA, 1% sodium dodecyl sulfate (SDS) pH 8.0, at 55°C. Precipitated DNA was screened by PCR for the presence of the SV40 tsTAg (simian virus 40 temperature-sensitive large tumor antigen) transgene using the following primers: 5'-CAC TGC CAT CCA AAT AAT CCC-3' and 5'-CAG CCC AGC CAC TAT AAG TAC C-3'. Amplification was performed for 30 cycles of 93° C for 1 min, 55° C for 1 min, and 72° C for 1 min.
Analysis by fluorescence-activated cell sorter (FACS)
Cells (105–106) were washed extensively in PBS and incubated in the presence of 0.2% bovine serum albumin (BSA) with the 429 (MVCAM.A) monoclonal antibody (PharMingen) for 20 min at 4°C. After being washed in PBS (3 times), cells were incubated with a fluorescein isothiocyanate-conjugated anti-rat secondary antibody (Southern Biotechnology Associates, Birmingham, AL, USA) for 20 min at 4°C in the dark, washed, and resuspended in 1 ml of PBS and analyzed with a FACSCalibur™ cytometer.
RNA extraction and differential display RT-PCR
Total RNA was extracted from subconfluent (70–80%) cultured SFs with the RNAwiz reagent (Ambion Inc, Austin, TX, USA), in accordance with the manufacturer's instructions. For Affymetrix gene chip hybridizations, RNA was extracted using the guanidinium isothiocynate/acid phenol protocol [21], followed by single passage through an RNeasy column from QIAGEN GmbH (Hilden, Germany), in accordance with the manufacturer's instructions. RNA integrity was assessed by electrophoresis on denaturing 1.2% agarose/formaldehyde gels. DNase treatment, first-strand cDNA synthesis, and differential-display PCR were executed with the Delta Differential Display kit PT1173-1 from Clontech/BD Biosciences (Palo Alto, CA, USA), in accordance with the manufacturer's instructions [22]. The (α-32P)dATP-labeled (Amersham Pharmacia Biotech GmbH, Freiburg, Germany) PCR products were analyzed on 5% polyacrylamide (19:1)/8 M urea denaturing gels run at a constant power of 60 W. Gels were dried and exposed to film (X-omat AR, Kodak, Hannover, Germany). Differentially expressed bands were located, excised from the gel, amplified by PCR, and cloned in the pT/Adv vector using the AdvanTage PCR cloning kit (Clontech), in accordance with the manufacturer's instructions. Positive plasmid clones were selected on LB/X-gal/IPTG plates containing 100 μg/ml ampicillin.
Reverse Northern slot blot and Northern blot analysis
0.5–1 μg of 4–6 positive plasmid clones for each differentially expressed band were denatured in 0.4 N NaOH for 15 min and slot blotted to nitrocellulose filter in duplicates (Protran, Schleicher & Schuell Biosciences GmbH, Dassel/Relliehausen, Germany) after the addition of 1 volume of cold 2 M ammonium acetate. After washing with 1 M ammonium acetate, the nitrocellulose filter was air-dried and baked for 2 hours at 80°C. The two sets of filters were then hybridized separately with the two different DD-RT-PCR reactions from where the differentially expressed band was detected. Hybridization was performed at 65°C for 12–17 h, in 3 × standard saline citrate (SSC), 0.1% SDS, 10 × Denhardt's solution, 10% (w/v) dextran sulfate, 100 μg/ml single-stranded salmon-sperm DNA. Filters were sequentially washed with 3 × SSC/0.1% SDS, 1 × SSC/0.1% SDS, and 0.3 × SSC/0.1% SDS for 15 min at 65°C and exposed to film (Kodak X-omat AR). For Northern blot analysis, 15 μg of total RNA was electrophoresed on denaturing 1.2% agarose/formaldehyde gels alongside a ribosomal RNA marker and visualized by ethidium bromide staining (0.5 μg/ml). The gel was then soaked sequentially in: H2O for 20 min (twice), 50 mM NaOH/150 mM NaCl for 20 min, 100 mM Tris-HCl pH 7.6/150 mM NaCl for 20 min, and 6 × SSC for 20 min and was transferred to nylon membranes (Hybond, Amersham Pharmacia Biotech GmbH) with 20 × SSC for 12–17 hours. Membranes were prehybridized at 65°C for 60 min in 5 × standard sodium phosphate EDTA (SSPE)/5 × Denhardt's solution/0.5% SDS in the presence of 20 μg/ml single-stranded salmon-sperm DNA. The denatured radiolabelled probe (α-32P dATP, Amersham Pharmacia Biotech GmbH; random primers/Klenow fragment of DNA polymerase, Fermentas UAB, Vilnius, Lithuania) was then added and hybridization was carried on at 65°C for 17–20 hours. Membranes were washed sequentially in 1 × SSPE/0.1% SDS at 65°C for 10 min, 0.3 × SSPE/0.1% SDS at 65°C for 10 min, and 0.1 × SSPE/0.1% SDS at 65°C for 10 min, depending on the probe, and exposed to film (Kodak X-omat AR).
RT-PCR
First-strand cDNA synthesis was performed with an oligo (dT)15 primer and the M-MLV reverse transcriptase from PROMEGA Biosciences Inc (Mannheim, Germany), in accordance with the manufacturer's instructions. PCR was performed on a thermal cycler (PTC-200, MJ Research, Waltham, MA, USA) using 25–30 cycles (depending on the primers) of 93°C for 1 min, 55°C for 1 min, and 72°C for 1 min with a custom-made Taq polymerase.
High-density oligonucleotide array hybridization
cRNA probes were generated and hybridized to the Mu11K (A,B) chip set in accordance with the manufacturer's instructions (Affymetrix, Santa Clara, CA, USA) and as previously described [23]. Data were normalized on the basis of total intensity with the Affymetrix GeneChip software, and data analysis was performed with the Affymetrix GeneChip and the Microsoft Excel software.
Proliferation assay
2 × 103 SFs, grown in monolayers and harvested by trypsinization, were placed in 24-well tissue-culture plates in DMEM medium (Gibco/Invitrogen) supplemented with 10% FCS and 100 Units/ml of penicillin/streptomycin. After 3 hours at 37%, 5% CO2, for cell attachment, 0.5 μCi of [3H]thymidine was added and incubation was continued for 24 and/or 48 hours. Cells were then washed, harvested by trypsinization, transferred to glass-fiber filters, and counted in a liquid scintillation counter.
Adhesion, migration, and wound-healing assays
Adhesion assays were performed on Cytometrix adhesion strips (Chemicon International, Temecula, CA, USA) coated with human fibronectin, vitronectin, laminin, and collagen I, in accordance with the manufacturer's instructions. Assays of cell migration were performed by using modified Boyden chambers with 8-μm pores (Transwell polycarbonate, Corning/Costar, Corning, NY, USA). The lower surface of the membrane was coated with 10 μg/ml human fibronectin (Becton and Dickinson) for 2 hours at 37°C. The lower chamber was filled with 0.6 ml of DMEM with 10% fetal bovine serum (FBS) or 0.5% BSA. Cells were harvested with trypsin/EDTA, washed with PBS, and resuspended to 1 × 106 cells per ml. The suspension (100 μl) was added to the upper chamber, and the cells were allowed to migrate at 37°C, 5% CO2, for 2–4 hours. The upper surface of the membrane was wiped with a cotton bud to mechanically remove nonmigratory cells. The migrant cells attached to the lower surface were extensively washed with PBS and stained with 0.2% crystal violet in 10% ethanol for 10 min. After extensive washing in H2O, the cells were lysed in 1% SDS for 5 min. The absorbance at 550 nm was determined on a microplate reader (SPECTRAmax PLUS384, Molecular Devices, Sunnyvale, CA, USA). Assays of wound healing were performed by scraping a confluent culture of cells (in DMEM supplemented with 10% FCS and 100 Units/ml of penicillin/streptomycin at 37%, 5% CO2), with the edge of a pipette tip, forming a straight line. Cells were then allowed to continue to grow and a picture was taken at each of 0, 12, 24, and 48 hours after the scraping.
Results
Generation of conditionally immortalized synovial fibroblasts
In order to create an in vitro cell system for analysis of the functional properties of the activated SF, we first generated conditionally immortalized SFs. The hTNF-expressing transgenic mice (Tg197) and their normal littermates were mated with the H-2Kb-tsA58 SV40-TAg (simian virus 40 large tumor antigen) transgenic mice [24]. This system has become a standard tool for isolation of specific conditionally immortalized cell lines and has proved useful for isolating diverse cell lines such as lung epithelial [25], osteoblast [26], osteoclast [27], and neuronal [27] cell lines. Adult mice carrying both transgenes or just the SV40 tsTAg transgene were identified by PCR as described previously for hTNF [9]. SFs were isolated from ankle joints and cultured under permissive conditions, as described in Materials and methods. All the isolated SFs were able to grow indefinitely without a change in the morphology and exhibited no signs of terminal differentiation, senescence, or death (after more than 40 passages). All the isolated SFs corresponded, most likely, to the intimal subpopulation of SFs [15], since they all expressed VCAM-1 (vascular cell adhesion molecule 1), as shown by FACS analysis (Fig. 1). Immortalized SFs were expanded by limiting dilution (under conditions that guarantee clonicity, as described in Materials and methods), and a number of hTNF/TAg SF clones, along with wt/TAg SF clones, were selected for the study. All selected clones were stained homogeneously with various surface markers (MHC class I, VCAM, data not shown), thus confirming that they were indeed monoclonal. Production of bioactive human TNF from hTNF/TAg SF clones was confirmed by hTNF-specific ELISA (Fig. 2) and L929 cytotoxicity assay (data not shown). Because of the lack of a definitive cellular marker for murine SFs, all clones were confirmed as SFs based on culture conditions (adherence for a minimum of 21 days/4 passages), morphology (spindle shape), and absence of specific cellular markers (F4/80, CD11b/Mac-1, MOMA-2, CD45), as determined by immunocytochemical and FACScan analysis (data not shown).
Figure 1.
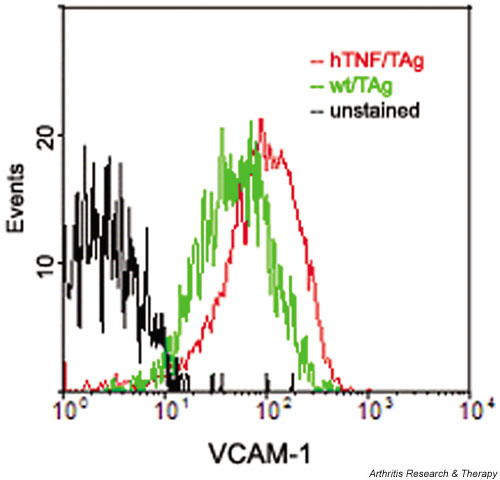
Expression of VCAM-1 by all the isolated synovial fibroblasts, as detected by FACS analysis. Similar results were obtained whether the cells were grown in permissive or nonpermissive conditions. FACS = fluorescence-activated cell sorter; VCAM = vascular cell adhesion molecule.
Figure 2.
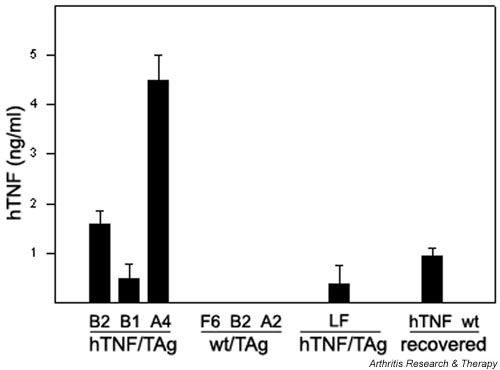
Expression of human TNF by SF clones. Anti-hTNF enzyme-linked immunosorbent assay from cell-culture supernatants was carried out as described in Materials and methods. Values are normalized for hTNF production per 1 × 106 cells/ml over a 24-hour period. Mean averages of triplicates with t-test P values less than 0.01. 'Recovered' refers to SFs derived from the diseased ankle of hTNF/TAg SF B2 injected mice (hTNF) or the nondiseased ankle of wt/TAg SF F6 injected mice (wt). hTNF production was assayed after 20 days/4 passages in culture. hTNF = human tumor necrosis factor; SF = synovial fibroblast; TNF = tumor necrosis factor; tsTAg = temperature-sensitive large tumor antigen.
Transfer of hTNF/TAg SFs into normal murine joints induces a T/B-cell-independent, SF-specific, TNF-driven form of arthritis
Isolated human RA SFs were shown to be able to induce arthritis upon transfer to the knee of healthy SCID mice – that is, even in the absence of a functioning immune system [28]. In order to examine if the established SF clones have similar functional properties, age-matched female nontransgenic F1 (C57BL6 × CBA) mice were injected intra-articularly in the right knee with cloned SFs. Animals were humanely killed 4 to 8 weeks after the injection. Clinical manifestations were usually not detectable. However, histological analysis of injected joints revealed a high incidence of disease transfer (Table 1), characterized by variable degrees of synovitis, soft-tissue inflammation, synovial hyperplasia, cartilage disruption characterized by pyknotic chondrocytes, and bone erosion. None of the control TAg-injected mice showed disease by the end of the study period. In addition, histological examination of other tissues such as liver, lung, spleen, and kidney failed to show evidence of tissue injury.
Table 1.
Summary of arthritis induction by transfer of TNF-expressing SFs
| Derived | Transgene | Clone | Host genotype | Incidence of arthritis |
| Synovium | hTNF/TAg | B2 | wt | 31/65 (47.6%) |
| Synovium | hTNF/TAg | B1 | wt | 5/8 (62.5%) |
| Synovium | hTNF/TAg | A4 | wt | 4/8 (50.0%) |
| Synovium | TAg | F6 | wt | 0/54 |
| Synovium | TAg | B2 | wt | 0/8 |
| Synovium | TAg | A2 | wt | 0/8 |
| Synovium | hTNF/TAg + Aba | B2 | wt | 0/16 |
| Synovium | hTNF/TAg | B2 | RAG-/- | 6/10 (60.0%) |
| Synovium | TAg | F6 | RAG-/- | 0/9 |
| Lungb | hTNF/TAg | LFs | wt | 0/6 |
| Lungb | hTNF/TAg | LFs | RAG-/- | 0/5 |
Mice were classified as arthritic upon positive confirmation by histological analysis. a+Ab denotes group injected with arthritogenic clone B2 and then treated with anti-hTNF antibody 2 weeks after injection. b 'Lung' refers to a population of hTNF-secreting lung fibroblasts derived from hTNF/TAg double transgenic mice. hTNF, human tumor necrosis factor; RAG, recombinant activating gene; TAg, large tumor antigen; wt, wild-type.
Despite the similar genetic backgrounds of the donor and recipient mice (C57BL6 × CBA), the presence of the human transgene might be expected to elicit an immune response, which might account for disease development. To assess this, we repeated our transfer procedure into immunodeficient RAG-/- mice [29]. We observed disease induction in the host mice, with incidence (see Table 1) and pathology similar to those in the previous experiments in immunocompetent animals.
Remarkably, the levels of transgenic TNF production by the transferred SFs do not alter the efficiency of disease transfer in these experiments. The three hTNF-expressing clones, although expressed different levels of hTNF (see Fig. 2), all gave similar incidences of disease (see Table 1). To investigate whether the transferred disease was driven by transgene expression, an additional group of mice was injected with the arthritogenic hTNF/TAg SF clone B2 and then treated with a neutralising, nondepleting anti-hTNF antibody 2 weeks after transfer (see Table 1). Antibody treatment was continued weekly for a further 6 weeks before the mice were humanely killed for histopathological analysis. The absence of histological evidence of disease in any of these mice at the end of the study period shows that hTNF blockade was able to block disease progression. The ability of anti-hTNF therapy to block disease suggested that disease pathology is TNF-driven. To investigate whether TNF-mediated disease could be induced by a mere transfer of locally produced TNF or, rather, involves an imprinted property of SFs, we isolated hTNF expressing (see Fig. 2) lung fibroblasts from double transgenic hTNF/TAg mice and injected them intra-articularly into both immunocompetent and immunodeficient hosts of similar genetic backgrounds (C57BL6 × CBA). We did not observe any pathology in recipient mice at any time point examined (see Table 1).
Synovial fibroblasts migrate to cause disease in distal joints
Remarkably, noninjected hind ankles from mice injected with hTNF/TAg SFs, both draining and opposing, as well as other distal joints such as the wrist joints, showed manifestations characteristic of arthritis in most cases. Histopathological examination of the affected joints showed variably synovitis, soft-tissue inflammation (mostly polymorphonuclear leukocytes), synovial hyperplasia, and cartilage disruption characterized by pyknotic chondrocytes (Fig. 3a).
Figure 3.
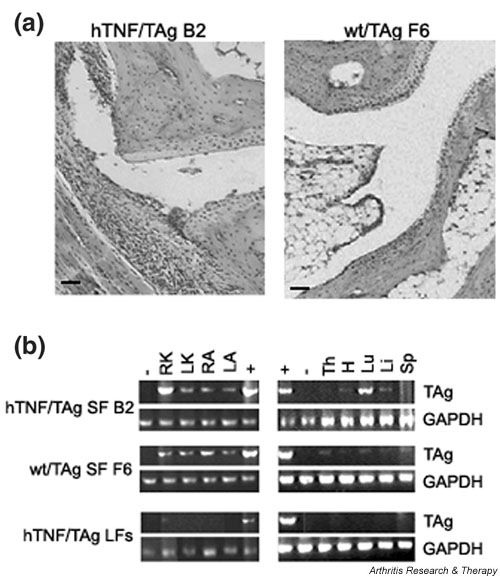
Transfer of arthritis into distal joints with hTNF-expressing SFs. (a) Histopathological analysis (H & E) of an ankle + 4 weeks after injection with hTNF/TAg SF clone B2 or wt/TAg SF clone F6. Representative diseased ankle joint shows arthritic features of synovitis and signs of chondrocyte loss. Original magnification × 95. (b) PCR amplification of TAg transgene from various tissue samples taken from mice injected in the right knee with the hTNF/TAg SF clone B2, the wt/TAg SF clone F6, and hTNF/TAg lung fibroblasts. +/- = positive and negative controls, respectively; GAPDH = glyceraldehyde-3-phosphate dehydrogenase; H = heart; hTNF = human tumor necrosis factor; LA = left ankle; Li = liver; LK = left knee; Lu = lung; RA = right ankle; RK = right knee; SF = synovial fibroblast; Sp = spleen; TAg = large tumor antigen; Th = thymus; tsTAg = temperature-sensitive large tumor antigen; wt = wild-type. Bars: 100 μm.
In order to confirm that transfer of disease to distal joints involves the physical presence of the arthritic input cells, mice injected intra-articularly with either the arthritogenic hTNF/TAg SF clone B2 or the control SF clone wild-type (wt)/TAg F6, as well as with hTNF/TAg lung fibroblasts (LFs), were humanely killed 4 weeks after transfer and total genomic DNA was isolated from all joints and various tissues. Samples were then screened by PCR for the presence of the TAg transgene, as described in Materials and Methods. In mice injected with SFs (both hTNF/TAg B2 and wt/TAg F6) the presence of the transgene was detected in almost all tissues examined, including injected and noninjected joints (Fig. 3b), suggesting that input SFs survive for at least 4 weeks after transfer and that they migrate throughout the body. In contrast, in mice injected with TNF-expressing lung fibroblasts (hTNF/TAg LFs) the presence of the transgene could be detected in only the injected knee. Careful analysis of the fibroblast-containing organs did not show any evidence of tissue pathology; this finding suggests that the ability of the input (hTNF/TAg) fibroblasts to cause disease is specific to joints.
In order to confirm that the induced disease observed in the hind paws was initiated by the transferred hTNF-expressing SFs, ankle joints showing clinical signs of disease 4 weeks after injection with hTNF/TAg SF clone B2 were used to generate primary cellular cultures in vitro and supernatants were tested for the presence of the transgene product by anti-hTNF ELISA. Only those cells derived from the diseased hTNF/Tag-injected mice were able to secrete detectable hTNF in culture (see Fig. 2), an observation providing strong evidence that the transferred SFs had migrated to the ankle joint.
Identification of differentially expressed genes and pathways
In order to understand on a molecular level the differences between the arthritic and normal SF clones and identify cellular pathways that govern SF activation, total RNA extracted from the arthritic (hTNF/TAg) SF clone B2 and the corresponding wt (wt/TAg) SF clone F6 was used for analysis of differential gene expression by differential display, as described in Materials and Methods. The selection of the clone was arbitrary, since the levels of TNF production did not alter the efficiency of disease transfer (see Table 1). The disease induction potential of the SF clone B2 and the up-regulation of matrix metalloproteinase 1 (MMP1) and MMP9 (a hallmark of SF activation in RA) (Fig. 4) indicate that our in vitro (ex vivo) system has functional in vivo characteristics, thus validating the system for the discovery of new genes and/or pathways.
Figure 4.
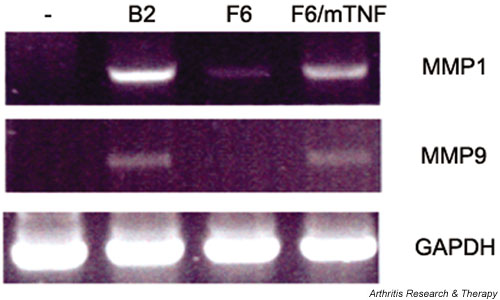
MMP1 and MMP9 are up-regulated in arthritic SF clone B2. RT-PCR of hTNF/TAg SF clone B2 and wt/TAg SF clone F6, as described in Materials and methods. F6/mTNF: wt/TAg SF clone F6 stably transfected with mouse TNF, acting as positive control. hTNF = human tumor necrosis factor; MMP = matrix metalloproteinase; RT-PCR = reverse transcriptase polymerase chain reaction; SF = synovial fibroblast; TAg = large tumor antigen; TNF = tumor necrosis factor; wt = wild-type.
Two different RNA preparations, which were isolated from cells that were cultured for different times (10 and 20 passages, resepctively), were used as duplicates. We performed a total of 80 reactions, using 35 different combinations of primers [22]. A representative reaction, with one set of primers, is shown in Fig. 4a. DD-RT-PCR products (50–100/reaction) ranged from 100 to 2000 nucleotides. On average, 1 to 3 differentially displayed bands were selected per reaction, based on the following criteria:
1) differential expression between B2 (arthritic) versus F6 (normal);
2) expression in both serial dilutions a and b of the sample (Fig. 5a); and
Figure 5.
(a) Representative differential display RT-PCR of the arthritic SF (hTNF/TAg) clone B2 versus the normal (wt/TAg) SF clone F6. I and II are duplicate experiments; b is a duplicate reaction of a, starting with a 1:5 dilution of RNA/cDNA sample. Representative (b) reverse Northern blot, (c) Northern blot, and (d) RT-PCR respectively, as described in Materials and methods. hTNF = human tumor necrosis factor; RT-PCR – reverse transcriptase polymerase chain reaction; SF = synovial fibroblast; TAg = large tumor antigen; wt = wild-type.
3) expression in both duplicate samples (Fig. 5a, I, II) isolated from different cell-culture passages/RNA preparations.
Before sequencing, cloning of the differentially displayed bands (and not of some underlying ones in the gel) was verified by reverse Northern slot blot, as described in Materials and Methods (Fig. 5b). The differential expression of most of the selected clones (Table 2) was verified by Northern blot and/or in some cases RT-PCR (Fig. 5c and 5d, respectively) as described in Materials and Methods. Of the 73 selected differentially expressed genes, 13 were found to be false positives (17%) and 11 clones were found redundant (after sequencing). Overall, 49 genes were identified, 39 up-regulated in arthritis (SF clone B2) and 10 down-regulated (see Table 2).
Table 2.
Deregulated genes in the arthritic SF as revealed by differential display RT-PCR
| Clone no. | RN | N | Deregulationa | RT-PCR | Gene ID | Accession no. |
| 6 | + | + | > 1 | NADH dehydrogenase S2 | M22756 | |
| 7 | + | + | 1.3 | (+) | FIN 13 | U42383 |
| 8 | + | + | > 1 | Aldose Reductase | U93231.1 | |
| 9,17,19 | + | + | 2.1/4.8/8.6 | Ribosomal protein L3 | U89417 | |
| 10 | + | + | 2.7 | NADH dehydrogenase S4 | AF100726 | |
| 13 | + | 4 | SPARC/osteonectin | X04017 | ||
| 14,39,71 | + | + | -5 | MHC-1b H2-T23 (Qa1-like) | U12822 | |
| 15,35 | + | + | 2.2 | EST~GMP reductase | AA240130 | |
| 16 | + | + | - 70 | mt. cytochrome b | AF159396.1 | |
| 18 | + | + | > 1 | ATP-specific succinyl-CoA synthetase β | AF058955 | |
| 20,25 | + | + | 1.6/5 | Hsp70 | M34561 | |
| 21,58 | + | + | 1.8/6.3 | HnRNP D-like / JKTBP | AB017020 | |
| 22,24 | + | + | 2.26 | + | ZO-2 | U75916 |
| 23 | + | + | 10 | + | HSPC194 | AF151028 |
| 27 | + | + | 4 | Unknown | ||
| 28 | + | + | -14.7 | Smoothelin large isoform L2 | AF064236.1 | |
| 29 | + | ? | < -1 | + | EST~RNA binding protein | L17076/S72641 |
| 29b | + | ? | < -1 | EST | AI013881 | |
| 31 | (+) | + | > 1 | + | Unknown | |
| 32,33 | + | + | 3 | + | Unknown | |
| 34 | + | (+) | > 1 | Unknown | ||
| 36 | + | ? | > 1 | Unknown | ||
| 37 | + | ? | > 1 | Hypothalamus protein HT001 | AF113539 | |
| 38 | + | ? | < -1 | Ran-GTP binding protein | Y08890 | |
| Karyopherin b3 | NM002271.1 | |||||
| 41 | + | (+) | > 1 | Huntingtin int. prot. 1 family | AF049613 | |
| HSPC136 | AF161485 | |||||
| 42 | + | + | > 1 | Unknown | ||
| 43 | (+) | + | < -1 | Pyruvate kinase (PK3)-M2 subunit | NM011099 | |
| 44 | + | > 1 | HSPC030 | AF170920 | ||
| 45,46,47 | + | + | 2.1 | Ribosomal protein L7a | X15013.1 | |
| 48 | + | + | 36 | (+) | Homologue to eIF6/integrin b4 int. prot. | AF081140 |
| 49 | + | + | 8.8 | + | HSPC249 (from CD34+ stem cells) | AF151083 |
| 50 | + | > 1 | Unknown | |||
| 51 | + | ? | > 1 | Unknown | ||
| 52 | + | + | 72? | (+) | EAP330 of ELL | NM007241 |
| 53 | + | + | > 1 | Ferritin heavy chain | NM010239 | |
| 54 | (+) | + | - 7.9 | Ly-6E.1 alloantigen | X04653 | |
| TAP (T cells activating pr) | M59713.1 | |||||
| 55 | (+) | ? | < -1 | E124 (etoposide induced/+p53) | U41751 | |
| 56 | + | ? | > 1 | EST | AA960119 | |
| 57 | (+) | ? | > 1 | mt DNA polymerase γ | U53584 | |
| 60 | + | ? | > 1 | EST | AA963457 | |
| 61 | (+) | + | 2.3 | Karyopherin a4 (importin a3)? | NM002268 | |
| 62 | + | + | 3.5 | + | LIM-protein? | AF037208 |
| 65 | + | + | 22 | MEKK4? | NM011948 | |
| 66 | + | ? | > 1 | Human mRNA expr. in thyroid gland | D83198 | |
| 66b | + | ? | > 1 | Human cDNA FLJ20657 fis | AK000664 | |
| 67 | + | ? | > 1 | Unknown | ||
| 68 | + | ? | > 1 | Unknown | ||
| 69 | + | (+) | > 1 | Unknown | ||
| 70 | + | ? | < -1 | MHC class II | AF110520 |
aFold of up/down-regulation, as calculated from Northern blots after normalization against glyceraldehyde-3-phosphate dehydrogenase. N, Northern; RN, reverse Northern; RT-PCR, reverse transcriptase polymerase chain reaction.
Total RNA extracted from the same clones (hTNF/TAg SF B2, wt/TAg SF F6) used for the differential display, grown under identical conditions, was used to hybridize the Mu11K (A,B) high-density oligonucleotide chip set from Affymetrix. The hybridizations were repeated twice from different cell-culture passages/RNA preparations. 91% of the genes gave similar intensities between the two samples and all genes represented more than once on the chip always gave similar values (data not shown). The gene expression levels of the duplicate samples were plotted against each other in order to find a reliable range of hybridization signal intensity and fold induction levels. Such a range lay above signal intensities of 3500 (arbitrary hybridization signal units) and above 4-fold induction levels (data not shown). On the basis of the above criteria and of various significance criteria from Afffymetrix (absolute call, difference call, baseline call), 85 up-regulated and 287 down-regulated genes were selected. The known genes (26 up-regulated and 118 down-regulated) are shown in Table 3. All genes that were tested by RT-PCR for confirmation of deregulation (11 expressed sequence tags) were found to have been correctly predicted by the DNA chip hybridization (data not shown). Only 11 of the genes selected by differential display were included in the DNA chips (five with the same accession number). Of these 11, six fell within the noninformative range of deregulation (< 2), three were in the doubtful range of 2- to 5-fold deregulation (and gave the same prediction of deregulation), and two were on the listed of those selected by the DNA chip method (> 5-fold deregulation). Of these last two, MEKK4 was predicted to be up-regulated with both platforms, while SPARC (secreted protein acidic and rich in cysteine) was predicted to be up-regulated by DD (and Northern blot) and down-regulated by DNA chip hybridization.
Table 3.
Deregulated genes in the arthritic synovial fibroblast as revealed by DNA microarrays
| Fold change | ||||
| A | B | Pa | Gene ID | Accesion no. |
| 25 | 20 | 0.099 | peroxisome proliferator activated protein-gamma-2 | U09138 |
| 14 | 20 | 0.119 | c-erbA alpha2 for thyroid hormone receptor | X07751 |
| 17 | 16 | 0.000 | clusterin | L08235 |
| 12 | 12 | 0.085 | matricin | L20509 |
| 8 | 13 | 0.031 | laminin B1 | M15525 |
| 8 | 10 | 0.064 | RNA-binding protein AUF1 | U11274 |
| 11 | 7 | 0.304 | ribosomal protein L41 | U93862 |
| 9 | 8 | 0.065 | type II DNA topoisomerase beta isoform | D38046 |
| 8 | 8 | 0.122 | ZO-1 | D14340 |
| 7 | 6 | 0.012 | Ca2+-dependent activator protein for secretion | D86214 |
| 8 | 5 | 0.317 | ryanodine receptor type 3 | X83934 |
| 4 | 8 | 0.051 | serine/threonine-protein kinase PRP4m (PRP4m) | U48737 |
| 5 | 6 | 0.038 | multifunctional aminoacyl-tRNA synthetase | AA048927 |
| 6 | 5 | 0.017 | p53-associated cellular protein PACT | U28789 |
| 5 | 6 | 0.021 | alpha-adaptin (C) | X14972 |
| 5 | 5 | 0.007 | splicing factor; arginine/serine-rich 7 (SFRS7) | AA408185 |
| 5 | 5 | 0.053 | Y box transcription factor (MSY-1) | M62867 |
| 5 | 5 | 0.014 | ASF | X66091 |
| 4 | 6 | 0.053 | stromelysin PDGF responsive element binding protein transcription factor | U20282 |
| 6 | 4 | 0.127 | translation initiation factor (Eif4g2) | U63323 |
| 5 | 4 | 0.153 | ubiquitin-conjugating enzyme UbcM2 | AF003346 |
| 5 | 4 | 0.065 | DNA topoisomerase I | D10061 |
| 6 | 3 | 0.082 | calcyclin | M37761 |
| 5 | 4 | 0.055 | activin receptor (ActR) | M65287 |
| 5 | 3 | 0.112 | putative RNA helicase and RNA dependent ATPase (mDEAH9) | AF017153 |
| 3 | 5 | 0.051 | small nuclear RNA (Rnu1a-1) | L15447 |
| -5 | -3 | 0.014 | gC1qBP gene | AJ001101 |
| -5 | -3 | 0.013 | primase small subunit | D13544 |
| -5 | -3 | 0.071 | T-cell specific protein s | L38444 |
| -5 | -3 | 0.064 | complement receptor (Crry) gene | M34173 |
| -2 | -6 | 0.168 | primary response gene B94 | L24118 |
| -5 | -3 | 0.033 | ferritin L-subunit | L39879 |
| -2 | -6 | 0.414 | C/EBP delta | X61800 |
| -5 | -4 | 0.092 | tropomyosin isoform 2 | M22479 |
| -6 | -3 | 0.136 | serine proteinase inhibitor (SPI3) | U25844 |
| -7 | -2 | 0.145 | interferon beta (type 1) | V00755 |
| -3 | -6 | 0.019 | TSC-22 mRNA | X62940 |
| -3 | -7 | 0.013 | novel GTP-binding protein | D10715 |
| -4 | -6 | 0.039 | core-binding factor | L03279 |
| -8 | -2 | 0.125 | G-protein-like LRG-47 | U19119 |
| -5 | -5 | 0.016 | SIG41 | X80232 |
| -5 | -5 | 0.017 | BAP31 | X81816 |
| -6 | -5 | 0.000 | Rat translational initiation factor (eIF-2) alpha subunit | AA408104 |
| -3 | -8 | 0.048 | latent TGF-beta binding protein-2 | AF004874 |
| -7 | -4 | 0.011 | endothelial monocyte-activating polypeptide I | U41341 |
| -7 | -4 | 0.007 | beta proteasome subunit (Lmp3) | U65636 |
| -7 | -4 | 0.075 | histone H2A.Z (H2A.Z) | U70494 |
| -9 | -3 | 0.029 | NAD-dependent methylenetetrahydrofolate dehydrogenase-methenyltetrahydrofolate cyclohydrolase | J04627 |
| -7 | -5 | 0.059 | fibrillin (Fbn-1) | L29454 |
| -7 | -5 | 0.031 | calumenin | U81829 |
| -8 | -4 | 0.023 | TIMP-3 gene for metalloproteinase-3 tissue inhibitor | Z30970 |
| -7 | -5 | 0.006 | Cctb mRNA for CCT (chaperonin containing TCP-1) beta subunit | Z31553 |
| -7 | -6 | 0.036 | Nedd5 mRNA for DIFF6- or CDC3,10,11,12-like | D49382 |
| -8 | -5 | 0.060 | cadherin-associated protein (CAP102/alpha catenin) | D90362 |
| -9 | -4 | 0.338 | OTS-8 | M73748 |
| -7 | -6 | 0.183 | 20S proteasome subunit Lmp7 (Lmp7d allele) | U22031 |
| -5 | -8 | 0.202 | ornithine aminotransferase | X64837 |
| -4 | -10 | 0.050 | Chromosome segregation protein CUT3 | AA241064 |
| -7 | -7 | 0.183 | small heat-shock protein (HSP25) | L07577 |
| -5 | -9 | 0.009 | triosephosphate isomerase | X53333 |
| -10 | -4 | 0.024 | Sec61 protein complex gamma subunit | U11027 |
| -9 | -5 | 0.010 | SPARC | X04017 |
| -11 | -4 | 0.025 | Mer | D73368 |
| -8 | -7 | 0.002 | IFN-gamma induced (Mg11) | U15635 |
| -15 | -2 | 0.129 | lysyl oxidase | L04262 |
| -9 | -8 | 0.100 | proteasome (lmp2) | L11613 |
| -13 | -4 | 0.059 | DNA topoisomerase II | D12513 |
| -9 | -9 | 0.006 | cytochrome c gene (MC1) | X01756 |
| -12 | -6 | 0.004 | destrin | W08453 |
| -12 | -7 | 0.027 | deleted in split hand/split foot 1 homologue (Dss1) | U41626 |
| -13 | -7 | 0.013 | adenine nucleotide translocase-1 (Ant1) | U27315 |
| -18 | -2 | 0.215 | major excreted protein (MEP). | X06086 |
| -15 | -5 | 0.137 | osteopontin | X51834 |
| -11 | -11 | 0.075 | integral membrane phosphoprotein band | U17297 |
| -18 | -4 | 0.027 | phosphatase 2A B' alpha3 regulatory subunit | U59418 |
| -11 | -11 | 0.038 | p85SPR | U96634 |
| -12 | -11 | 0.036 | ubiquitinating enzyme E2-20K | U19854 |
| -11 | -12 | 0.006 | brain factor-1 (Hfhbf1) | U36760 |
| -13 | -11 | 0.072 | synaptonemal complex protein Sc65 | AA028785 |
| -12 | -12 | 0.009 | mevalonate pyrophosphate decarboxylase | AA059528 |
| -14 | -11 | 0.061 | MO15-associated kinase (MO15) | U11822 |
| -12 | -13 | 0.031 | overexpressed and amplified in teratocarcinoma cell line ECA39 | X17502 |
| -13 | -12 | 0.109 | DNA-polymerase delta catalytic subunit. | Z21848 |
| -12 | -14 | 0.114 | alanyl-tRNA synthetase | AA254996 |
| -15 | -11 | 0.035 | pituitary tumor-specific transforming factor | AA711028 |
| -19 | -7 | 0.041 | neural precursor mRNA | D85414 |
| -15 | -11 | 0.107 | protective protein (Mo54) | J05261 |
| -15 | -11 | 0.044 | C57BL/6 Ly-49D-GE antigen | U10090 |
| -12 | -15 | 0.169 | histone H2A.Z | AA285607 |
| -11 | -16 | 0.031 | bcl-2 binding protein BAG-1 | U17162 |
| -17 | -11 | 0.005 | antigen (homologue of human CD9 antigen) | C80730 |
| -18 | -10 | 0.002 | adenine nucleotide translocase-1 (Ant1) | U27315 |
| -18 | -11 | 0.080 | laminin | J02870 |
| -14 | -15 | 0.036 | protective protein (Mo54) | J05261 |
| -11 | -18 | 0.210 | keratinocyte lipid-binding protein | X70100 |
| -14 | -16 | 0.253 | manganese superoxide dismutase (MnSOD) | X04972 |
| -12 | -18 | 0.155 | MyD118, a myeloid differentiation primary response gene | X54149 |
| -15 | -16 | 0.007 | glutamate dehydrogenase | X57024 |
| -14 | -18 | 0.505 | cytosolic aspartate aminotransferase isoenzyme1 | J02623 |
| -16 | -16 | 0.010 | voltage-dependent anion channel 1 mRNA | U30840 |
| -17 | -15 | 0.059 | fractalkine | U92565 |
| -17 | -15 | 0.044 | alpha glucosidase II beta subunit | U92794 |
| -15 | -17 | 0.038 | glutamate dehydrogenase. | X57024 |
| -21 | -12 | 0.069 | p53 cellular tumor antigen | K01700 |
| -19 | -14 | 0.020 | bcl-2 binding protein BAG-1 | U17162 |
| -22 | -12 | 0.051 | FKBP65 binding protein mRNA, complete cds | L07063 |
| -18 | -16 | 0.022 | hepatoma transmembrane kinase ligand | L38847 |
| -19 | -16 | 0.022 | hypothetical E. coli protein | AA271603 |
| -23 | -12 | 0.091 | talin | X56123 |
| -24 | -12 | 0.086 | alpha-1 type IV collagen (Col4a-1) | J04694 |
| -19 | -17 | 0.007 | S-adenosyl homocysteine hydrolase (ahcy) | L32836 |
| -16 | -20 | 0.054 | UBcM4 protein | X97042 |
| -21 | -16 | 0.098 | cadherin-11 | AA184551 |
| -17 | -20 | 0.012 | YL-1 protein | D43643 |
| -23 | -14 | 0.199 | alpha-B crystallin | M63170 |
| -23 | -14 | 0.155 | TDAG51 | U44088 |
| -18 | -19 | 0.088 | chop-10 | X67083 |
| -21 | -17 | 0.033 | 3-oxoacyl-CoA thiolasee | C79215 |
| -23 | -15 | 0.171 | alpha-B2-crystallin | M73741 |
| -23 | -15 | 0.134 | reduced folate carrier (RFC1) | U32469 |
| -28 | -11 | 0.079 | Ubiquinol-cytochrome C reductase complex 6.4 KD protein | AA198790 |
| -27 | -13 | 0.255 | epimorphin | D10475 |
| -22 | -18 | 0.004 | alpha-2 type IV collagen | J04695 |
| -21 | -19 | 0.027 | acrogranin | M86736 |
| -29 | -11 | 0.013 | endogenous murine leukemia virus modified polytropic provirus DNA | M17327 |
| -20 | -20 | 0.023 | C3H cytochrome P450 (Cyp1b1) | U03283 |
| -21 | -21 | 0.005 | delta-aminolevulinate dehydratase | X13752 |
| -22 | -21 | 0.058 | U1 snRNP-specific protein C | U70315 |
| -27 | -16 | 0.058 | HN1 | U90123 |
| -28 | -17 | 0.145 | mMIS5 | D86726 |
| -37 | -11 | 0.104 | proteasome subunit MECL1. | D85561 |
| -22 | -26 | 0.063 | growth factor-induced delayed early response protein | L02914 |
| -21 | -27 | 0.121 | TAP2-d | U60087 |
| -27 | -23 | 0.007 | RNA polymerase I 40 kD subunit | D31966 |
| -16 | -34 | 0.212 | Ma | X62742 |
| -38 | -13 | 0.282 | argininosuccinate synthetase (Ass) | M31690 |
| -31 | -22 | 0.087 | Gas 5 growth arrest specific protein | X59728 |
| -27 | -26 | 0.006 | mama | X67809 |
| -26 | -27 | 0.031 | Cctz mRNA for CCT (chaperonin containing TCP-1) zeta | Z31557 |
| -34 | -20 | 0.185 | hypothetical 28.4 KD protein | AA617493 |
| -22 | -37 | 0.148 | H2-M alpha, H2-M beta 2, H2-M beta 1, Lmp2 | U35323 |
| -39 | -35 | 0.056 | metaxin | L36962 |
| -41 | -39 | 0.060 | Ubiquinol-cytochrome C reductase complex 7.2 KD protein | AA237529 |
| -49 | -43 | 0.022 | hypothetical 26.5 KD protein | AA122622 |
Deregulation is expressed as fold change of gene expression after global normalization, which is an estimate of the transcript abundance between the control and experimental samples, as determined by the Affymetrix software http://www.affymetrix.com. Negative values indicate downregulation A and B refer to duplicate samples that differ by 10 passages. aCalculated with paired t-test for the average difference changes between the samples http://www.affymetrix.com.
Functional clustering of deregulated genes
Known genes whose expression was found to be deregulated in either differential display or DNA chip hybridizations were clustered collectively, where possible, according to their function (Table 4) to reveal deregulated functions or cellular pathways of the arthritogenic SFs. Classifications were redundant, since some genes were included in more than one class of functions. The most prominent deregulated cellular functions of the arthritic SF, equally predicted by both methods, include stress response, energy production, transcription, RNA processing, protein synthesis, protein degradation, growth control, adhesion, cytoskeletal organization, Ca++ binding, and antigen presentation.
Table 4.
Functional clustering of deregulated genes in the arthritic synovial fibroblasts
| Stress response | Protein synthesis |
| DD(M34561) Hsp70 | DDU89417) Ribosomal protein L3 (x3) |
| DD(AF170920) HSPC030 | DD(X15013.1) Ribosomal protein L7a (x3) |
| DD(AA240130) EST~GMP reductase Reductase | MA(U93862) Ribosomal protein L41 |
| DD(U93231.1) Aldose | MA(AA048927) Aminoacyl-tRNA synthetase |
| DD(NM010239) Ferritin heavy chain | DD(AF081140) Homologue to eIF6/integrin b4 int. prot. |
| Energy production | MA(U63323) Translation initiation factor (Eif4g2) |
| DD,.MA(U53584) Mt DNA polymerase γ | MA(AA408104) Translational initiation factor (eIF-2) α |
| DD(M22756) NADH dehydrogenase S2 | MA(AA254996) Alanyl-tRNA synthetase |
| DD(AF100726) NADH dehydrogenase S4 | Protein degardation |
| DD(AF058955) Succinyl-CoA synthetase β | MA(U19854) ubiquitinating enzyme E2-20K |
| DD(AF159396.1) Mt. Cytochrome b | MA(X97042) UBcM4 |
| MA(X01756) Cytochrome c (MC1) | MA(AA198790)ubiquinol-cytochrome c reductase (6.4) |
| MA(U03283) C3H cytochrome P450 (Cyp1b1) | MA(AA237529)ubiquinol-cytochrome c reductase (7.2) |
| Transcription | Growth control |
| DD(NM007241) EAP330 of ELL | DD, MA(U42383) FIN 13 |
| MA(U09138) PPAR γ2 | MA(M65287) ActR |
| MA(M62867) MSY-1 | MA(K01700) p53 |
| MA(U20282) SPBP | MA(U28789) PACT |
| MA(D31966) RNA polymerase I 40 kDa subunit | MA(X59728) gas5 |
| MA(X67083) Chop-10 | MA(X54149) MyD118 |
| MA(X61800) C/EBP δ | MA(L02914) Aquaporin |
| MA(X62940) TSC-22 | MA(M86736) Acrogranin |
| MA(L03279) CBF | ECM/Adhesion |
| MA(D43643) YL-1 | MA(M15525) Laminin B1 |
| RNA processing | MA(M65287) ActR |
| DD(AB017020) HnRNP D-like/JKTBP (x2) | MA(L08235) Clusterin |
| MA(U11274) AUF1 | DD(X040170) SPARC |
| MA(AA408185) SFRS7 | MA(D14340) ZO-1 |
| MA(X66091) ASF | DD(U75916) ZO-2 |
| MA(L15447) Rnu1a-1 | MA(X56123) Talin |
| MA(U48737) PRP4m | MA(L02918) procollagen type V alpha 2 |
| MA(AF017153) mDEAH9 | MA(J04694) α-1 type IV collagen |
| MA(X80232) SIG41 | MA(J04695) α-2 type IV collagen |
| MA(U70315) U1 snRNP- C | MA(L29454) Fibrillin |
| Calcium binding | MA(Z30970) TIMP-3 |
| MA(M37761) Calcyclin | MA(AA184551) Cadherin-11 |
| MA(D86214) Ca2+ dep. activated. Prot. for secretion | MA(D90362) CAP102/alpha catenin |
| MA(X83934) Ryanodine receptor type 3 | Antigen presentation |
| DD(X04017) SPARC | DD(M35244) MHC-1b H2-TL-T10-129 |
| MA(U81829) Calumenin | MA(U10090) C57BL/6 Ly-49D-GE antigen |
| MA(L29454) Fibrillin | DD,MA(AF110520) MHC class II |
| Cytoskeleton organization | DD(X04653) T-cells activating protein (TAP) |
| MA(L20509) Matricin | MA(U60087) TAP2-d |
| Nβ-actin | MA(X62742) Ma |
| DD(AF064236.1) Smoothelin large isoform L2 | MA(U35323) H2-M α, H2-M β2, H2-M β1, Lmp2 |
| MA(X56123) Talin | MA(U65636) beta proteasome subunit (Lmp3) |
| MA(Z31553) Cctz (Chaperonin containing TCP-1 β z) | MAU22031) 20S proteasome subunit (Lmp7) |
| MA(D49382) Nedd5 (Septin) | MA(L11613) proteasome (lmp2) |
| MA(W08453) Destrin | MA(D85561) proteasome subunit MECL1 |
Numbers in parentheses refer to accession numbers; superscript prefixes indicate the method of gene selection, as follows: DD, differential display; MA, microarrays; N, Northern blot. Shading denotes down-regulated genes.
Decreased ECM adhesion of the arthritic SF clone correlates with increased proliferation and migration in vitro
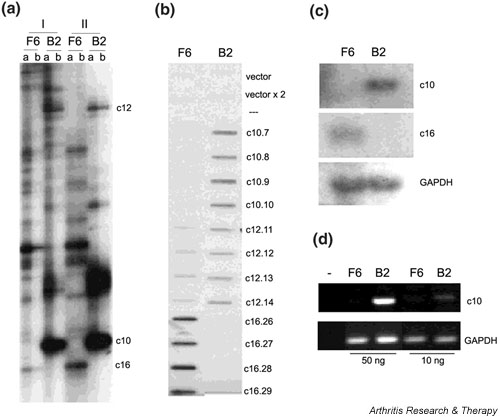
The most prominent functional class of genes found to be deregulated with both differential display and DNA chip hybridization is a class comprising genes encoding for proteins involved in either the ECM, cell–subtratum and cell–cell adhesion, or the cytoskeleton (see Table 4, ECM/Adhesion, Cytoskeleton organization). Several genes involved in cell–cell and cell–ECM adhesion were found to be deregulated, suggesting deregulated adhesion of the arthritogenic SF clone. In order to test the hypothesis functionally, the adherence of both the RA SF clone (B2) and the normal SF clone (F6) to various ECM proteins (fibronectin, vitronectin, laminin, and collagen I) was tested in vitro. The arthritic SF clone adhered less well to all ECM proteins tested than did the normal SF clone (Fig. 6).
Figure 6.
The arthritogenic SF clone B2 exhibits diminished adhesion to proteins of the extracellular matrix. Adhesion assays as described in Materials and methods. Mean averages of triplicates with mean background (adhesion to BSA) values subtracted. Representative experiment out of three. t-test P values were always less than 0.05. BSA = bovine serum albumin; hTNF = human tumor necrosis factor; SF = synovial fibroblast; TAg = large tumor antigen; wt = wild-type.
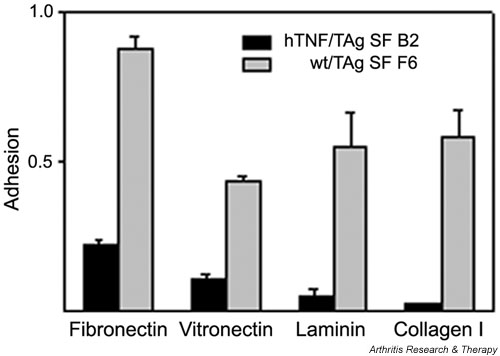
The ability of cells to adhere to the ECM is a critical determinant of cytoskeletal organization and cellular morphology [30], as well as of the ability of a cell to proliferate and migrate [31]. Several genes that control the proliferation rate of the cell were found to be deregulated upon differential gene expression analysis (see Table 4, Growth control) suggesting an altered proliferation capacity. In order to test the hypothesis functionally, the proliferation rate of the two SF clones (arthritic versus normal) was examined in vitro by the [3H]thymidine incorporation/DNA synthesis assay. The arthritogenic SF clone was indeed found to proliferate faster, confirming the expression-based hypothesis (Fig. 7a).
Figure 7.
The arthritogenic SF clone B2 has a higher proliferation rate and exhibits increased migration to fibronectin. (a) DNA synthesis/proliferation assay as described in Materials and methods in the presence or absence of 10% FBS. Values represent [3H]thymidine incorporation (× 103). Mean averages of triplicates. Representative experiment out of three. t-test P values were always less than 0.01. (b) Migration assays as described in Materials and methods. Migration shown was for 2 hours; similar results were found for 4 hours. Mean averages of triplicates with mean background (adhesion to BSA) values subtracted. Representative experiment out of three. t-test P values were always less than 0.05. (c) Assay of wound healing, as described in Materials and methods. Pictures were taken at 0 and 48 hours after the wound. BSA = bovine serum albumin; FBS = fetal bovine serum; hTNF = human tumor necrosis factor; SF = synovial fibroblast; TAg = large tumor antigen; wt = wild-type.
Because it has been suggested that an intermediate state of adhesion (as opposed to strong adhesion or none at all) favors cell motility [32], we investigated the motility of the arthritogenic SF clone by studying its ability to migrate to fibronectin. The arthritic SF clone migrated to fibronectin (through Boyden chambers) more efficiently than its normal counterpart (Fig. 7b). Moreover, the ability of the two clones to 'heal a wound' was also assayed; this is a combined measure of both migration and proliferation. The arthritic SF clone was able to heal the wound much more efficiently, further confirming its increased rate of proliferation and migration (Fig. 7c).
Discussion
Fibroblasts are ubiquitous connective tissue cells of mesenchymal origin, whose primary function is to provide mechanical strength to tissues by secreting a supporting framework of ECM. Chemokines secreted by fibroblasts are an important link between the innate and acquired immune responses and play a crucial role in determining the nature and magnitude of the inflammatory infiltrate. As a result of their activation and inappropriate production of chemokines and matrix components during inflammation and disease, fibroblasts actively define tissue microenvironments and are thought to be responsible for the transition from acute to chronic inflammation and/or acquired immunity [33].
In RA, several potential mechanisms independent of T and B cells have been suggested as the mechanism for disease induction, including those involving macrophage or SF-driven disease [12,34]. SFs derived from RA patients display unique properties and secrete a distinct pattern of cytokines, chemokines, matrix proteases and many other effector molecules. Isolated human RA SFs were able to induce arthritis upon transfer to the knee of healthy SCID mice even in the absence of a functioning immune system [28]. Similarly, in the present study, transfer to the knee joint of immortalized SFs from an immune-independent animal model of RA [9,5] induced an SF-specific, T/B-cell independent, TNF-dependent, arthritis-like disease in healthy mice. An intriguing finding in this report is the ability of SFs to migrate to most tissues throughout the body, including peripheral joints, where only the hTNF-activated/expressing SFs induced signs of the disease. The observed migratory potential could provide an alternative explanation of the origins of the polyarticular nature of RA.
SF activation in arthritis leads to alterations in a number of signalling pathways, which stem from changes in gene expression. The study of differences in gene expression patterns is one of the most promising approaches for understanding mechanisms of differentiation, development, and disease pathogenesis. In the present study, we have used two of the most prominent methods, DD-RT-PCR and DNA microarrays, to compare the gene expression of an arthritogenic SF clone with that of the corresponding wt clone, in a search for novel genes and/or pathways involved in the pathogenesis of RA. We chose to analyze a single clone rather than a population of cells because we needed to establish a monoclonal in vitro cell system for functional validation of gene expression studies, because the SF clone we used was able to transfer the disease to healthy animals and, most importantly, because of the suggested heterogeneity of SFs [13,35]. In accordance with the suggested SF subpopulations in the synovium [13,35], we noticed functional differences (in adhesion to ECM, proliferation rates, cytoskeletal organization) between SF and LF populations and SF clones, as well as between SF clones themselves (unpublished results). TNF-expressing populations of SFs (as well as LFs) adhered more strongly to ECM than their wt counterparts (and than the arthritogenic clone analyzed in this study), suggesting that only a subpopulation of SFs have a pathogenic potential characterized by diminished adhesion. In accordance, the observed down-regulation of MHC class II in the arthritogenic clone (see Table 4, Antigen presentation), which was confirmed by FACS analysis (utilizing the M5/114 antibody; data not shown), was also discovered in a fraction (44%) of the SF population. A large number of SF clones from two different animal models of RA have been prepared and are currently being analyzed in order to correlate gene expression with functional characteristics and the ability to transduce the disease, as means to functionally define subpopulations in the (arthritic or not) synovium.
A number of genes already known to be involved in arthritis or to be regulated by TNF were selected in the differential screen, thus validating our system and approach, together with the discovery of large number of novel genes. Genes, which were found to be deregulated with a high degree of confidence in both DD-RT-PCR and microarray hybridizations, were clustered together according to function (see Table 4), to reveal deregulation of particular cellular functions. Activated SFs have been shown to constitutively up-regulate the expression of transcription factors such as AP-1 (activating protein-1) and nuclear factor kappa B (NF-κB) [36], which are known to control the expression of genes involved in RA such as the matrix metalloproteinases [37,38]. It is expected that up- or down-regulation of numerous other transcription factors during SF activation will be discovered, and that such discovery which will lead indirectly to important genes that get activated or deactivated during pathogenesis. Various transcription and splicing factors were found to be either up-regulated or down-regulated in the arthritogenic SF clone, in accord with the observed massive reprogramming of gene expression. Among these factors, stromelysin-1 platelet-derived growth factor-responsive element binding protein (SPBP) is a putative transcription factor that binds to the promoter region of stromelysin [39], a metalloproteinase known to be up-regulated in the arthritic synovium [40]. Surprisingly, peroxisome proliferator-activated receptor-γ (PPAR-γ), a transcription factor involved in the differentiation of adipocytes [41] (which originate from the same progenitor cells as SFs), was found to be up-regulated in the arthritogenic SF.
A number of stress-response genes were found to be up-regulated in the arthritogenic SF clone, where most of them can be linked directly or indirectly to TNF-mediated cytotoxicity and have been reported in the literature. Elevated levels of ferritin heavy chain have been found to be elevated in the synovial fluid, but not in the serum, of RA patients [42,43]. Ferritin heavy chain, an iron homeostasis protein, was shown to be specifically induced by TNF in fibroblasts, most likely through NF-κB activation, in response to oxidative stress [44,45]. Aldose reductase is a NADPH-dependent aldo-keto reductase, implicated in cellular osmoregulation and detoxification. TNF has been shown to induce aldose reductase through NF-κB binding to the osmotic response elements [46]. Heat-shock protein 70 is also a stress-response protein, which was reported to protect cells against TNF-mediated cytotoxicity [47]. Similarly, the deregulation of several mitochondrial genes or enzymes can be imputed to TNF cytotoxicity, which is known to be mediated by early damage of mitochondrial functions through generation of free radicals [48].
The up-regulation of ribosomal protein mRNA, which is seen in the arthritogenic SF clone, has been reported in various pathological states, with unknown etiology. Interestingly, a number of genes involved in ubiquitination, which targets proteins for degradation, were found to be down-regulated. Therefore, it seems that there is increased protein synthesis and stability in the arthritogenic SF clone. A gross look at the proteome of the two cell types did indeed reveal massive differences (data not shown).
The deregulation of several regulators of calcium levels (ryanodine receptor) or calcium-binding proteins (calumenin) might indicate changes in the intracellular levels of calcium. An increase in the intracellular calcium ion concentration controls a diverge range of cellular functions, including adhesion, motility, gene expression, and proliferation [49]. Calcium crystals have been implicated in the pathogenesis of disease through various pathways [50]. In lymphocytes, calcium plays an essential role in signal transduction upon receptor cross-linking, through activation of many transcription factors, including nuclear factor of activated T cells (NFAT), NF-κB, c-Jun N-terminal kinase 1 (JNK1), myocyte enhancer factor-2 (MEF-2) and cAMP-response-element-binding protein (CREB) [51]. A similar situation could also envisaged for fibroblasts.
Constant tissue remodeling is a characteristic feature of the synovium. The phenomenon is based on, among other factors (i.e. osteoblast/osteoclast ratio), a balance between production of matrix-degrading enzymes (metalloproteinases, cathepsins) on the one hand and, on the other hand, their inhibitors (TIMPs [tissue inhibitors of metalloproteinases]) and matrix components (collagens, fibronectin). In RA, the balance is tilted towards the former side, resulting in tissue destruction. The down-regulation of the expression of a number of collagen genes, fibrillin, and TIMP-3 (see Table 4) and the up-regulation of the expression of MMP1 and MMP9 (see Fig. 4) show that the arthritic SF clone has functional characteristics typical of an RA SF. Moreover, laminin B1, a basement-membrane-specific glycoprotein found to be up-regulated in the arthritic synovium [52], was also found to be up-regulated in the arthritogenic SF clone. In addition, several genes involved in cell–ECM adhesion were found to be deregulated, suggesting decreased adhesion of the arthritic SF clone, a hypothesis functionally confirmed in vitro (see Fig. 6). SPARC (or BM-40 or osteonectin), a matricellular glycoprotein that modulates the interaction of cells with the ECM [53] and whose levels were found to be elevated in the synovial fluids of RA patients [54], was also found to be up-regulated in the arthritogenic SF clone. Transient transfection of the arthritogenic clone with a plasmid over-expressing the antisense mRNA of SPARC did not have any appreciable effects on cell adhesion to ECM proteins (data not shown). Similarly, overexpression of SPARC in the normal SF clone had no effects either. Moreover, in order to decipher the role of SPARC in the transgenic animal model of RA, the hTNF-overexpressing arthritic mouse was crossed with a SPARC-deficient mouse [55]. Knocking out SPARC expression did affect the severity or onset of disease (data not shown).
The ability of cells to adhere to the ECM is a critical determinant of cytoskeletal organization and cellular morphology [30], and of the ability of a cell to proliferate and migrate [31]. Several cytoskeletal genes were found to be deregulated in the arthritic SF clone (see Table 4). Given the differences in the cell shape between the arthritic and normal clones (with the arthritic clone being more rounded, less spread; Fig. 7c and data not shown), the data suggest that the demonstrated altered adhesion to ECM most likely results in reorganization of the cytoskeleton, which in turn is reflected in the altered cellular morphology of the arthritic SF clone. On the other hand, several genes that control the proliferation status of the cell were found to be deregulated in the arthritogenic SF clone. FIN13, a phosphatase inducible by fibroblast growth factor, is found predominately in tissues undergoing proliferation [56]. Levels of activin, a cytokine with potential effects on fibroblast proliferation and structural remodeling [57,58], were found to be elevated in the synovial fluid of RA patients [59]. Both FIN13 and the receptor for activin were found to be up-regulated in the arthritogenic RA SF clone, indicating an enhanced proliferative status. It is consistent with this hypothesis that a number of genes that negatively regulate cell growth (p53, MyD118, gas5, aquaporin, acrogranin) were found to be down-regulated. Most importantly, the hypothesis was tested functionally in vitro, where the arthritogenic SF clone was found to proliferate faster than its normal counterpart (see Fig. 7), thus correlating decreased adhesion with increased proliferation. Moreover, the arthritic SF clone was found to migrate to ECM in vitro much faster than its normal counterpart (see Fig. 7). Since both SFs were able to migrate throughout the body when injected at the knee (see Fig. 3), differential migration to ECM could explain their observed differential pathogenic potential.
The discovery in activated RA SFs of somatic mutations in key regulatory genes, such as H-ras and that for p53, has led to alternative perspectives on synovial biology [12]. Accumulating evidence suggests that SFs exhibit characteristics of transformed cells that contribute to the pathogenesis of RA, namely, expression of several oncogenes (i.e. c-myc) [60,61], anchorage-independent growth and loss of contact inhibition [62], and increased proliferation and reduced apoptosis [63-65]. In agreement, the observed correlation of decreased adhesion with increased proliferation and migration in the arthritic SF has been recently observed in highly metastatic melanoma cells, where a very similar set of genes was found to be deregulated [66]. This analogy suggests that activated fibroblasts utilize similar mechanisms to those of metastatic cancer cells, strengthening the view that the SF has a transformed-like character.
Conclusion
Our results demonstrate an autonomous pathogenic role for TNF-expressing synovial fibroblasts (SFs) in the development of polyarthritis, by showing that these cells have the capacity to migrate throughout the body and cause pathology specifically in joints. These findings provide a possible explanation for the polyarticular nature of rheumatoid arthritis and introduce a novel, simplified model system, which may facilitate the functional dissection of the SF's contribution to RA. Moreover, expression analysis of the arthritogenic SF clone and functional clustering of the deregulated genes revealed a number of cellular pathways that get deregulated in RA. Of these, decreased adhesion to ECM was shown to correlate with increased proliferation and migration, extending the analogies of activated SFs to cancer cells. Expression analysis combined with functional clustering and validation proved to be an indispensable tool in identifying pathogenic mechanisms. Clearly, extending the analysis to include various points in the development of the disease and cross-reference to samples from human patients will undoubtedly lead to deciphering of pathogenic mechanisms and implicate other specific cellular pathways and genes.
Competing interests
None declared.
Abbreviations
BSA = bovine serum albumin; DD = differential display; DD-RT-PCR = differential display reverse transcriptase polymerase chain reaction; DMEM = Dulbecco's modified Eagle's medium; ECM = extracellullar matrix; ELISA = enzyme-linked immunosorbent assay; FACS = fluorescence-activated cell sorter; FBS = fetal bovine serum; FCS = fetal calf serum; H & E = hematoxylin and eosin; hTNF = human tumor necrosis factor; LF = lung fibroblast; MHC = major histocompatibility complex; MMP = matrix metalloproteinase; PBS = phosphate-buffered saline; PCR = polymerase chain reaction; RA = rheumatoid arthritis; RT = reverse transcriptase; SCID = severe combined immunodeficiency; SDS = sodium dodecyl sulfate; SF = synovial fibroblast; SPARC = secreted protein acidic and rich in cysteine; SSC = standard saline citrate; SSPE = standard sodium phosphate EDTA; SV40 = simian virus 40; TAg = large tumor antigen; TIMP = tissue inhibitor of metalloproteinases; TNF = tumor necrosis factor; tsTAg = temperature-sensitive large tumor antigen; VCAM = vascular cell adhesion molecule; wt = wild-type.
Acknowledgments
Acknowledgements
The authors would like to thank Celltech Ltd for providing the CB0006 anti-hTNF antibody and Dr Wim Buurman (University of Limburg, The Netherlands) for providing the hTNF ELISA. VA would like to thank Dr Dimitris Kontoyiannis for critical reading of the manuscript and support. A special thank-you to Ms S Papandreou and Mr S Lalos for technical assistance. This work was supported by European Commission grants QLG1-CT-1999-00202 and QLG1-CT-2001-01407. DP was a holder of a Marie Curie Research Fellowship.
Contributor Information
Vassilis Aidinis, Email: V.Aidinis@Fleming.gr.
George Kollias, Email: G.Kollias@Fleming.gr.
References
- Choy EH, Panayi GS. Cytokine pathways and joint inflammation in rheumatoid arthritis. N Engl J Med. 2001;344:907–916. doi: 10.1056/NEJM200103223441207. [DOI] [PubMed] [Google Scholar]
- Feldmann M. Pathogenesis of arthritis: recent research progress. Nat Immunol. 2001;2:771–773. doi: 10.1038/ni0901-771. [DOI] [PubMed] [Google Scholar]
- Arai KI, Lee F, Miyajima A, Miyatake S, Arai N, Yokota T. Cytokines: coordinators of immune and inflammatory responses. Annu Rev Biochem. 1990;59:783–836. doi: 10.1146/annurev.bi.59.070190.004031. [DOI] [PubMed] [Google Scholar]
- Locksley RM, Killeen N, Lenardo MJ. The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell. 2001;104:487–501. doi: 10.1016/s0092-8674(01)00237-9. [DOI] [PubMed] [Google Scholar]
- Kollias G, Douni E, Kassiotis G, Kontoyiannis D. On the role of tumor necrosis factor and receptors in models of multiorgan failure, rheumatoid arthritis, multiple sclerosis and inflammatory bowel disease. Immunol Rev. 1999;169:175–194. doi: 10.1111/j.1600-065x.1999.tb01315.x. [DOI] [PubMed] [Google Scholar]
- Saxne T, Palladino MA, Jr, Heinegard D, Talal N, Wollheim FA. Detection of tumor necrosis factor alpha but not tumor necrosis factor beta in rheumatoid. Arthritis Rheum. 1988;31:1041–1045. doi: 10.1002/art.1780310816. [DOI] [PubMed] [Google Scholar]
- Di Giovine FS, Nuki G, Duff GW. Tumour necrosis factor in synovial exudates. Ann Rheum Dis. 1988;47:768–772. doi: 10.1136/ard.47.9.768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler DM, Piccoli DS, Hart PH, Hamilton JA. Stimulation of human synovial fibroblast DNA synthesis by recombinant human cytokines. J Rheumatol. 1988;15:1463–1470. [PubMed] [Google Scholar]
- Keffer J, Probert L, Cazlaris H, Georgopoulos S, Kaslaris E, Kioussis D, Kollias G. Transgenic mice expressing human tumour necrosis factor: a predictive genetic model of. EMBO J. 1991;10:4025–4031. doi: 10.1002/j.1460-2075.1991.tb04978.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kontoyiannis D, Pasparakis M, Pizarro TT, Cominelli F, Kollias G. Impaired on/off regulation of TNF biosynthesis in mice lacking TNF AU-rich elements: implications for joint and gut-associated immunopathologies. Immunity. 1999;10:387–398. doi: 10.1016/s1074-7613(00)80038-2. [DOI] [PubMed] [Google Scholar]
- Moreland LW, Baumgartner SW, Schiff MH, Tindall EA, Fleischmann RM, Weaver AL, Ettlinger RE, Cohen S, Koopman WJ, Mohler K, Widmer MB, Blosch CM. Treatment of rheumatoid arthritis with a recombinant human tumor necrosis factor receptor (p75)-Fc fusion protein. N Engl J Med. 1997;337:141–147. doi: 10.1056/NEJM199707173370301. [DOI] [PubMed] [Google Scholar]
- Firestein GS. Invasive fibroblast-like synoviocytes in rheumatoid arthritis. Passive responders or transformed aggressors? Arthritis Rheum. 1996;39:1781–1790. doi: 10.1002/art.1780391103. [DOI] [PubMed] [Google Scholar]
- Kontoyiannis D, Kollias G. Fibroblast biology: Synovial fibroblasts in rheumatoid arthritis: leading role or chorus line? Arthritis Res. 2000;2:342–343. doi: 10.1186/ar109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pap T, Muller-Ladner U, Gay RE, Gay S. Fibroblast biology: Role of synovial fibroblasts in the pathogenesis of rheumatoid arthritis. Arthritis Res. 2000;2:361–367. doi: 10.1186/ar113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards JC. Fibroblast biology: Development and differentiation of synovial fibroblasts in arthritis. Arthritis Res. 2000;2:344–347. doi: 10.1186/ar110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konttinen YT, Li TF, Hukkanen M, Ma J, Xu JW, Virtanen I. Fibroblast biology: Signals targeting the synovial fibroblast in arthritis. Arthritis Res. 2000;2:348–355. doi: 10.1186/ar111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchlin C. Fibroblast biology: Effector signals released by the synovial fibroblast in arthritis. Arthritis Res. 2000;2:356–360. doi: 10.1186/ar112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan FM, Chantry D, Jackson A, Maini R, Feldmann M. Inhibitory effect of TNF alpha antibodies on synovial cell interleukin-1 production in rheumatoid arthritis. Lancet. 1989;2:244–247. doi: 10.1016/s0140-6736(89)90430-3. [DOI] [PubMed] [Google Scholar]
- Bemelmans MH, Gouma DJ, Buurman WA. LPS-induced sTNF-receptor release in vivo in a murine model. Investigation of the role of tumor necrosis factor, IL-1, leukemia inhibiting factor, and IFN-gamma. J Immunol. 1993;151:5554–5562. [PubMed] [Google Scholar]
- Alexopoulou L, Pasparakis M, Kollias G. A murine transmembrane tumor necrosis factor (TNF) transgene induces arthritis by cooperative p55/p75 TNF receptor signaling. Eur J Immunol. 1997;27:2588–2592. doi: 10.1002/eji.1830271018. [DOI] [PubMed] [Google Scholar]
- Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- Clonetech Delta Differential Display kit PT1173-1 Palo Alto, CA, USA: Clonetech Laboratories Inc. 2000.
- Ibrahim SM, Mix E, Bottcher T, Koczan D, Gold R, Rolfs A, Thiesen HJ. Gene expression profiling of the nervous system in murine experimental autoimmune encephalomyelitis. Brain. 2001;124:1927–1938. doi: 10.1093/brain/124.10.1927. [DOI] [PubMed] [Google Scholar]
- Jat PS, Noble MD, Ataliotis P, Tanaka Y, Yannoutsos N, Larsen L, Kioussis D. Direct derivation of conditionally immortal cell lines from an H-2Kb-tsA58 transgenic mouse. Proc Natl Acad Sci USA. 1991;88:5096–5100. doi: 10.1073/pnas.88.12.5096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- deMello DE, Mahmoud S, Padfield PJ, Hoffmann JW. Generation of an immortal differentiated lung type-II epithelial cell line from the adult H-2K(b)tsA58 transgenic mouse. In Vitro Cell Dev Biol Anim. 2000;36:374–382. doi: 10.1290/1071-2690(2000)036<0374:GOAIDL>2.0.CO;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Divieti P, Lanske B, Kronenberg HM, Bringhurst FR. Conditionally immortalized murine osteoblasts lacking the type 1 PTH/PTHrP receptor. J Bone Miner Res. 1998;13:1835–1845. doi: 10.1359/jbmr.1998.13.12.1835. [DOI] [PubMed] [Google Scholar]
- Takeshita S, Kaji K, Kudo A. Identification and characterization of the new osteoclast progenitor with macrophage phenotypes being able to differentiate into mature osteoclasts. J Bone Miner Res. 2000;15:1477–1488. doi: 10.1359/jbmr.2000.15.8.1477. [DOI] [PubMed] [Google Scholar]
- Lehmann J, Jungel A, Lehmann I, Busse F, Biskop M, Saalbach A, Emmrich F, Sack U. Grafting of fibroblasts isolated from the synovial membrane of rheumatoid arthritis (RA) patients induces chronic arthritis in SCID mice-A novel model for studying the arthritogenic role of RA fibroblasts in vivo. J Autoimmun. 2000;15:301–313. doi: 10.1006/jaut.2000.0435. [DOI] [PubMed] [Google Scholar]
- Spanopoulou E, Roman CA, Corcoran LM, Schlissel MS, Silver DP, Nemazee D, Nussenzweig MC, Shinton SA, Hardy RR, Baltimore D. Functional immunoglobulin transgenes guide ordered B-cell differentiation in Rag-1-deficient mice. Genes Dev. 1994;8:1030–1042. doi: 10.1101/gad.8.9.1030. [DOI] [PubMed] [Google Scholar]
- Gumbiner BM. Cell adhesion: the molecular basis of tissue architecture and morphogenesis. Cell. 1996;84:345–357. doi: 10.1016/s0092-8674(00)81279-9. [DOI] [PubMed] [Google Scholar]
- Lelievre SA, Bissell MJ, Pujuguet P. Cell nucleus in context. Crit Rev Eukaryot Gene Expr. 2000;10:13–20. doi: 10.1615/critreveukargeneexpr.v10.i1.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palecek SP, Loftus JC, Ginsberg MH, Lauffenburger DA, Horwitz AF. Integrin-ligand binding properties govern cell migration speed through cell-substratum adhesiveness. Nature. 1997;385:537–540. doi: 10.1038/385537a0. [DOI] [PubMed] [Google Scholar]
- Buckley CD, Pilling D, Lord JM, Akbar AN, Scheel-Toellner D, Salmon M. Fibroblasts regulate the switch from acute resolving to chronic persistent inflammation. Trends Immunol. 2001;22:199–204. doi: 10.1016/s1471-4906(01)01863-4. [DOI] [PubMed] [Google Scholar]
- Burmester GR, Stuhlmuller B, Keyszer G, Kinne RW. Mononuclear phagocytes and rheumatoid synovitis. Mastermind or workhorse in arthritis? Arthritis Rheum. 1997;40:5–18. doi: 10.1002/art.1780400104. [DOI] [PubMed] [Google Scholar]
- Jelaska A, Strehlow D, Korn JH. Fibroblast heterogeneity in physiological conditions and fibrotic disease. Springer Semin Immunopathol. 1999;21:385–395. [PubMed] [Google Scholar]
- Han Z, Boyle DL, Manning AM, Firestein GS. AP-1 and NF-kappaB regulation in rheumatoid arthritis and murine collagen-induced arthritis. Autoimmunity. 1998;28:197–208. doi: 10.3109/08916939808995367. [DOI] [PubMed] [Google Scholar]
- Vincenti MP, Coon CI, Brinckerhoff CE. Nuclear factor kappaB/p50 activates an element in the distal matrix metalloproteinase 1 promoter in interleukin-1beta-stimulated synovial fibroblasts. Arthritis Rheum. 1998;41:1987–1994. doi: 10.1002/1529-0131(199811)41:11<1987::AID-ART14>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Benbow U, Brinckerhoff CE. The AP-1 site and MMP gene regulation: what is all the fuss about? Matrix Biol. 1997;15:519–526. doi: 10.1016/s0945-053x(97)90026-3. [DOI] [PubMed] [Google Scholar]
- Sanz L, Moscat J, Diaz-Meco MT. Molecular characterization of a novel transcription factor that controls stromelysin expression. Mol Cell Biol. 1995;15:3164–3170. doi: 10.1128/mcb.15.6.3164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson LL, Dyer R, Hupe DJ. Matrix metalloproteinases. Curr Opin Chem Biol. 1998;2:466–471. doi: 10.1016/s1367-5931(98)80122-1. [DOI] [PubMed] [Google Scholar]
- Tontonoz P, Hu E, Graves RA, Budavari AI, Spiegelman BM. mPPAR gamma 2: tissue-specific regulator of an adipocyte enhancer. Genes Dev. 1994;8:1224–1234. doi: 10.1101/gad.8.10.1224. [DOI] [PubMed] [Google Scholar]
- Ota T, Katsuki I. Ferritin subunits in sera and synovial fluids from patients with rheumatoid arthritis. J Rheumatol. 1998;25:2315–2318. [PubMed] [Google Scholar]
- Kumon Y, Suehiro T, Nishiya K, Hashimoto K, Nakatani K, Sipe JD. Ferritin correlates with C-reactive protein and acute phase serum amyloid A in synovial fluid, but not in serum. Amyloid. 1999;6:130–135. doi: 10.3109/13506129909007314. [DOI] [PubMed] [Google Scholar]
- Miller LL, Miller SC, Torti SV, Tsuji Y, Torti FM. Iron-independent induction of ferritin H chain by tumor necrosis factor. Proc Natl Acad Sci USA. 1991;88:4946–4950. doi: 10.1073/pnas.88.11.4946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak EL, Larochelle DA, Beaumont C, Torti SV, Torti FM. Role for NF-kappa B in the regulation of ferritin H by tumor necrosis factor-alpha. J Biol Chem. 1995;270:15285–15293. doi: 10.1074/jbc.270.25.15285. [DOI] [PubMed] [Google Scholar]
- Iwata T, Sato S, Jimenez J, McGowan M, Moroni M, Dey A, Ibaraki N, Reddy VN, Carper D. Osmotic response element is required for the induction of aldose reductase by tumor necrosis factor-alpha. J Biol Chem. 1999;274:7993–8001. doi: 10.1074/jbc.274.12.7993. [DOI] [PubMed] [Google Scholar]
- Jaattela M, Wissing D, Bauer PA, Li GC. Major heat shock protein hsp70 protects tumor cells from tumor necrosis factor cytotoxicity. EMBO J. 1992;11:3507–3512. doi: 10.1002/j.1460-2075.1992.tb05433.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze-Osthoff K, Bakker AC, Vanhaesebroeck B, Beyaert R, Jacob WA, Fiers W. Cytotoxic activity of tumor necrosis factor is mediated by early damage of mitochondrial functions. J Biol Chem. 1992;267:5317–5323. [PubMed] [Google Scholar]
- Berridge MJ, Lipp P, Bootman MD. The versatility and universality of calcium signalling. Nat Rev Mol Cell Biol. 2000;1:11–21. doi: 10.1038/35036035. [DOI] [PubMed] [Google Scholar]
- Cheung HS. Calcium crystal effects on the cells of the joint: implications for pathogenesis of disease. Curr Opin Rheumatol. 2000;12:223–227. doi: 10.1097/00002281-200005000-00012. [DOI] [PubMed] [Google Scholar]
- Dolmetsch RE, Lewis RS, Goodnow CC, Healy JI. Differential activation of transcription factors induced by Ca2+ response amplitude and duration. Nature. 1997;386:855–858. doi: 10.1038/386855a0. [DOI] [PubMed] [Google Scholar]
- Konttinen YT, Li TF, Xu JW, Tagaki M, Pirila L, Silvennoinen T, Santavirta S, Virtanen I. Expression of laminins and their integrin receptors in different conditions of synovial membrane and synovial membrane-like interface tissue. Ann Rheum Dis. 1999;58:683–690. doi: 10.1136/ard.58.11.683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane TF, Sage EH. The biology of SPARC, a protein that modulates cell-matrix interactions. FASEB J. 1994;8:163–173. [PubMed] [Google Scholar]
- Nakamura S, Kamihagi K, Satakeda H, Katayama M, Pan H, Okamoto H, Noshiro M, Takahashi K, Yoshihara Y, Shimmei M, Okada Y, Katu Y. Enhancement of SPARC (osteonectin) synthesis in arthritic cartilage. Increased levels in synovial fluids from patients with rheumatoid arthritis and regulation by growth factors and cytokines in chondrocyte cultures. Arthritis Rheum. 1996;39:539–551. doi: 10.1002/art.1780390402. [DOI] [PubMed] [Google Scholar]
- Gilmour DT, Lyon GJ, Carlton MB, Sanes JR, Cunningham JM, Anderson JR, Hogan BL, Evans MJ, Colledge WH. Mice deficient for the secreted glycoprotein SPARC/osteonectin/BM40 develop normally but show severe age-onset cataract formation and disruption of the lens. EMBO J. 1998;17:1860–1870. doi: 10.1093/emboj/17.7.1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guthridge MA, Bellosta P, Tavoloni N, Basilico C. FIN13, a novel growth factor-inducible serine-threonine phosphatase which can inhibit cell cycle progression. Mol Cell Biol. 1997;17:5485–5498. doi: 10.1128/mcb.17.9.5485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohga E, Matsuse T, Teramoto S, Ouchi Y. Activin receptors are expressed on human lung fibroblast and activin A facilitates fibroblast-mediated collagen gel contraction. Life Sci. 2000;66:1603–1613. doi: 10.1016/s0024-3205(00)00480-x. [DOI] [PubMed] [Google Scholar]
- Ohga E, Matsuse T, Teramoto S, Katayama H, Nagase T, Fukuchi Y, Ouchi Y. Effects of activin A on proliferation and differentiation of human lung fibroblasts. Biochem Biophys Res Commun. 1996;228:391–396. doi: 10.1006/bbrc.1996.1672. [DOI] [PubMed] [Google Scholar]
- Yu EW, Dolter KE, Shao LE, Yu J. Suppression of IL-6 biological activities by activin A and implications for inflammatory arthropathies. Clin Exp Immunol. 1998;112:126–132. doi: 10.1046/j.1365-2249.1998.00522.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller-Ladner U, Kriegsmann J, Gay RE, Gay S. Oncogenes in rheumatoid arthritis. Rheum Dis Clin North Am. 1995;21:675–690. [PubMed] [Google Scholar]
- Cutolo M, Sulli A, Barone A, Seriolo B, Accardo S. Sex hormones, proto-oncogene expression and apoptosis: their effects on rheumatoid synovial tissue. Clin Exp Rheumatol. 1996;14:87–94. [PubMed] [Google Scholar]
- Lafyatis R, Remmers EF, Roberts AB, Yocum DE, Sporn MB, Wilder RL. Anchorage-independent growth of synoviocytes from arthritic and normal joints. Stimulation by exogenous platelet-derived growth factor and inhibition by transforming growth factor-beta and retinoids. J Clin Invest. 1989;83:1267–1276. doi: 10.1172/JCI114011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima T, Aono H, Hasunuma T, Yamamoto K, Shirai T, Hirohata K, Nishioka K. Apoptosis and functional Fas antigen in rheumatoid arthritis synoviocytes. Arthritis Rheum. 1995;38:485–491. doi: 10.1002/art.1780380405. [DOI] [PubMed] [Google Scholar]
- Hashiramoto A, Sano H, Maekawa T, Kawahito Y, Kimura S, Kusaka Y, Wilder RL, Kato H, Kondo M, Nakajima H. C-myc anti-sense oligodeoxynucleotides can induce apoptosis and down-regulate Fas expression in rheumatoid synoviocytes. Arthritis Rheum. 1999;42:954–962. doi: 10.1002/1529-0131(199905)42:5<954::AID-ANR14>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Qu Z, Garcia CH, O'Rourke LM, Planck SR, Kohli M, Rosenbaum JT. Local proliferation of fibroblast-like synoviocytes contributes to synovial hyperplasia. Results of proliferating cell nuclear antigen/cyclin, c-myc, and nucleolar organizer region staining. Arthritis Rheum. 1994;37:212–220. doi: 10.1002/art.1780370210. [DOI] [PubMed] [Google Scholar]
- Clark EA, Golub TR, Lander ES, Hynes RO. Genomic analysis of metastasis reveals an essential role for RhoC. Nature. 2000;406:532–535. doi: 10.1038/35020106. [DOI] [PubMed] [Google Scholar]