

Abstract
Tumor necrosis factor (TNF) has been implicated in the development and pathogenicity of infectious diseases and autoimmune disorders, such as septic shock and arthritis. The zinc-finger protein tristetraprolin (TTP) has been identified as a major regulator of TNF biosynthesis. To define its intracellular location and examine its regulation of TNF, a quantitive intracellular staining assay specific for TTP was developed. We establish for the first time that in peripheral blood leukocytes, expression of endogenous TTP is confined to the cytoplasm. Baseline expression of TTP was higher in monocytes than in lymphocytes or neutrophils. After in vitro incubation with lipopolysaccharide (LPS), leukocyte TTP levels increased rapidly, peaking after approximately 2 hours. Monocytes showed the greatest response to LPS stimulation and lymphocytes the least. TTP levels were also studied in leukocytes isolated from healthy volunteers infused with a bolus dose of LPS. TTP expression and initial upregulation in response to LPS infusion were consistent with the in vitro data. Neutrophil TTP levels responded first, reaching an initial peak within 1 hour, monocyte levels peaked next at 2 hours, followed by lymphocytes at 4 hours. This response paralleled plasma TNF levels, which peaked 2 hours after infusion and were no longer detectable after 12 hours. A second rise in intracellular TTP levels, which did not parallel plasma TNF levels, was observed in all leukocyte populations, starting 12 hours after infusion. These data establish the cytoplasmic location of TTP, supporting a major role for this protein in regulating TNF production, and suggest that TTP levels are not regulated solely by TNF.
Keywords: endotoxic shock, inflammation, lipopolysaccharide, tristetraprolin
Introduction
Tumor necrosis factor (TNF) has been implicated in the development and pathogenicity of infectious diseases and autoimmune disorders, such as septic shock, arthritis, and hypotension [1-4]. It plays a pivotal role in the inflammatory cascade and, together with IL-1 and IL-6, has been shown to mediate the acute-phase response [5,6]. Confirmation of its significance in disease comes from the success of clinical trials using anti-TNF therapy to treat rheumatoid arthritis and Crohn's disease [7-10].
Bacterial lipopolysaccharide (LPS), or endotoxin, is found only in Gram-negative bacterial cell walls; it can on its own induce the acute inflammatory response and the clinical characteristics associated with sepsis. After administration of LPS, TNF and other inflammatory mediators are released [11,12]. Infusion of TNF initiates an inflammatory process leading to a phenotype that is indistinguishable from bacterial sepsis [13,14].
Recent work on tristetraprolin (TTP) has provided a new perspective on the regulation of TNF biosynthesis. TTP (also referred to as NuP475, TIS11, or GOS24) is a member of the family of zinc-finger proteins that was originally identified through genetic screens designed to identify immediate early response genes [15-18]. TTP was shown to be an unusually proline-rich protein with a predicted molecular weight of 33.6 kDa. Its name derives from the presence of three PPPPG amino acid repeats that are conserved across species. There are two Cys-Cys-Cys-His (CCCH) zinc fingers, which are responsible for RNA binding [17,19]. There are no studies of the subcellular localization or the regulation of expression of native TTP in freshly isolated cells or tissue. Transfection studies using nonhematopoietic cell lines or prolonged cell culture have suggested that TTP is found in both cytoplasm and the nucleus [20-24].
A deficiency of TTP is associated with a profound proinflammatory phenotype. TTP-deficient mice exhibit cachexia, with dermatitis, medullary and extramedullary hyperplasia, erosive polyarthritis, and autoimmunity with autoantibodies [25]. This phenotype is similar to that observed in mice that overexpress TNF [26]. In agreement with these data, TTP-deficient mice were shown to overproduce TNF, and weekly injections of a neutralizing antibody to TNF eliminated the predominant features of this phenotype. In addition, mice with the double deletion of TTP and TNF receptor (TNFR)-1 do not manifest the aggressive inflammation observed in TTP-deficient mice. In contrast, mice deficient in TTP and TNFR-2 show a more severe phenotype, suggesting a protective role for this receptor [27].
The increase in TNF biosynthesis in TTP-deficient mice is associated with increased mRNA stability [23]. TTP binds to the AU-rich element (ARE) in the 3' untranslated region of TNF mRNA, which regulates TNF mRNA turnover in vivo [28]. The interaction between TTP/TNF and the ARE promotes the turnover of TNF mRNA. TTP also regulates the stability of GM-CSF (granulocyte/macrophage-colony-stimulating factor) mRNA through the ARE, and therefore TTP deficiency is associated with increased production of GM-CSF and with myeloid hyperplasia. This is not a TNF-dependent effect, since the phenotype is not affected by the removal of either TNFR-1 or -2 or by the addition of TNF antibody [27]. Recent studies have suggested a potential interaction between TTP and both IL-3 and IL-8 via the ARE in their mRNA [29,30].
All studies of TTP expression to date have relied on transfected cells and cell lines; there have been no studies on the localization of expression of human TTP, or on its regulation in vivo or in freshly isolated normal cells. To examine the kinetic relation between TNF production and TTP expression in models of septic shock, we developed an intracellular flow cytometric assay. By this method, expression of TTP was determined in peripheral blood leukocytes after the response to in vitro stimulation with LPS. TTP was initially upregulated in all the cells in response to LPS, as shown by flow cytometry and confocal microscopy. The contribution of TNF to this LPS-induced upregulation was confirmed using a humanized monoclonal antibody to TNF. The kinetics of TTP production was also examined in four individuals infused with LPS. After in vivo exposure to LPS, the initial rise in TTP expression correlated with the plasma TNF level. We also observed secondary increases in some individuals, without parallel increments in TNF. These observations suggest that there are multiple factors involved in the regulation of TTP.
Materials and methods
Generation of antihuman TTP polyclonal antibody
A peptide corresponding to the C terminus (amino acids 302–328) of human TTP was synthesized, purified by HPLC, and coupled to KLH (keyhole limpet hemocyanin; Macromolecular Resources, Fort Collins, CO, USA). Anti-human TTP polyclonal antibody was generated by immunization of New Zealand white rabbits with the KLH-coupled TTP C-terminal peptide (GeneMed, San Francisco, CA, USA). The rabbit antiserum obtained was affinity-purified by passage over a Sulfolink column (Pierce, Rockford, IL, USA) to which disulfide linkage of the peptide had been used to immobilize the peptide. Specificity in Western blotting and flow cytometry was evaluated by detection of exogenous TTP in transfected 293 cells and by blocking this activity with peptide [24].
Generation of TTP vector constructs
Human TTP cDNA was generated by RT-PCR from THP-1 RNA and cloned into the pcDNA 3.1His-C vector (Invitrogen, Carlsbad, CA, USA), which encodes a histidine and epitope (EXPRESS-TAG) at the N-terminus of the protein. Orientation of the cloned DNA and the integrity of the DNA sequences were confirmed by sequencing with the ABI Prism Dye Terminator Cycle Sequencing kit (PerkinElmer, Wellesley, MA, USA), and searched against the published sequence (TTP accession no. M63625) on the NCBI Database using the BLAST search program.
Transient transfection
Human embryonic kidney 293 cells (ATCC, Manassas, VA, USA) were plated at a density of 160,000 cells per cm2 in 6-well plates (Costar, Cambridge, MA, USA). TTP (1 μg) or the TTP parental vector pcDNA3.1 His-C (control) was mixed with 50 μl of serum-free RPMI (Roswell Park Memorial Institute) medium. The DNA was mixed with 2 μl of Lipofectamine 2000 reagent (Life Technologies, Rockville, MD, USA) in a total volume of 50 μl of serum-free RPMI per cm2 of cells, incubated for 3 hours, and followed by the addition of an equal volume of media containing 20% fetal calf serum.
Cell preparation
THP-1 (generously provided by Dr P Guyre, Dartmouth Medical School), Jurkat (ATCC), and 293 cell lines were cultured in RPMI complete medium (CM), consisting of RPMI 1640 with 10% heat-inactivated fetal bovine serum, 0.1 ml of sodium pyruvate, 2.0 mmol/l of L-glutamine, 25 mmol/l of 1 M HEPES (N-2-hydroxyethylpiperazine-N'-2-ethanesulfonic acid), 0.1 mmol/l of nonessential amino acids, 0.25 μg/ml of amphotericin, 50 μg/ml gentamicin (all from BioWhittaker, Walkersville, MD, USA) and 5 × 10-5M 2-mercaptoethanol (Sigma, St Louis, MO, USA). Transfected and nontransfected 293 cells were harvested using trypsin (Life Technologies), washed twice with CM, resuspended to a concentration of 5 × 106 cells/ml, and fixed with 4% methanol-free formaldehyde (Polysciences Inc, Warrington, PA, USA). THP-1 cells were incubated with and without LPS, 0.1 μg/ml (Escherichia coli 055:B5, Sigma), or with and without TNF (100 U/ml). In addition, infliximab (Remicade®), 100 μg/ml (Centocor Inc, Malvern, Pennsylvania, USA), was also incubated with and without LPS or TNF to determine how much effect TNF has on TTP induction. Preliminary studies on TNF-induced IκBα phosphorylation determined the optimum concentration for inhibition with infliximab. After incubation, the cells were washed in staining buffer and fixed with 1% formaldehyde.
Leukocytes from healthy donors were isolated by dextran sedimentation from heparinized peripheral blood. In some experiments, blood was combined 1:1 with RPMI CM and incubated with or without 0.1 μg/ml LPS. At the indicated times, blood was mixed 1:1 with 2% dextran (150 kDa; Polysciences) in prewarmed RPMI and left at 37°C for 20 min to sediment the red blood cells preferentially. The white-cell supernatant was then aspirated, centrifuged, and mixed 1:5 with CM. After centrifugation, the cell pellet was resuspended with staining buffer (prechilled to 4°C) consisting of phosphate-buffered saline solution with 0.2% BSA and 1 μg/ml sodium azide. The white cells were then counted using Turks solution (0.01% crystal violet and 3% acetic acid in distilled water), washed as before, and resuspended to a concentration of 5 × 106 cells/ml with staining buffer.
Immunofluorescence staining
To detect extracellular antigens, cells were pipetted into a 96-well polypropylene plate (Costar), and the plate was centrifuged at 400 g for 5 min. The cells and reagents were kept at 4°C for the remainder of the experiment. The supernatant was aspirated, and the cells were resuspended in 6% human IgG (Sigma) in staining buffer to block nonspecific binding. Allophycocyanin (APC)-conjugated CD14 (Becton, Dickinson and Co, San Jose, CA, USA) was added, and the cells were incubated for a further 20 min in the dark. The cells were then washed once in staining buffer and residual red blood cells lysed with FACS' Lysing solution (Becton Dickinson). After 5 min the cells were washed twice with staining buffer and fixed overnight in 1% formaldehyde.
To detect intracellular TTP, fixed cells were washed twice in permeabilization buffer (consisting of staining buffer with the addition of 1 μg/ml saponin), centrifuged as before, and incubated for 30 min at 4°C in blocking buffer, consisting of 3% IgG with 3% nonfat powdered milk for leukocytes in permeabilization buffer. The plate was centrifuged, the supernatant was aspirated, and 3.8 μg per 5 × 105 cells of anti-TTP rabbit polyclonal or control antibody (DAKO A/S, Glostrup, Denmark) was added for 45 min more. After incubation, the cells were washed in permeabilization buffer and resuspended in FITC-conjugated F(ab')2 (antigen-binding fragment) donkey antirabbit IgG (25 μg/ml, Jackson ImmunoResearch, West Grove, PA, USA) for 45 min at 4°C. Cells were washed with permeabilization buffer and then with staining buffer. They were resuspended with 1% formaldehyde and stored in the dark at 4°C until analysis. For the infliximab inhibition experiments, a goat polyclonal antibody to TTP was used (Santa Cruz Biotechnology Inc, Santa Cruz, CA) with a secondary swine antigoat FITC-conjugated IgG antibody (Sigma). Preliminary experiments showed that the affinity of this antibody for TTP was equivalent to that of the rabbit polyclonal antibody.
In the LPS infusion study, TruCount® calibration beads (Becton Dickinson) were quantitatively added to the stained cells. Enumeration of individual leukocyte counts was then completed using Cell Quest® software.
FACS analysis and confocal microscopy
Analysis by flow cytometry was completed using a FAC-SCalibur (Becton Dickinson). Contaminants, such as red cells and cell debris, were removed by forward and side scatter gating; in addition, CD14 versus side scatter was used to create gates to differentiate lymphocytes, monocytes, and neutrophils. Fluorescein fluorescence was detected through the FL-1 channel, APC fluorescence was assessed using the FL-4 channel, and data were acquired and analyzed using the CellQuest® application on a Power Macintosh G3 computer. For each sample, at least 2000 monocytes were collected and the mean fluorescence intensity was evaluated. Calibration was done using Rainbow Microspheres (Spherotech Inc, Libertyville, IL, USA), with final values expressed as the milliequivalents of soluble fluorescein (MESF) [31].
293 cells were sorted for the brightest FITC fluorescence using a FACStar Plus flow cytometer (Becton Dickinson). Cells were collected into tubes containing 100 μl fetal calf serum, centrifuged, and resuspended in 1% formaldehyde. The cells were stored overnight at 4°C in the dark. TTP/FITC-stained white blood cells or THP-1 cells were centrifuged and resuspended in formaldehyde as for 293 cells. Nuclear staining was done using propidium iodide 0.01 mg/ml (Sigma). For confocal microscopy, cells were pipetted from the base of the settled cell suspension and placed onto a glass slide with Prolong® Antifade (Molecular Probes, Eugene, OR, USA) to inhibit quenching. The cells were then covered with a cover slip protected with clear nail polish.
Microscopy for FITC-conjugated TTP analysis was completed using a BioRad MRC1024 laser scanning confocal microscope (BioRad, Hercules, CA, USA). Cells were scanned for fluorescence using the 488-nm line from a 15-mW Kr/Ar laser and two photodetectors (522/32-nm dichroic for FITC fluorescence and 585-nm longpass for propidium iodide fluorescence). A 63× Plan-APO 1.4 NA objective (Carl Zeiss Inc, Thornwood, NY, USA) was used in conjunction with an iris setting of 2.5, which allowed for detection of optical sections of the fluorescence image that were approximately 0.5 μm thick. Representative images were selected from the slices though the center of the cells after sectioning through the entire cell.
LPS infusion
Consenting volunteers were given an intravenous bolus of endotoxin in accordance with a protocol approved by the Institutional and Scientific Review Boards of the Dartmouth-Hitchcock Medical Center and Norris Cotton Cancer Center, Lebanon, NH, USA. One month before admission and again just before admission, the volunteers' physical wellbeing and health were assessed, and current medications were noted. The infusion was conducted in the intensive care unit at Dartmouth-Hitchcock Medical Center with continuous supervision for the first 12 hours. Blood pressure, heart rate, and body temperature were evaluated throughout this period.
Clinical Center reference endotoxin (CCRE), lot O:133, derived from E. coli (United States Pharmacopeia Convention Inc, Rockville, MD, USA [32]), was injected intravenously at a dose of 4 ng/kg. Three consecutive baseline blood samples were taken, at 15, 10, and 5 min before infusion, and samples were taken at 0.45, 1, 2, 4, 8, 12, 24, 48, and 72 hours after infusion, from a peripheral, indwelling, venous catheter directly into a heparinized syringe (Becton Dickinson). Blood was sedimented in dextran, extracellularly stained for CD14, and fixed with 1% formaldehyde. The cells were stored overnight at 4°C and then stained for intracellular TTP. A human TNF ELISA kit (R&D Systems, Minneapolis, MN, USA) was used to determine the concentration of TNF in plasma from peripheral blood, in accordance with the manufacturer's instructions. The TNF-α standard was made up as directed to read a maximum concentration of 1000 pg/ml and a minimum of 15.6 pg/ml.
Statistical analysis
Analysis was by one-way analysis of variance (ANOVA) with post hoc Bonferroni adjustment (all-means comparison) test, using the Graphpad® Prism version 3.00 for Windows (Graphpad Software, San Diego, CA, USA).
Results
Optimization and specificity of the TTP polyclonal antibody
To determine the optimal working concentration of the TTP antibody, 293 cells transfected with TTP were fixed, permeabilized, and stained at various concentrations of antibody or IgG control. There was a concentration-dependent increase in fluorescence (Fig. 1a), which was standardized using fluorescent microspheres in order to compare different experiments and cell types [31]. The optimum concentration of antibody was determined to be 3.8 μg per 5 × 105 cells, where the intensity curve began to flatten. At higher concentrations, the MESF value for the control sera increased concordantly, indicating nonspecific binding. Specificity of the TTP antibody was determined by blocking with the identical peptide sequence used to generate the polyclonal antibody. In the presence of blocking TTP peptide, at concentrations either 10 or 50 times the amount of TTP antibody used, the detection of the TTP protein within the transfected cells was nearly eliminated (Fig. 1b).
Figure 1.
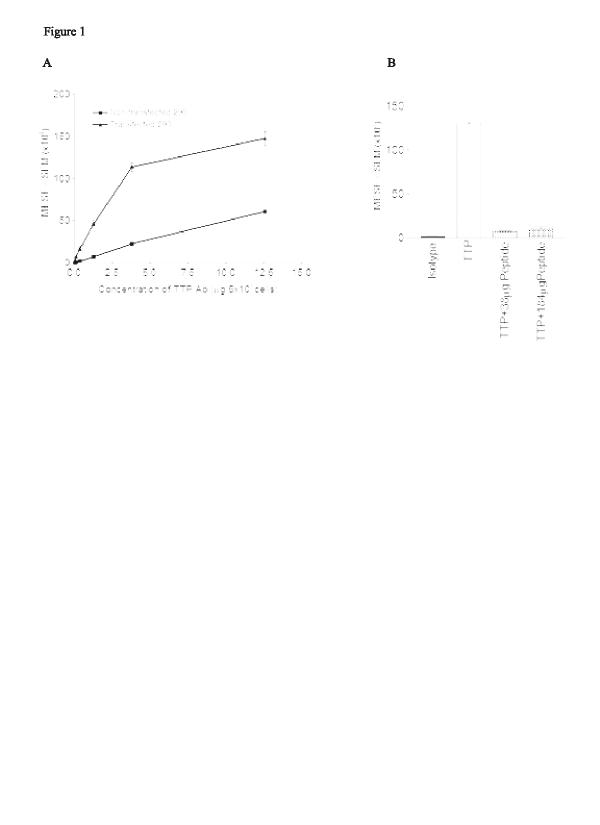
Saturation curve and specificity of the tristetraprolin (TTP) polyclonal antibody. Results are shown as milliequivalents of soluble fluorescein ± SE and are representative of two separate experiments. (a) Nontransfected and transfected 293 cells were stained with increasing concentrations of the rabbit-derived anti-TTP antibody, or a rabbit IgG control, and then with secondary FITC-conjugated donkey antirabbit IgG. (b) Specificity was demonstrated by introducing the TTP peptide at 10 and 50 times the concentration of the antibody used. All bars derive from the transfected phenotype. (c) Confocal microscopy of cells stained as described for TTP and nuclear staining with propidium iodide. Ab = antibodies; MESF = milliequivalents of soluble fluorescein; SE = standard error of the mean.
Confocal sectioning of transfected 293 cells showed that the presence of TTP protein was primarily in the cytoplasmic fraction (Fig. 1c). Z sections taken every 0.5 μm through the cell showed little, if any, TTP within the nucleus. In nontransfected 293 cells, a small amount of TTP was detected by flow cytometry and confirmed by confocal microscopy. Transfected 293 cells expressed nine times as much TTP as the untransfected cells at the optimal working concentration of antibody. Preincubation of the nontransfected 293 cells with TTP peptide blocked the activity, confirming its identity. These results confirmed our previously published data in which Western blot analysis showed that TTP was localized in the cytoplasmic extract of 293 cells [24].
Induction of TTP in the THP-1 cell line
Since TNF biosynthesis in macrophages is rapidly triggered by treatment with LPS, the effects of this endotoxin on TTP expression were examined. THP-1 cells, a human myelomonocytic cell line, were incubated with and without LPS (0.1 μg/ml) for up to 24 hours. At representative time points, a sample was removed and stained with either anti-TTP or the IgG control, as described in Materials and methods. The addition of LPS rapidly increased the expression of TTP by THP-1 cells (Fig. 2a). Increases in TTP were detectable 1 hour after stimulation and were still evident in the cytosol 4 hours after LPS treatment. Analysis of the cells by confocal microscopy (Fig. 2b) confirmed the upregulation and indicated that the TTP was expressed intracellularly, relatively homogeneously but confined to the cytoplasm. These data show that LPS stimulation rapidly increases human TTP protein expression in the cytoplasm. The kinetics of induction by LPS parallels that seen with TTP mRNA in bone-marrow-derived murine macrophages [23].
Figure 2.
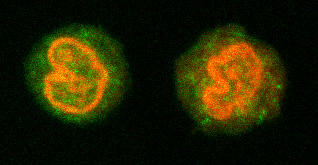
Lipopolysaccharide (LPS) induction of tristetraprolin (TTP) in resting THP-1 cells. Cells were incubated with and without LPS and then at specific times were permeabilized and stained with TTP antibody or a rabbit IgG control. (a) Induction was rapid, reaching a maximum at 1 hour. Results are expressed as the percentage change over baseline in MESF ± SE (n = 3). Significant increases are shown with respect to time 0 (*P < 0.05). (b) Confocal microscopy reveals the basal expression of TTP at time 0, and the upregulation at 2 hours after LPS treatment. The left panel shows the expression before LPS incubation, the right, after LPS stimulation. Nuclear staining was completed with propidium iodide. MESF = milliequivalents of soluble fluorescein; SE = standard error.
In vitro TTP induction with LPS in peripheral blood leukocytes
Modulation of TTP expression by LPS was subsequently examined in normal peripheral leukocytes. Whole blood from eight healthy volunteers was incubated at 37°C alone or with LPS (0.1 μg/ml) for up to 24 hours. At periodic time points, a sample was taken and leukocytes were purified by dextran sedimentation, fixed, permeabilized, and then stained with either anti-TTP or control antibody. In leukocytes, constitutive expression of TTP ranged from approximately 9 × 103 to 35 × 103 MESF. Monocytes constitutively expressed over three times as much TTP as either lymphocytes or neutrophils (Table 1). Stimulation with LPS in vitro produced an increase in TTP expression in all leukocyte subpopulations. This was maximal 2 hours after stimulation. TTP levels increased to a higher maximal level in monocytes than in other leukocytes (+8.5 × 103 MESF; Fig. 3a). Two-color flow cytometry indicated that CD4+ and CD8+ T lymphocytes expressed equivalent amounts of TTP (data not shown). The kinetics of TTP induction with LPS was consistent and monophasic in all leukocytes (Fig. 3i). The change in TTP expression was greatest in neutrophils and least in lymphocytes. Since lymphocytes lack Toll-like receptor 4 and express little or no CD14 [33], we conclude that the induction of TTP in these cells was occurring indirectly, presumably through stimulation with TNF, although other cytokines, including IL-1, IL-6, and IL-8, could contribute to this response.
Table 1.
Constitutive expression of tristetraprolin in peripheral blood leukocytes
| Cell type | MESF (in thousands) Baseline expressiona | Range |
| Lymphocyte | 9.5 (1.2) | 4.45–14.17 |
| Monocyte | 35.4 (3.3)* | 13.90–44.32 |
| Neutrophil | 13.8 (3.3) | 9.14–18.28 |
Intracellular TTP levels were detected using the TTP antibody in conjunction with a fluorescein-labelled antirabbit Ig. Background levels were subtracted from these results and CD14-staining plus forward and side scatter gating permitted specific analysis of TTP expression in the leukocyte populations. Monocytes have a significantly higher constitutive TTP expression than other leukocytes. aMean (SE); n = 8. *P < 0.001. MESF = milliequivalents of soluble fluorescein.
Figure 3.
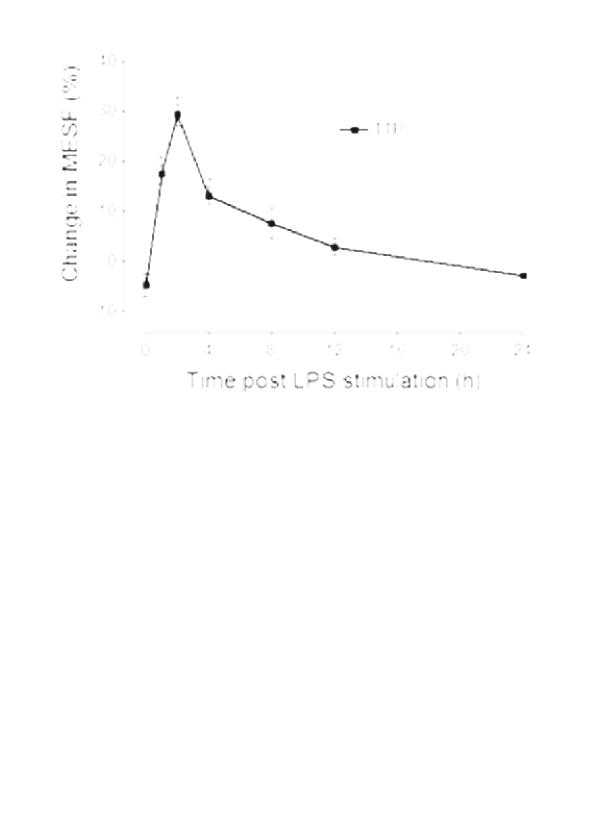
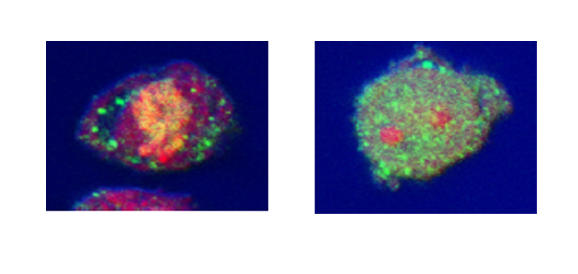
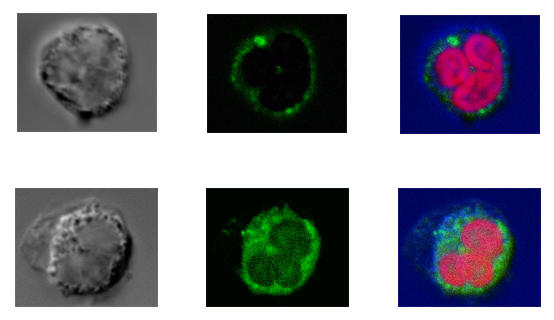
Changes in tristetraprolin (TTP) expression by leukocytes after lipopolysaccharide (LPS) stimulation in vitro. (a) LPS induced TTP expression in all leukocytes. Results are expressed as the LPS-induced increase in TTP specific staining over baseline expression. Significant increases are shown with respect to time 0 (*P < 0.05 monocytes or lymphocytes; #P < 0.05 neutrophils; n = 8). Monocyte recovery was poor at time points beyond 4 hours and these results are excluded. (b) Confocal microscopy shows the upregulation of TTP in neutrophils 2 hours after LPS stimulation. Neutrophils before (i-iii) and after (iv-vi) LPS stimulation. (i,iv) transmitted light (differential interference contrast) image of the cell; (ii,v) TTP (FITC); (iii,vi) composite image, nuclear material stained red with propidium iodide.
LPS-induced TTP upregulation was confirmed in all cell types by confocal microscopy. Analysis confirmed an increase in TTP in neutrophils 2 hours after stimulation (Fig. 3). Similar data were obtained for lymphocytes and monocytes (data not shown). Distribution in resting levels and after stimulation with LPS was primarily cytosolic, which is in accord with our data for 293 and THP-1 cells.
Correlation of in vivo TTP production with TNF after LPS bolus
To study the kinetics of TTP induction in vivo, we infused 4 ng/kg of clinical-grade endotoxin at time 0 in four volunteers. Blood was drawn before the infusion and at specified intervals afterwards, and aliquots were analyzed for leukocyte cell counts, TTP expression, and plasma TNF levels.
The cell counts of all the leukocyte subpopulations in all the volunteers fell rapidly, as seen 1 hour after infusion in all volunteers (Fig. 4a,4b,4c), in concordance with published data [34-36]. Beyond the 1-hour time point, the neutrophil cell count increased, reaching a peak at 12 hours. It then decreased to baseline values at 24 hours (Fig. 4a). Lymphocyte and monocyte data showed similar initial kinetics. The lymphocyte and monocyte curves were biphasic, peaking at 4 hours, dropping to baseline at 12 hours, and beginning a second increase at 48 hours.
Figure 4.
White blood cell count in four volunteers at various times after infusion with lipopolysaccharide. Cell numbers for (a) neutrophils, (b) lymphocytes, and (c) monocytes. Plasma TNF levels (d) were measured by ELISA. TNF = tumor necrosis factor.
Plasma TNF levels were analyzed throughout the study. The circulating TNF level increased to a peak at 2 hours after infusion (Fig. 4d). After 12 hours, there was no detectable TNF in any of the four volunteers. This monophasic peak was consistent with findings in other studies [37].
Analysis of TTP expression in leukocyte populations before and after LPS infusion revealed a biphasic induction, with an initial peak occurring within 4 hours and a second after 12 hours (Fig. 5a,5b,5c). Baseline and maximum TTP expression in each cell type for each volunteer is presented in Table 2. The expression and initial upregulation of TTP in response to LPS in all leukocyte populations was consistent with the data found in vitro. Neutrophils responded rapidly to LPS infusion, reaching an initial peak within an hour of infusion, although the average increase in the four volunteers appears to be moderate (50%). Analysis of monocytic TTP showed an increase in expression, peaking at 2 hours (Fig. 5b). Lymphocyte TTP expression peaked at 4 hours, which was later than the initial peak response of neutrophils and monocytes (Fig. 5c). A second, more dramatic, rise, which did not parallel changes in plasma TNF levels (Fig. 5d), was observed 12 hours after infusion in all leukocyte populations.
Figure 5.
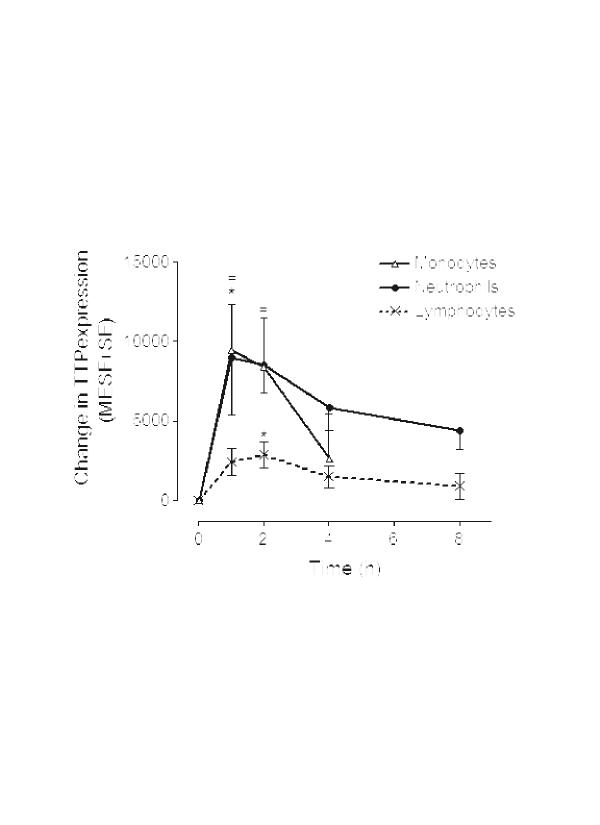
The effect of lipopolysaccharide (LPS) on leukocyte tristetraprolin (TTP) expression in vivo. Four human subjects were infused with a bolus dose of LPS at T = 0. Blood was drawn at specific times before and after infusion, the leukocytes were purified, and expression of TTP was determined. The graphs show the percentage change in TTP expression in (a) neutrophils, (b) monocytes, and (c) lymphocytes with respect to values before infusion, where a doubling in TTP expression is 100% (mean ± SE; n = 4).
Table 2.
Expression in vivo of tristetraprolin in volunteers
| Volunteer no. | Neutrophils | Lymphocytes | Monocytes | |
| 1 | Baseline | 16.5 (1.2) | 18.8 (1.0) | 15.6 (1.6) |
| Peak | 54.9 (2.0) | 90.4 (8.8) | 18.9 (2.4) | |
| 2 | Baseline | 15.6 (1.2) | 9.9 (1.0) | 23.2 (2.0) |
| Peak | 17.1 (3.6) | 13.1 (1.2) | 40.8 (4.3) | |
| 3 | Baseline | 9.7 (8.2) | 4.2 (0.5) | 20.8 (1.3) |
| Peak | 20.8 (1.4) | 9.4 (2.3) | 62.0 (29.0) | |
| 4 | Baseline | 13.6 (1.1) | 6.0 (0.6) | 16.6 (1.7) |
| Peak | 17.5 (1.1) | 19.1 (1.8) | 47.2 (1.6) |
Values are mean milliequivalents of soluble fluorescein (SE) (in thousands). Human subjects were infused with a bolus dose of lipopolysaccharide at T = 0. Blood was drawn at specific times before and after infusion, and the leukocytes were purified by dextran sedimentation. Expression of tristetraprolin was determined by flow cytometry.
TNF contribution in LPS induction of TTP
The human in vivo studies described here show increases in circulating plasma TNF that preceded TTP induction in all the leukocyte types investigated. This observation is consistent with the hypothesis that LPS mediates induction of TTP through the production of TNF. However, this hypothesis does not explain the second rise in TTP we observed in vivo, which was not associated with TNF. To examine this phenomena in more detail, THP-1 cells were cultured with LPS or TNF in the presence of blocking TNF antibody. The antibody sequesters TNF in the supernatant, preventing its interaction with TNF receptors. LPS induced greater TTP expression than TNF (Fig. 6a,6b). When THP-1 cells were stimulated with LPS in the presence of the blocking TNF antibody, the TTP response was reduced but not eliminated (Fig. 6a). In fact, the initial induction of TTP detectable during the first hour was unaffected, suggesting that TNF was not solely responsible for the LPS-induced rise. In contrast, TNF induction of TTP by THP-1 cells was completely blocked in the presence of the blocking TNF antibody (Fig. 6b).
Figure 6.
Contribution of tumor necrosis factor (TNF) to lipopolysaccharide (LPS) induction of tristetraprolin (TTP) in THP-1 cells. The cells were incubated with LPS (a) or TNF (b) in the absence or presence of blocking TNF antibody (infliximab [Remicade]), and TTP levels were measured at specific times. Results are expressed as percentage change from baseline expression (mean ± SE). Experiments were performed in duplicate.
Discussion
This study is the first to evaluate the expression and regulation of endogenous human TTP in myeloid and lymphoid cells. It is the first to examine the subcellular localization of native TTP protein in freshly isolated cells, since all previous studies relied on prolonged cell culture or transfection/overexpression approaches using cell lines [20-22,24,25]. We show that endogenous human TTP is expressed in neutrophils, lymphocytes, and monocytes. They are significant in their demonstration that native human TTP is totally cytoplasmic; there was little evidence of nuclear localization in these studies, an observation that is consistent with previous work with human TTP, in which immunoblotting showed cytoplasmic localization [24].
The discrepancy between our findings and findings of nuclear localization of TTP may be due to several factors [20-22,24,25]. First, most of those studies did not measure endogenous TTP but usually relied on transfection of cell lines, perhaps resulting in mislocalization due to the presence of an epitope tag or overexpression [20-22,24,25]. Second, by definition, those cells were largely cycling and thus were not representative of normal hematopoietic cells. Third, mouse or rat TTP was used in those studies [20-22,25]. Although highly conserved, it is possible that human TTP is subtly different from rodent TTP. We think this unlikely, as immunoprecipitation studies of bone-marrow-derived murine macrophages show murine TTP predominantly in the cytoplasmic fraction [23]. This matter requires further analysis once improved antibodies to murine TTP are generated.
Our studies clearly showed human TTP in lymphocytes and neutrophils, in addition to monocytes. Early work by Carballo and colleagues [38] showed the importance of macrophages, not lymphocytes, in the development of the phenotype associated with TTP deficiency. This highlights the importance of macrophages in the production of TNF, supported by current hypotheses on the pivotal role of this cell type in TNF overproduction in autoimmune diseases [39]. Thus, our findings suggest that TTP serves an entirely different role in other leukocytes, relative to their role in regulating cytokine production by macrophages, and that TNF production is not simply regulated by TTP.
Despite these potential differences in the function of human TTP based on cell type, our studies show a common pattern of induction by LPS in monocytes, neutrophils, and lymphocytes. The upregulation of human TTP occurs rapidly, within the first hour of stimulation. This is consistent with murine data demonstrating that upregulation occurs within 30 min of LPS stimulation, remaining high for 4 hours [23]. Using confocal imaging, we found TTP was primarily detected in the cytoplasmic fraction, confirming our earlier immunoblotting studies [24].
LPS stimulation of peripheral blood leukocytes in vitro revealed TTP-induction kinetics analogous to that observed for THP-1 cells. We used a whole blood assay, since the cytokines produced after in vitro incubation with LPS are analogous to those produced after in vivo infusion, providing an important model of sepsis [40]. Baseline expression of TTP was higher in monocytes than in either lymphocytes or neutrophils. Lymphocytes responded the least to LPS stimulation, whereas monocytes responded the most. Monocytes were not followed beyond 4 hours after stimulation with LPS, because of the low numbers of cells retrieved, probably due to upregulation of adhesion molecules (ICAM-1) [41].
The induction of TTP expression by either LPS or TNF has not been studied in an in vivo system before. In this study, four human volunteers were infused with LPS and the subsequent changes in leukocyte cell count, TTP expression in leukocyte subpopulations, and levels of TNF were followed over a 72-hour period. Although there was some individual variability in the overall kinetics, basal levels and the initial induction of TTP by LPS were within the range observed in vitro (Tables 1 and 2). All cell types showed an increase in TTP expression within the first hour. Interestingly, TTP was induced by LPS in vivo more slowly (after 4 hours vs 1–2 hours) and to a lesser degree in the lymphocyte population than in neutrophils or monocytes. The absence of LPS-signal transducers (CD14, Toll-like receptor 4) on most lymphocytes relative to monocytes and neutrophils may account for this altered expression [42,43]. Since lymphocytes express TNF receptors and TNF itself can induce TTP mRNA expression [38], these data are consistent with the interpretation that TTP induction in lymphocytes is transduced not by LPS binding, but by TNF produced by monocytes and/or neutrophils acting on lymphocytes.
The initial fall in leukocyte cell count observed in vivo is in accord with published data [34-36]. The numbers of monocytes and neutrophils dropped within the first hour, by approximately 98% and 76%, respectively, and of lymphocytes, by 80% after 4 hours. This decrease is consistent with data suggesting that the reduction in leukocytes results from a decrease in granulocytes and monocytes as they become activated, migrating and adhering to vascular endothelium [44]. This drop in cell numbers was followed by an increase in all leukocyte populations, beginning at 12 hours and continuing through the 72-hour time point. This raises the issue of whether the apparent changes in TTP expression were due, in part, to differential rates of margination by leukocyte subpopulations. Since the levels of TTP induction seen in the first hour in vivo approximate those seen in vitro, we believe that this induction reflects the increased synthesis and expression of TTP by all leukocyte subpopulations and is consistent with the kinetics of their mRNA induction [23].
TTP and TNF are reportedly involved in a negative-feedback loop, in which increases in TNF concentration are associated with induction of TTP [23]. In these studies, LPS, which induces the release of TNF [45], produced a rise in TTP within 4 hours of stimulation. Although the initial rise in TTP in vivo paralleled a rapid and considerable increase in plasma TNF, the second, late increases in intracellular TTP did not reflect TNF levels. This change may have been mediated by other cytokines (IL-1β, IL-8) whose levels have been shown to increase with kinetics that parallel this rise in TTP [40,46,47]. Alternatively, the late rise in cytoplasmic TTP may be due to release from the bone marrow of leukocytes that have high levels of TTP or an adaptive response to endotoxemia.
TNF overproduction mediates both sepsis and chronic inflammatory disorders such as RA. Its production may be modulated at transcriptional, post-transcriptional, and post-translational levels [48-50]. Many of the effects observed after LPS stimulation can be attributed to subsequent production of TNF. Moreover, infusion of TNF initiates inflammatory processes, with a resulting phenotype that is indistinguishable from bacterial sepsis [13,14]. We investigated the contribution of TNF in the LPS-mediated induction of TTP in THP-1 cells using a blocking TNF antibody. TNF induction of TTP expression was almost completely eliminated by incubation with the TNF inhibitor, proving the direct effect of TNF on TTP induction. LPS, however, induced much greater levels of TTP in THP-1 cells than TNF did. In the presence of blocking TNF antibody, LPS induction of TTP was initially unaffected but began to decline after 1 hour, suggesting that LPS itself may mediate the initial increases in TTP and showing that the prolonged induction of TTP is dependent on TNF.
Conclusion
We have shown through an intracellular staining assay that human TTP expression is modulated by LPS and TNF. LPS-induction of TTP was partly dependent on the production of TNF. The ability of LPS to induce TTP independently of TNF may account for the earlier response observed in neutrophils and monocytes that have LPS-signaling transduction molecules. In contrast, lymphocytes demonstrate a delayed and lower TTP response and are probably responding to TNF, or other cytokines, produced by myeloid cells. In addition, a second rise in TTP expression was observed in vivo, which did not correlate with any detectable rise in TNF concentration, implying that there are additional factors involved in the regulation of this intracellular protein. TNF is a predominant proinflammatory cytokine, particularly in sepsis and chronic inflammatory disorders suggesting that the role of its negative regulator, TTP, may be of functional importance in the development and pathogenesis of disease. Investigating any potential abnormalities in expression or regulation may prove to be a valuable therapeutic intervention.
Competing interests
None declared.
Abbreviations
APC = allophycocyanin; ARE = AU-rich element; CM = complete media; ELISA = enzyme-linked immunosorbent assay; FACS = fluorescence-activated cell sorting; FITC = fluorescein isothiocyanate; IL = interleukin; KLH = keyhole limpet hemocyanin; LPS = lipopolysaccharide; MESF = milliequivalents of soluble fluorescein; RPMI = Roswell Park Memorial Institute [medium]; RT-PCR = reverse transcriptase polymerase chain reaction; TNF = tumor necrosis factor; TNFR = TNF receptor; TTP = tristetraprolin; WBC = white blood cell.
Acknowledgments
Acknowledgements
Supported in part by grants from NH-IRC (PKW), NIH Grant P20 RR16437 from the COBRE Program of the National Center for Research Resources (PKW), Hitchcock Foundation Grant 90461 (MY, AJR), NRSA Predoctoral Training grants 02T32 AI07363 and AR07576 (KH, JC), and the Grimshaw-Gudewicz Foundation and Joint Research Board of St Bartholomew's Hospital (XMSR) (AMF). Flow cytometry and laser scanning confocal microscopy were performed at Dartmouth Medical School in the Herbert C Englert Cell Analysis Laboratory, which was established with equipment grants from the Fannie E Rippel Foundation and the NIH Shared Instrument Program and is supported in part by the core grant of the Norris Cotton Cancer Center (CA-23108).
References
- Waage A, Brandtzaeg P, Halstensen A, Kierulf P, Espevik T. The complex pattern of cytokines in serum from patients with meningococcal septic shock. Association between interleukin 6, interleukin 1, and fatal outcome. J Exp Med. 1989;169:333–338. doi: 10.1084/jem.169.1.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tracey KJ, Beutler B, Lowry SF, Merryweather J, Wolpe S, Milsark IW, Hariri RJ, Fahey TJ, 3rd, Zentella A, Albert JD, et al. Shock and tissue injury induced by recombinant human cachectin. Science. 1986;234:470–474. doi: 10.1126/science.3764421. [DOI] [PubMed] [Google Scholar]
- Houssiau FA, Devogelaer JP, Van Damme J, de Deuxchaisnes CN, Van Snick J. Interleukin-6 in synovial fluid and serum of patients with rheumatoid arthritis and other inflammatory arthritides. Arthritis Rheum. 1988;31:784–788. doi: 10.1002/art.1780310614. [DOI] [PubMed] [Google Scholar]
- Miesel R, Hartung R, Kroeger H. Priming of NADPH oxidase by tumor necrosis factor alpha in patients with inflammatory and autoimmune rheumatic diseases. Inflammation. 1996;20:427–438. doi: 10.1007/BF01486744. [DOI] [PubMed] [Google Scholar]
- Badolato R, Oppenheim JJ. Role of cytokines, acute-phase proteins, and chemokines in the progression of rheumatoid arthritis. Semin Arthritis Rheum. 1996;26:526–538. doi: 10.1016/s0049-0172(96)80041-2. [DOI] [PubMed] [Google Scholar]
- Brennan FM, Maini RN, Feldmann M. TNF alpha – a pivotal role in rheumatoid arthritis? Br J Rheumatol. 1992;31:293–298. doi: 10.1093/rheumatology/31.5.293. [DOI] [PubMed] [Google Scholar]
- Keating GM, Perry CM. Infliximab: an updated review of its use in Crohn's disease and rheumatoid arthritis. BioDrugs. 2002;16:111–148. doi: 10.2165/00063030-200216020-00005. [DOI] [PubMed] [Google Scholar]
- O'Quinn RP, Miller JL. The effectiveness of tumor necrosis factor alpha antibody (infliximab) in treating recalcitrant psoriasis: a report of 2 cases. Arch Dermatol. 2002;138:644–648. doi: 10.1001/archderm.138.5.644. [DOI] [PubMed] [Google Scholar]
- Moreland LW, Baumgartner SW, Schiff MH, Tindall EA, Fleischmann RM, Weaver AL, Ettlinger RE, Cohen S, Koopman WJ, Mohler K, Widmer MB, Blosch CM. Treatment of rheumatoid arthritis with a recombinant human tumor necrosis factor receptor (p75)-Fc fusion protein. N Engl J Med. 1997;337:141–147. doi: 10.1056/NEJM199707173370301. [DOI] [PubMed] [Google Scholar]
- Elliott MJ, Maini RN, Feldmann M, Long-Fox A, Charles P, Katsikis P, Brennan FM, Walker J, Bijl H, Ghrayeb J, et al. Treatment of rheumatoid arthritis with chimeric monoclonal antibodies to tumor necrosis factor alpha. Arthritis Rheum. 1993;36:1681–1690. doi: 10.1002/art.1780361206. [DOI] [PubMed] [Google Scholar]
- Michie HR, Manogue KR, Spriggs DR, Revhaug A, O'Dwyer S, Dinarello CA, Cerami A, Wolff SM, Wilmore DW. Detection of circulating tumor necrosis factor after endotoxin administration. N Engl J Med. 1988;318:1481–1486. doi: 10.1056/NEJM198806093182301. [DOI] [PubMed] [Google Scholar]
- Martich GD, Danner RL, Ceska M, Suffredini AF. Detection of interleukin 8 and tumor necrosis factor in normal humans after intravenous endotoxin: the effect of antiinflammatory agents. J Exp Med. 1991;173:1021–1024. doi: 10.1084/jem.173.4.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichacker PQ, Hoffman WD, Farese A, Banks SM, Kuo GC, MacVittie TJ, Natanson C. TNF but not IL-1 in dogs causes lethal lung injury and multiple organ dysfunction similar to human sepsis. J Appl Physiol. 1991;71:1979–1989. doi: 10.1152/jappl.1991.71.5.1979. [DOI] [PubMed] [Google Scholar]
- Tracey KJ, Lowry SF, Fahey TJ, 3rd, Albert JD, Fong Y, Hesse D, Beutler B, Manogue KR, Calvano S, Wei H, et al. Cachectin/tumor necrosis factor induces lethal shock and stress hormone responses in the dog. Surg Gynecol Obstet. 1987;164:415–422. [PubMed] [Google Scholar]
- DuBois RN, McLane MW, Ryder K, Lau LF, Nathans D. A growth factor-inducible nuclear protein with a novel cysteine/histidine repetitive sequence. J Biol Chem. 1990;265:19185–19191. [PubMed] [Google Scholar]
- Varnum BC, Lim RW, Sukhatme VP, Herschman HR. Nucleotide sequence of a cDNA encoding TIS11, a message induced in Swiss 3T3 cells by the tumor promoter tetradecanoyl phorbol acetate. Oncogene. 1989;4:119–120. [PubMed] [Google Scholar]
- Varnum BC, Ma QF, Chi TH, Fletcher B, Herschman HR. The TIS11 primary response gene is a member of a gene family that encodes proteins with a highly conserved sequence containing an unusual Cys-His repeat. Mol Cell Biol. 1991;11:1754–1758. doi: 10.1128/mcb.11.3.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heximer SP, Forsdyke DR. A human putative lymphocyte G0/G1 switch gene homologous to a rodent gene encoding a zinc-binding potential transcription factor. DNA Cell Biol. 1993;12:73–88. doi: 10.1089/dna.1993.12.73. [DOI] [PubMed] [Google Scholar]
- Lai WS, Kennington EA, Blackshear PJ. Interactions of CCCH zinc finger proteins with mRNA: non-binding tristetraprolin mutants exert an inhibitory effect on degradation of AU-rich element-containing mRNAs. J Biol Chem. 2002;277:9606–9613. doi: 10.1074/jbc.M110395200. [DOI] [PubMed] [Google Scholar]
- Johnson BA, Blackwell TK. Multiple tristetraprolin sequence domains required to induce apoptosis and modulate responses to TNFalpha through distinct pathways. Oncogene. 2002;21:4237–4246. doi: 10.1038/sj.onc.1205526. [DOI] [PubMed] [Google Scholar]
- Murata T, Yoshino Y, Morita N, Kaneda N. Identification of nuclear import and export signals within the structure of the zinc finger protein TIS11. Biochem Biophys Res Commun. 2002;293:1242–1247. doi: 10.1016/S0006-291X(02)00363-7. [DOI] [PubMed] [Google Scholar]
- Phillips RS, Ramos SB, Blackshear PJ. Members of the tristetraprolin family of tandem CCCH zinc finger proteins exhibit CRM1-dependent nucleocytoplasmic shuttling. J Biol Chem. 2002;277:11606–11613. doi: 10.1074/jbc.M111457200. [DOI] [PubMed] [Google Scholar]
- Carballo E, Lai WS, Blackshear PJ. Feedback inhibition of macrophage tumor necrosis factor-alpha production by tristetraprolin. Science. 1998;281:1001–1005. doi: 10.1126/science.281.5379.1001. [DOI] [PubMed] [Google Scholar]
- Brooks SA, Connolly JE, Diegel RJ, Fava RA, Rigby WF. Analysis of the function, expression, and subcellular distribution of human tristetraprolin. Arthritis Rheum. 2002;46:1362–1370. doi: 10.1002/art.10235. [DOI] [PubMed] [Google Scholar]
- Taylor GA, Thompson MJ, Lai WS, Blackshear PJ. Mitogens stimulate the rapid nuclear to cytosolic translocation of tristetraprolin, a potential zinc-finger transcription factor. Mol Endocrinol. 1996;10:140–146. doi: 10.1210/mend.10.2.8825554. [DOI] [PubMed] [Google Scholar]
- Keffer J, Probert L, Cazlaris H, Georgopoulos S, Kaslaris E, Kioussis D, Kollias G. Transgenic mice expressing human tumour necrosis factor: a predictive genetic model of arthritis. Embo J. 1991;10:4025–4031. doi: 10.1002/j.1460-2075.1991.tb04978.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carballo E, Blackshear PJ. Roles of tumor necrosis factor-alpha receptor subtypes in the pathogenesis of the tristetraprolin-deficiency syndrome. Blood. 2001;98:2389–2395. doi: 10.1182/blood.V98.8.2389. [DOI] [PubMed] [Google Scholar]
- Kontoyiannis D, Pasparakis M, Pizarro TT, Cominelli F, Kollias G. Impaired on/off regulation of TNF biosynthesis in mice lacking TNF AU-rich elements: implications for joint and gut-associated immunopathologies. Immunity. 1999;10:387–398. doi: 10.1016/s1074-7613(00)80038-2. [DOI] [PubMed] [Google Scholar]
- Stoecklin G, Ming XF, Looser R, Moroni C. Somatic mRNA turnover mutants implicate tristetraprolin in the interleukin-3 mRNA degradation pathway. Mol Cell Biol. 2000;20:3753–3763. doi: 10.1128/MCB.20.11.3753-3763.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W, Brauchle MA, Di Padova F, Gram H, New L, Ono K, Downey JS, Han J. Gene suppression by tristetraprolin and release by the p38 pathway. Am J Physiol Lung Cell Mol Physiol. 2001;281:L499–508. doi: 10.1152/ajplung.2001.281.2.L499. [DOI] [PubMed] [Google Scholar]
- Guyre PM, Graziano RF, Vance BA, Morganelli PM, Fanger MW. Monoclonal antibodies that bind to distinct epitopes on Fc gamma RI are able to trigger receptor function. J Immunol. 1989;143:1650–1655. [PubMed] [Google Scholar]
- Suffredini AF, Fantuzzi G, Badolato R, Oppenheim JJ, O'Grady NP. New insights into the biology of the acute phase response. J Clin Immunol. 1999;19:203–214. doi: 10.1023/A:1020563913045. [DOI] [PubMed] [Google Scholar]
- Beutler B, Poltorak A. Sepsis and evolution of the innate immune response. Crit Care Med. 2001;29(suppl 7):S2–6. doi: 10.1097/00003246-200107001-00002. [DOI] [PubMed] [Google Scholar]
- Elin RJ, Wolff SM, McAdam KP, Chedid L, Audibert F, Bernard C, Oberling F. Properties of reference Escherichia coli endotoxin and its phthalylated derivative in humans. J Infect Dis. 1981;144:329–336. doi: 10.1093/infdis/144.4.329. [DOI] [PubMed] [Google Scholar]
- Jilma B, Blann A, Pernerstorfer T, Stohlawetz P, Eichler HG, Vondrovec B, Amiral J, Richter V, Wagner OF. Regulation of adhesion molecules during human endotoxemia. No acute effects of aspirin. Am J Respir Crit Care Med. 1999;159:857–863. doi: 10.1164/ajrccm.159.3.9805087. [DOI] [PubMed] [Google Scholar]
- Suffredini AF, Reda D, Banks SM, Tropea M, Agosti JM, Miller R. Effects of recombinant dimeric TNF receptor on human inflammatory responses following intravenous endotoxin administration. J Immunol. 1995;155:5038–5045. [PubMed] [Google Scholar]
- Hintz KA, Rassias AJ, Wardwell K, Moss ML, Morganelli PM, Pioli PA, Givan AL, Wallace PK, Yeager MP, Guyre PM. Endotoxin induces rapid metalloproteinase-mediated shedding followed by upregulation of the monocyte hemoglobin scavenger receptor CD163. J Leukoc Biol. 2002;72:711–717. [PubMed] [Google Scholar]
- Carballo E, Gilkeson GS, Blackshear PJ. Bone marrow transplantation reproduces the tristetraprolin-deficiency syndrome in recombination activating gene-2 (-/-) mice. Evidence that monocyte/macrophage progenitors may be responsible for TNFalpha overproduction. J Clin Invest. 1997;100:986–995. doi: 10.1172/JCI119649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maini RN, Taylor PC. Anti-cytokine therapy for rheumatoid arthritis. Annu Rev Med. 2000;51:207–229. doi: 10.1146/annurev.med.51.1.207. [DOI] [PubMed] [Google Scholar]
- Oliver JC, Bland LA, Oettinger CW, Arduino MJ, McAllister SK, Aguero SM, Favero MS. Cytokine kinetics in an in vitro whole blood model following an endotoxin challenge. Lymphokine Cytokine Res. 1993;12:115–120. [PubMed] [Google Scholar]
- Heinzelmann M, Mercer-Jones MA, Gardner SA, Wilson MA, Polk HC. Bacterial cell wall products increase monocyte HLA-DR and ICAM-1 without affecting lymphocyte CD18 expression. Cell Immunol. 1997;176:127–134. doi: 10.1006/cimm.1997.1089. [DOI] [PubMed] [Google Scholar]
- Medzhitov R, Janeway CA., Jr Innate immunity: impact on the adaptive immune response. Curr Opin Immunol. 1997;9:4–9. doi: 10.1016/S0952-7915(97)80152-5. [DOI] [PubMed] [Google Scholar]
- Shimazu R, Akashi S, Ogata H, Nagai Y, Fukudome K, Miyake K, Kimoto M. MD-2, a molecule that confers lipopolysaccharide responsiveness on Toll-like receptor 4. J Exp Med. 1999;189:1777–1782. doi: 10.1084/jem.189.11.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolff SM, Rubenstein M, Mullholland JH, Alling DW. Comparison of hematologic and febrile response to endotoxin in man. Blood. 1965;26:190–201. [PubMed] [Google Scholar]
- Tracey KJ, Cerami A. Tumor necrosis factor: a pleiotropic cytokine and therapeutic target. Annu Rev Med. 1994;45:491–503. doi: 10.1146/annurev.med.45.1.491. [DOI] [PubMed] [Google Scholar]
- Creasey AA, Stevens P, Kenney J, Allison AC, Warren K, Catlett R, Hinshaw L, Taylor FB., Jr Endotoxin and cytokine profile in plasma of baboons challenged with lethal and sublethal Escherichia coli. Circ Shock. 1991;33:84–91. [PubMed] [Google Scholar]
- Redl H, Schlag G, Bahrami S, Schade U, Ceska M, Stutz P. Plasma neutrophil activating peptide-1/interleukin-8 and neutrophil elastase in a primate bacteremia model. J Infect Dis. 1991;164:383–388. doi: 10.1093/infdis/164.2.383. [DOI] [PubMed] [Google Scholar]
- Beutler B. Regulation of cachectin biosynthesis occurs at multiple levels. Prog Clin Biol Res. 1990;349:229–240. [PubMed] [Google Scholar]
- Kovacs EJ, Radzioch D, Young HA, Varesio L. Differential inhibition of IL-1 and TNF-alpha mRNA expression by agents which block second messenger pathways in murine macrophages. J Immunol. 1988;141:3101–3105. [PubMed] [Google Scholar]
- Vassalli P. The pathophysiology of tumor necrosis factors. Annu Rev Immunol. 1992;10:411–452. doi: 10.1146/annurev.immunol.10.1.411. [DOI] [PubMed] [Google Scholar]