

Abstract
The detection of autoantibodies in human sera is an important approach to the diagnosis and management of patients with autoimmune conditions. To meet market demands, manufacturers have developed a wide variety of easy to use and cost-effective diagnostic kits that are designed to detect a variety of human serum autoantibodies. A number of studies over the past two decades have suggested that there are limitations and concerns in the use and clinical application of test results derived from commercial kits. It is important to appreciate that there is a complex chain of users and circumstances that contributes to variations in the apparent reliability of commercial kits. The goal of this review is to identify the principal links in this chain, to identify the factors that weaken the chain and to propose a plan of resolution. It is suggested that a higher level of commitment and partnership between all of the participants is required to achieve the goal of improving the quality of patient care through the use of autoantibody testing and analysis.
Keywords: autoantibodies, autoimmunity, diagnosis, diagnostic kits, quality assurance, standardization
Introduction
Over the past five decades, the detection of autoantibodies in human sera has become an increasingly important approach to the diagnosis and management of patients with a variety of autoimmune conditions. The need for supplies and reagents to perform these assays has become a major entry point for manufacturers and commercial vendors to develop and market a wide variety of diagnostic kits. Although some diagnostic laboratories still use assays that are developed in house, commercial kits have gained a significant foothold in many areas of autoimmune serology. The reasons that the use of commercial kits has become so widespread are that they are cost-effective, are easy to use and can satisfy criteria for accreditation. Kits usually provide all the necessary reagents and a cookbook approach to performance of the assay.
Commercial autoantibody assay kits employ a variety of technologies that include indirect immunofluorescence (IIF), immunodiffusion, immunoblotting, ELISA and, more recently, addressable laser beads and antigen arrays. One of the more popular technology platforms is based on the ELISA because it offers sensitivity, high throughput and relatively low cost on the background of only modest equipment needed to perform the assay. Unfortunately, little has been done to standardize these kits [1], and postmarketing surveillance and quality assurance is largely left to the manufacturers.
A number of studies have evaluated the performance characteristics of antinuclear antibody (ANA) [2-13] and antineutrophil cytoplasmic antibody (ANCA) [14] kits. Studies that compared enzyme immunoassay (EIA) kits from different manufacturers with conventional assays such as IIF and immunodiffusion concluded that there was significant discordance between conventional assays and EIAs [3,5] and significant discordance between kits from different manufacturers [4]. In one study EIAs were found to be more sensitive than immunodiffusion [2], and another study that used a cross-section of serum referred to a rheumatology laboratory found moderate to good agreement between ANA-IIF and anti-DNA results with two commercial EIA kits [6]. Analysis of the design of some studies suggests that the lack of agreement between EIA and conventional assays may depend on the diagnosis and/or the selection bias of the patients under study [2-4,6].
A study by Tan and colleagues [15] focused on the EIA kits themselves, and in particular highlighted deficiencies in intrinsic properties of the kits (sensitivities and specificities). A more recent study focused on the clinical laboratories themselves and, although one might expect academic laboratories to be rather proficient in the implementation of the EIA kits, that was not the case [12]. As in other studies [1], it was suggested that quality control procedures for daily performance of tests in the clinical laboratory setting should not be ignored, and that a minimal performance target of coefficients of variation in EIA assays should be established [12].
If the conclusions of these studies can be briefly summarized, it could be said that there are significant concerns with the accuracy (sensitivity, specificity), reliability, and quality of many commercial kits. In many cases the manufacturer has been assumed the root cause for these shortcomings; however, the use of commercial kits and their appropriate application in a clinical setting involves a rather complex chain of constituencies and events. The factors that have disconnected the highly regulated and good laboratory practices of the manufacturer and the assumptions of the client clinician are numerous. The goal of the present review is to identify the principal links in this chain, to identify the factors that weaken the chain and to propose a plan of resolution. It is suggested that a higher level of commitment and partnership between all of the participants is required to achieve the goal of improving the quality of patient care through the use of autoantibody testing and analysis.
The matrix of manufacturing and application of autoantibody diagnostic kits (Fig. 1)
Figure 1.
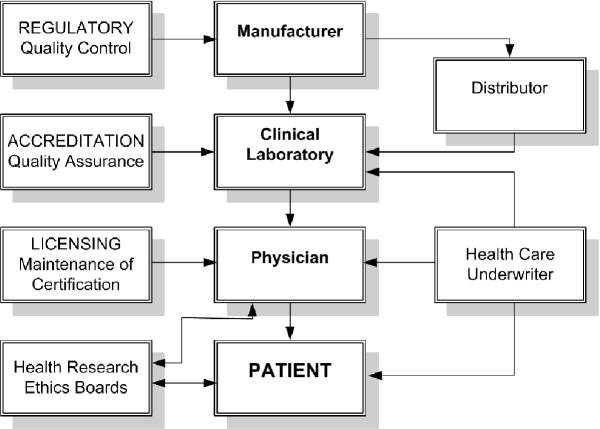
The chain of production, use and application of autoantibody test kits.
The delivery of health care in many countries has been 'streamlined' through consolidation of services, including those provided by diagnostic laboratories. This has often translated into higher workloads and increased pressure to improve the interval between when the test is requested and the time the test result is reported (turnaround time). These factors have prompted and necessitated the search for and adoption of economical, high-throughput assays. This in turn has provided fertile soil for the germination and growth of the diagnostic kit manufacturing and distributor industries. Unfortunately, the rush for a product to solve the problem of increasing throughput and decreasing costs has lead to a fast track approach in the development, the validation and the approval of commercial kits. Regulatory agencies such as the Food and Drug Administration in the USA, through the 510 K approval mechanism, have attempted to maintain a reasonable level of quality before allowing the release and marketing of diagnostic kits by manufacturers. At a minimum this has required proof of performance that is often missing when diagnostic laboratories develop and use inhouse assays. It is then left to the diagnostic laboratories to evaluate the products that are available and to make an 'informed' decision about which kit is suited to that laboratory's particular environment so that cost–performance issues are adequately addressed.
Downstream to the production, marketing, distributing, adopting and performance of commercial kits are the 'clients' or physicians that are the drivers in this economy. The availability of meaningful, accurate and dependable assays is often assumed by the client who is acting on behalf of the patient. Unfortunately, as many studies have shown, the assumption of the client cannot be validated.
Manufacturers
In the area of autoantibody diagnostic kits there are at least 15 manufacturing companies involved worldwide. Those with a significant market share are presented in Table 1. Most manufacturers operate under a high level of security, of scrutiny and of standardization, and under good laboratory practices and quality control. Many of these companies subscribe to and offer postmarketing quality assurance programs. However, the threat of lost markets becomes the driver that dictates continual changes to enhance kit performance. This jockeying and marketing strategy becomes a significant preoccupation. In this constantly changing environment, it is the responsibility of the clinical diagnostic laboratory to assure that the day-to-day and month-to-month performance of kits is adequate because accreditation, reputation and payment for service are at risk.
Table 1.
Autoantibody kit manufacturers and distributors
| Manufacturer | Location | Website |
| Axis Shield Diagnostics | Dundee, UK | http://www.axis-shield.com |
| Biomedical Diagnostics | Marne la Vallée, France | http://www.bmd-net.com |
| Cambridge Life Sciences | Cambridge, UK | http://www.cambridgelifesciences.co.uk |
| Euro-Diagnostica | Arnhem, Netherlands | http://www.eurodiagnostica.com |
| Euroimmun AG | Lubeck, Germany | http://www.euroimmun.de |
| BioRad/Helix Diagnostics | Hercules, CA, USA | http://www.helixdx.com |
| IMMCO Diagnostics | Buffalo, NY, USA | http://www.immcodiagnostics.com |
| ImmunoConcepts | Sacramento, CA, USA | http://www.immunoconcepts.com |
| IMTEC Immundiagnostika | Zepernick, Germany | http://www.imtec-berlin.de |
| INOVA Diagnostics Inc. | San Diego, CA, USA | http://www.inovadx.com |
| Medical and Biological Laboratories Co. Ltd | Nagoya, Japan | http://www.mbl.co.jp |
| ORGENTEC Diagnostika GmbH | Mainz, Germany | http://www.orgentec.com |
| Pharmacia | Peapack, NJ, USA | http://www.pharmacia.com/products/diagnostics.asp |
| Wieslab AB | Lund, Sweden | http://www.wieslab.se |
| Zeuss Scientific Inc. | Raritan, NJ, USA | http://www.zeusscientific.com |
The manufacturer is, understandably, continually scanning the horizon, looking for new technologies and new assays that will give them a novel niche and marketing advantage. Entry into these markets is rarely exclusive, so the challenge is to manufacture a test kit that can be validated and can adhere to regulatory controls (510 K approval in the USA). This then clears the pathway to market a kit that recovers research and development costs, and hopefully makes a profit. It is in the context of this activity that some problems with kit performance occur because kit validation at this level may not always live up to postmarketing performance. The validation studies often include a collection of normal and disease cohort sera obtained from a consulting clinician or a clinical laboratory that are used in an internal evaluation of kit performance and in establishing appropriate levels of positive and negative boundaries. In this setting, the validation is often performed on state of the art equipment, using the freshest reagents and kits just off the assembly line. A second level of validation involves providing the kits to laboratories that are willing and able (often without compensation) to 'beta test' the kit. If the results from the external beta tests are in agreement with internal data, the kit is submitted for approval by regulatory agencies and, having achieved that, marketing and manufacturing begins.
Another source of variation is that manufacturers tend to purchase kit components from a wide variety of suppliers and that not all kit manufacturers use the same supplier. The decision to produce or to purchase critical components such as purified antigens and secondary antibodies is based on cost and performance. When a reagent that serves both of these features satisfactorily is found, manufacturers tend to buy large lots of these reagents to minimize variability between production lots.
In summary, at the level of the manufacturer, the sources of potential variability in the performance and quality of a kit include the equipment used by the manufacturer to evaluate the kit, the reagents and materials that are purchased or manufactured to become components of the kit, the reliability of the serum samples used to validate and develop performance characteristics (cutoff points, upper, middle and lower levels), and the stability of the final product. The certification and regulatory systems under which manufacturers operate are designed to minimize variability in quality and performance of the assay. Clinical evaluation is generally performed by expert laboratories with defined patient samples that result in coefficients of variation and receiver operator analyses to determine performance characteristics.
Health care underwriter
The health care underwriter or paymaster (health insurance companies, government-based health care systems) is often regarded as the weakest link in the chain. Their admirable goal is to improve quality of care while constraining health care costs, but there are widespread concerns among health care economists that this may be unachievable. For example, as much as 80% of health care costs occur in the final 2–3 years of the patients' lives. The principles of cost-containment are often transmitted to diagnostic laboratory services that include the laboratories conducting the autoantibody testing. In response, the diagnostic laboratory tries to find a kit that offers the highest throughput on the background of lowest cost. In effect, the quality of the kit can become a secondary or even tertiary consideration. To complicate things further, when a new technology that achieves higher accuracy comes along it cannot be adopted because cost constraints are the major driver.
In fairness to health care underwriters, although the costs of laboratory diagnostic studies can easily be calculated, very little is known about the actual costs incurred through inappropriate laboratory testing. In Scandinavia, the estimated cost of all types of in vitro laboratory diagnostic testing is 2–3% of the total health care budget. In some jurisdictions, diagnostic imaging techniques and other relatively invasive procedures are being used much earlier in patient evaluations in spite of their much higher expense. Frequently, their clinical value with regard to the long-term prognosis has not been clearly elucidated.
Since early diagnosis is thought to be a key factor in outcome, and hence downstream health care costs, it is reasonable to assume that health care providers might want to ensure that funds are available for this important aspect of 'preventative medicine'. In light of the significant health care (renal dialysis, stroke and cardiovascular events, musculoskeletal rehabilitation) and consumer expense (lost wages, decreased productivity) that attends the delayed diagnosis of a systemic rheumatic disease, it is difficult to understand why low-cost and high-quality autoimmune serology is not given a higher priority by the health care system.
It is appreciated that inappropriate laboratory testing (e.g. indiscriminate panel testing) is both costly and potentially misleading in the diagnostic workup. An estimation of long-term costs related to early accurate diagnosis and therapeutic intervention compared with a missed or wrong diagnosis with or without treatment is required to reinforce the value of high-quality laboratory diagnostic. This begs another question, however: What are the costs attended by not providing the best possible autoantibody diagnostic service possible? The significant lag time in recognizing and budgeting for useful new analytes such as Saccharomyces cerivisiae, tissue transglutaminase, cyclic citrullinated peptide and chromatin are among recent examples that are of concerns to clinicians.
Clinical diagnostic laboratory
The clinical diagnostic laboratory is usually dependent on the integrated and optimal performance of a number of individuals that perform different tasks, ranging from the laboratory manager, to the clinical laboratory specialist (usually a clinical laboratory immunologist with a PhD or MD degree and/or certification qualifications), to the technologist and support staff that handle the specimens and perform the assay. As noted earlier, the decision to adopt a particular kit is often based on fiscal matters and is left to the discretion of the manager in consultation with the laboratory specialist.
In a biomedical world where knowledge and highly specialized technologies are rapidly expanding and specialized products are being produced, the clinical laboratory specialist is expected to be an expert. It does not take much introspection to appreciate that, if you were the patient or physician, you would much prefer the diagnostic results from a laboratory that had the highest level of expertise in your particular disease and the technologies relevant to that disease. Laboratory clinicians are doing their best to maintain and advance their level of competence, while carrying out burdensome and necessary quality assurance and accreditation standards. Meanwhile the industry is advancing quickly to adapt, to modify, and to advance diagnostic technologies as well as offer new diagnostic kits to make the laboratory clinician's life more manageable.
The characteristics of diagnostic laboratories that perform well are not clearly understood. Even though manufacturers provide clear directions on how to optimally use their kit and perform a certain assay, some laboratories adapt kits to their own protocols [12]. Some studies have shown that when the laboratories are a part of, or affiliated with, academic institutions they might perform better, but this is not always the case [12]. It is clear that laboratories that perform well engage in a process of internal validation of new kits before they are adopted. However, clinically defined serum samples are a rare commodity and make the evaluation process that much more difficult. As noted earlier, new assays are frequently marketed before it has been demonstrated that their diagnostic specificity has not suffered due to attempts to increase sensitivity. Even in ideal manufacturing circumstances after internal evaluations and external beta testing, the capability of the assay to accurately predict a specific diagnosis is not fully known. Therefore, it is incumbent on the clinical diagnostic laboratory to evaluate each new kit with test sera from local patients with inflammatory rheumatic diseases. This process needs to be attended by close collaboration of diagnostic laboratories, by experienced clinicians who strive for an accurate diagnosis and by patients that willingly donate their blood for testing and research purposes.
Screening sera for ANA by ELISA plates that have adsorbed complex mixtures of native and/or recombinant autoantigens or nuclear extracts is used by many laboratories instead of screening ANA by IIF on HEp-2 or other substrates. A considerable body of data shows that a significant proportion of sera from Sjögren's syndrome, from scleroderma and from polymyositis/dermatomyositis score negative using composite ANA-ELISA techniques [16,17]. Further examination of these false-negative sera by IIF screening reveals that they contain antibodies to nucleolar components, nuclear matrix, proliferating cell nuclear antigen, nuclear envelope and nuclear pore complexes, coiled bodies, promyelocytic leukemia domains, SS-A/Ro 52, centromeres and other mitotic spindle apparatus antigens, as well as a variety of cytoplasmic organelles and structures (e.g. mitochondria, Golgi apparatus, endosomes, a signal recognition particle or ribosomes) [18].
Many of these autoantibodies are readily recognized by IIF techniques when experienced technicians read the slides [16], and there is a growing body of evidence showing that these autoantibodies are useful in clinical diagnostics [19]. A recent study that found false negative results when systemic rheumatic disease sera were screened by an ANA-ELISA suggested that this test should still be used as a screening test and that negative sera could then be tested by the IIF technique [17]. A problem with this approach is that sera from patients with systemic rheumatic diseases commonly have multiple autoantibodies, including antibodies to antigens not found in the ANA-ELISA. Since many of these autoantibodies have clinical and diagnostic relevance, this approach would provide ANA results that could be misleading and the approach would lack uniformity with respect to all sera tested. Furthermore, the need to perform a second test on negative sera has the potential to add significant costs to laboratory operations. Until further improvements in the ANA-screening ELISA can be achieved, IIF-ANA should be considered the gold standard [4,6,16,20]. A problem with both IIF and the ELISA, and other assays, is represented by sera that give borderline or weakly positive results, making the clinical utility of the test of questionable value.
Once a kit is selected and internally validated, presumably through a combination of effective marketing and internal validation of performance of a sample kit, the diagnostic laboratory embarks on an adventure that is fraught with even more potential problems. The first is the equipment used to perform the test. This includes the entire spectrum of equipment, but of pivotal importance are the microscopes used for IIF and the spectrophotometers and plate readers used for ELISA. These instruments vary in performance, not only with respect to intralaboratory configurations, but also with respect to interlaboratory configurations. A key element related to microscopy is the use of a transmitted UV light source rather than incident (epifluorescence) light. In addition, the quality of microscope objective lenses (numerical aperture) varies from manufacturer to manufacturer, and the choice of objectives is often dictated by cost. Some laboratories use oil immersion objectives while others insist that dry objectives are adequate. Some laboratories use their own mounting medium and cover slips even when these are supplied as components of kits.
ELISA and dot blot kits are likewise victimized by equipment that bears little resemblance to the equipment used by manufacturers for production, and they have varying capacity and photonics. In addition, equipment is becoming more sophisticated, which generally results in assays becoming more sensitive. As that occurs, the diagnostic specificity decreases and cutoff points must be adjusted. For years it was thought that the cutoff point and appropriate screening dilution for ANA on HEp-2 substrates was 1/40 or 1/80. However, after a multicenter study showed that 32% of normal sera were positive at 1/40, it was recommended that a cutoff point of 1/160 is more appropriate [21].
Variations in equipment are compounded when the test requires a subjective assessment by the technologist or laboratory immunologist, leading to a wide range of results. A typical example of this variation is the discrepant results that may occur when a technologist performs and interprets the results one day and a different technologist in the same laboratory performs and interprets the results on weekends. It might be concluded that this results in a complete breakdown of interlaboratory and intertest performance. Thankfully, that is not always the case because, despite these variables, performance characteristics might be considered remarkably high. This is in large part due to quality assurance and accreditation programs (Table 2).
Table 2.
Recommendations for problem areas in antinuclear antibody kit use and application
| Principal | Problem | Recommendations |
| Manufacturer | Variable reagents and analytes | Standardized or common sources of reagents and analytes |
| Variable secondary antibodies | Standardized or common sources of secondary antibodies | |
| Premarketing, beta testing and selling kits | Wider testing in clinical laboratories before marketing and selling | |
| Quality control and postmarketing surveillance | Willingness and ability to adjust and to improve kit performance | |
| Clinical laboratory | Equipment | Upgrading and use of equipment to standards |
| Protocols and standard operating procedures | Follow manufacturer's protocols | |
| Utilize international reference sera | ||
| Utilize local sera from clinically defined and normal controls to assess performance of kit before adopting into practice | ||
| Training and maintenance of competence | Technologist required to demonstrate competence and to participate in appropriate educational forums | |
| Test requisitions and reports | Should be clear and concise | |
| Provide clear algorithm of tests provided and approach to use (Fig. 2) | ||
| Design and adopt a universal format | ||
| Report graded positive results when appropriate or give clinically meaningful cutoff | ||
| Physician liaison | Staff with specific skills serve as primary physician liaison to provide advice and to assist with test interpretation | |
| Physician | Ordering tests | Aware of clinical laboratory capabilities |
| Utilize graded approach to ordering tests (Figure 2) | ||
| Receiving tests | In cooperation with ethics boards, move to digital or electronic receipt of test results while maintaining patient confidentiality | |
| Interpreting result | If result not understood contact physician liaison at laboratory | |
| Communication of results | Timely action after report received because diseases can progress rapidly | |
| Patient advocate and educator | If appropriate, enquire about patient's willingness to participate in research | |
| Seek informed consent | ||
| Patient | Accurate historian | Possess and protect record of current medical history and record important symptoms |
| Participation | Consider participation in research studies | |
| Review and provide signed informed consent | ||
| Allow serum to be used for research | ||
| Regulatory bodies | Quality assurance | Attention to quality of samples provided for ongoing quality assurance programs |
| Utilize international standards and reference sera | ||
| Health care underwriters | Budgeting | Ensure that budget keeps pace with appropriate advances in serology |
| Support the use of tests that have been proven to have an impact on patient care and outcome | ||
| Alignment of services | Ensure that laboratory services are optimized | |
| Consider impacts of laboratory consolidation on quality of care |
One of the key problems in the interpretation of the ANA result is the content and design of the test report. The information contained in a report varies tremendously from laboratory to laboratory. Even when the same kits are used to perform a test, different laboratories generate very different reports. Clinicians must be able to understand the reported results without having to pore over the report and 'read between the lines'. If the report is not easily understood, it can be erroneously interpreted or the clinician may take unnecessary time to contact the laboratory to request an interpretation or may file the report as useless information. The results must be clearly expressed and must be an indication of whether they represent a high, moderate, low, borderline or negative result.
In most cases, there is little clinical value in reporting numerical results such as the optical density values for ELISA. Clinicians usually want to know whether the result is positive, whether it is highly abnormal or borderline, and what a general interpretation of the result might be. Some laboratories also provide current literature references to support the result so that clinicians can inform themselves further if they desire. When an autoantibody is found, the positive result is communicated to the clinician as a printed report or as a secured digital report sent directly to the doctor. Other general information to aid in the interpretation, such as the sensitivity and specificity of a positive result, should be tabulated in a printed or an Internet-based guide or handbook.
Physician
The test has been ordered and the result is reported. Now comes the next critical event in the chain: interpretation of the result. Many factors contribute to how a test is interpreted. First and foremost, the result must be interpreted in the context of the patient's symptoms and/or clinical findings. An autoantibody test on its own rarely establishes a diagnosis because systemic rheumatic diseases involve multiple organ systems and, particularly early in the disease course, there is rarely a pathogonomic feature. Multiple criteria have therefore been developed and must be fulfilled to confirm a particular diagnosis. Importantly, each disease is associated with different autoantibody profiles and specificities [22-27], and there are ongoing efforts to develop improved criteria for particular groups of patients. The goal is to increase the likelihood that a diagnosis or tentative classification of the disease is correct. The more knowledgeable the clinician is with regards to the clinical and laboratory characteristics of diseases, the greater the chance that a diagnosis is correct. To achieve this physicians are encouraged, and in some countries are required, to maintain a high level of competence through continuing education.
A survey of physicians showed that advances in diagnostic testing were regarded as among the most significant medical innovations in recent years [28]. With the proliferation of diagnostic tests, however, clinical interpretations and decisions have become more complex. The physician finds it imperative to decide on the best test for a patient and, at the same time, to consider the best sequence of tests that follow. The need for a practical, evidence-based and reasoned approach for diagnostic testing has never been greater. Hence, clinical practice guidelines (CPGs) are needed for a rational, judicious and economic use of serologic testing [29-33].
A large number of variables must be considered before CPGs are developed. First, CPGs should take into full account evidence-based research. Second, CPGs should be based on consensus of experts in the field. The conclusions in some studies are dependent on the quality of the data, and it is necessary to develop inclusion and exclusion criteria to evaluate a large body of data [29]. The value and application of conclusions based on a grouped literature review in the setting of rapidly changing technology is open to debate. There is no point in developing CPGs when high-performance laboratories are grouped with poor performance laboratories. Finally, the application of CPGs is difficult to apply in all settings. In small laboratories and clinical service environments it is easier to achieve consensus on testing strategies than it is for large laboratories that provide service to more extensive populations.
A CPG may include a guide to clinicians suggesting which screening tests should be used for a particular patient with a tentative diagnosis (Fig. 2 and Table 2) and those tests that are useful to monitor disease activity or progression. This guide also provides a flow diagram of tests that are necessary to support a diagnosis and estimate prognosis. A positive screening test may lead to the referral of a patient to a specialist, who will then initiate further serological testing. It is important to realize that the pretest probability of detecting a useful diagnostic laboratory result increases with each clinical feature that has been incorporated into the tentative diagnosis [34].
Figure 2.
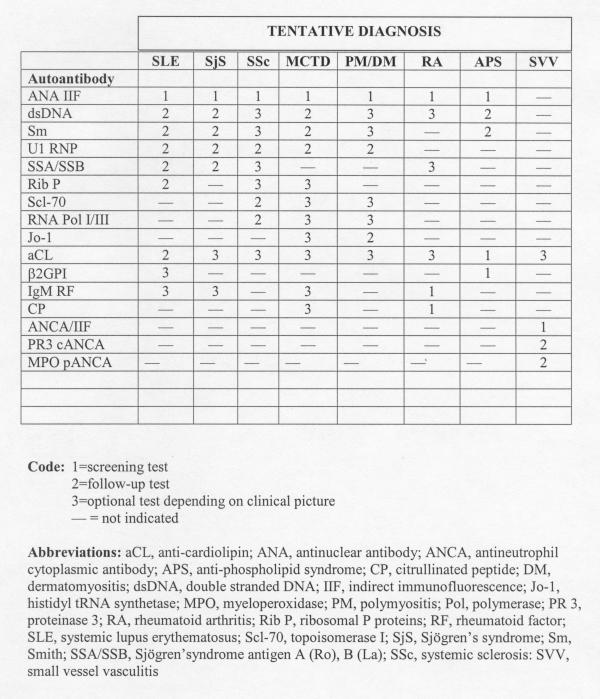
A clinician's guide to autoantibody test requests.
Autoantibodies that occur in one disease, and thus are regarded as disease-specific markers, tend to be rare [19]. Autoantibodies that were previously assumed to relate specifically to one disease are now found to be associated with a variety of autoimmune diseases [35]. Data related to certain autoantibodies and their disease specificity relied on old technology and on studies published several decades ago that may be outdated and may need revision. In addition, the presence of multiple disease-related autoantibodies occurring in a single serum (antibody profile) may be more indicative that a certain diagnosis is correct than the presence of a single antibody [19,36]. Another controversy arises when discrepant results are obtained by different anti-double-stranded DNA assays, or when a positive anti-double-stranded DNA result is reported for a serum that is negative on the HEp-2 substrate [37-39]. Accordingly, the strengths and shortcomings of new tests must be monitored and there must be agreement on how to interpret discrepant results before the test is introduced for clinical use. Positive or negative results that are out of context with known clinical parameters suggest that a test system may be unreliable and requires re-evaluation. These issues all emphasize the importance of developing CPGs that are current and in tune with conventional autoantibody testing.
Finally, physicians should be aware of the performance issues raised in previous studies [12]. They should be involved in these issues by asking their laboratory director how that laboratory assesses its own performance and the performance of the commercially available testing systems.
Patient
The ongoing demands of quality control and quality assurance are highly dependent on the availability of prototype sera and on the participation of patients who are willing to donate blood. The serum from patients with 'monospecific' and 'polyspecific' autoantibody reactivity is critical in research and development of new diagnostics products. There appears to be increasing reluctance of patients to participate because of concerns about confidentiality that have been complicated by heightened awareness of misadventures reported in the press. Equally important is the long-term participation of patients in clinical studies so that the accurate and reliable correlations of diagnostic markers to clinical outcome can be studied and validated. The difficulties in obtaining and exchanging human sera have been increased in many jurisdictions because of reluctant human ethics research boards.
Institutional ethics review boards
In the past decade, ethics review boards have become a pivotal part in the participation of patients in efforts to maintain quality control, quality assurance, the generation of clinical practice guidelines and the development of better diagnostic kits. The ability to obtain and exchange patient sera between manufacturers and laboratories that are involved in ANA kit development and monitoring has become increasingly difficult. In some cases, this has come to a complete halt because of increasing paranoia and intrusion of ethics review boards. There is a critical need to achieve a much higher level of understanding between the parties concerned so that a more balanced approach can be taken. The key issues of patient confidentiality and protection of identity are often used as a reason that exchange of sera or participation in protocols cannot be approved. There is no reason for this, since the identity of the individual is not required for these studies. The next critical issue appears to be the compiling and communication of clinical information (age, gender, race, social and family history, clinical and laboratory parameters). This should not constitute a break in confidentiality if the anonymity of the patient is guaranteed.
How can patient anonymity be guaranteed? In the past, many laboratories engaged in the development of diagnostic kits have acquired sera through a number of channels: physicians, service providers, exchange from other laboratories. The concern that samples include information that could directly identify a patient is easily resolved when the primary source of the sera 'strips', codes and repackages the serum sample that is then stored or transmitted for these studies. In the setting of most modern clinical databases, key information such as age, gender, race, diagnosis or international classification of disease codes can readily be extracted without breach of confidentiality. In many centers this may require reorganization of sample collection, but given the constraints implied in protection of patient confidentiality there are few options. Finally, in contemporary clinical environments, it is highly recommended that each patient be asked to sign a consent form that will permit their serum and plasma to be used for research and development.
Regulatory and quality assurance agencies
Both the industry and diagnostic laboratories have access to and utilize a set of standardized sera provided by the International Union of Immunology Societies/Arthritis Foundation/World Health Organization/Center for Diseases Control Serology Committee and made available through the Center for Diseases Control in Atlanta [40]. The reference sera available through this program are continuously monitored and are currently being expanded to include antibodies to cardiolipin, fibrillarin, RNA polymerase I/III, ribosomal P proteins, and c-ANCA and p-ANCA. At this time standardized secondary antibodies are not widely available. Also, sera used as standards for a particular methodology need re-evaluation from time to time, as exemplified by the re-evaluation of Arthritis Foundation/Center for Diseases Control reference sera for immunoblotting purposes [41].
Many laboratories are required to participate in a number of improvement and quality assurance programs such as that administered by the College of American Pathologists (http://www.cap.org). The Clinical Laboratory Improvement Amendments of 1988 set standards for all laboratories engaged in clinical testing. These standards include requirements for trained and competent supervisory and testing personnel, for record keeping and instrument maintenance, for daily quality control practices, for result reporting, and for laboratory inspection and maintenance. It is not clear that these standards are being met in routine practice.
An interesting model of mandatory quality assurance and proficiency testing was initiated in France in 1998. This program is administered and implemented by the French Health Products Safety Agency. Serum samples that have achieved consensus testing results in up to nine reference laboratories are distributed annually. The quality assessment division of the Agency monitors all clinical laboratories through a computer database, so that the reported results can be related to the tests performed by each laboratory. After the distribution of a serum sample, the results are collated and a statistical evaluation is undertaken, culminating in an individual report. This permits each participant to verify the accuracy and precision of their results and to compare them with reference values.
Conclusions and summary
The principal goal of a clinician is to obtain an early and accurate diagnosis in patients that can present with a wide variety of symptoms and signs during development, progression or regression of systemic rheumatic and other autoimmune conditions. Over the past two decades, commercial autoantibody assay kits have had a tremendous impact on achieving this goal. However, there are still significant challenges in the manufacture, the marketing, and the clinical application of commercial autoantibody assay kits. Table 2 summarizes some of the principal issues that are discussed in the present review and that are believed to limit the effective use of commercial autoantibody detection kits. Certain actions are proposed so that there can be resolution of some of the current shortcomings and challenges. To achieve a higher level of effectiveness will require a commitment and coordination of all organizations and individuals in the network (Fig. 1).
Competing interests
MF is a consultant to ImmunoConcepts Inc., Sacramento, CA
Abbreviations
ANA = antinuclear antibody; ANCA = antineutrophil cytoplasmic antibody; CPG = clinical practice guideline; EIA = enzyme immunoassay; ELISA = enzyme-linked immunoassay; IIF = indirect immunofluorescence.
References
- Feltkamp TEW. Antinuclear antibody determination in a routine laboratory. Ann Rheum Dis. 1996;55:723–727. doi: 10.1136/ard.55.10.723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaskowski TD, Schroder C, Martins TB, Mouritsen CL, Hill HR. Comparison of three commercially available enzyme immunoassays for the screening of autoantibodies to extractable nuclear antigens. J Clin Lab Anal. 1995;9:166–172. doi: 10.1002/jcla.1860090304. [DOI] [PubMed] [Google Scholar]
- Jaskowski TD, Schroder C, Martins TB, Mouritsen CL, Litwin CM, Hill HR. Screening for antinuclear antibodies by enzyme immunoassay. Am J Clin Pathol. 1996;105:468–473. doi: 10.1093/ajcp/105.4.468. [DOI] [PubMed] [Google Scholar]
- Emlen W, O'Neill L. Clinical significance of antinuclear antibodies: comparison of detection with immunofluorescence and ELISA. Arthritis Rheum. 1997;40:1612–1618. doi: 10.1002/art.1780400910. [DOI] [PubMed] [Google Scholar]
- Fawcett PT, Rose CD, Gibney KM, Emerich MJ, Athreya BH, Doughty RA. Use of ELISA to measure antinuclear antibodies in children with juvenile rheumatoid arthritis. J Rheumatol. 1999;26:1822–1826. [PubMed] [Google Scholar]
- Ulvestad E, Kansetrom A, Madland TM, Thomasse E, Haga HJ, Vollset SE. Evaluation of diagnostic tests for antinuclear antibodies in rheumatological practice. Scand J Immunol. 2000;52:309–315. doi: 10.1046/j.1365-3083.2000.00781.x. [DOI] [PubMed] [Google Scholar]
- Bossuyt X. Evaluation of two automated enzyme immunoassays for detection of antinuclear antibodies. Clin Chem Lab Med. 2000;38:1033–1037. doi: 10.1515/CCLM.2000.153. [DOI] [PubMed] [Google Scholar]
- Jansen EM, Deng J, Beutner EH, Kumar V, Chorzelski TP. Comparison of commercial kits for the detection of anti-nDNA antibodies using Crithidia lucilae. Am J Clin Pathol. 1987;87:461–469. doi: 10.1093/ajcp/87.4.461. [DOI] [PubMed] [Google Scholar]
- Avina-Zubieta JA, Galindo-Rodriguez G, Kwan-Yeung L, Davis P, Russell AS. Clinical evaluation of various selected ELISA kits for the detection of anti-DNA antibodies. Lupus. 1995;5:370–374. doi: 10.1177/096120339500400507. [DOI] [PubMed] [Google Scholar]
- Cordiali FP, D'Agosto G, Ameglio F, Valesini G, Alessandri C, Farsi A, Domenighetti MP, Passaleva A, Scamardella F, Neri R, Bombardieri S, Quinzanini M, Franceschini F, Chiarotti F. Determination of antibodies to extractable nuclear antigens by commercial kits: a multicenter study. Int J Clin Lab Res. 1998;28:29–33. doi: 10.1007/s005990050014. [DOI] [PubMed] [Google Scholar]
- Kern P, Kron M, Hiesche K. Measurement of antinuclear antibodies: assessment of different test systems. Clin Diagn Lab Immunol. 2000;7:72–78. doi: 10.1128/cdli.7.1.72-78.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritzler MJ, Wiik A, Tan EM, Smolen JS, McDougal JS, Chan EKL, Gordon TP, Hardin JA, Kalden JR, Lahita RG, Maini RN, Reeves WH, Rothfield NF, Takasaki Y, Wilson M, Byrd M, Slivka L, Koziol JA. A critical evaluation of enzyme immunoassay kits for detection of antinuclear antibodies of defined specificities. III. Comparative performance characteristics of academic and manufacturers' laboratories. J Rheumatol. 2003. [PubMed]
- Bizzaro N, Tozzoli R, Tonutti E, Piazza A, Manoni F, Ghirardello A, Bassetti D, Villalta D, Pradella M, Rizzotti P. Variability between methods to determine ANA, anti-dsDNA and anti-ENA autoantibodies: a collaborative study with the biomedical industry. J Immunol Methods. 1998;219:99–107. doi: 10.1016/s0022-1759(98)00140-9. [DOI] [PubMed] [Google Scholar]
- Csernok E, Ahlquist D, Ullrich S, Gross WL. A critical evaluation of commercial immunoassays for antineutrophil cytoplasmic antibodies directed against proteinase 3 and myeloperoxidase in Wegener's granulomatosis and microscopic polyangiitis. Rheumatology (Oxford) 2002;41:1313–1317. doi: 10.1093/rheumatology/41.11.1313. [DOI] [PubMed] [Google Scholar]
- Tan EM, Smolen J, McDougal JS, Butcher BT, Conn D, Dawkins R, Fritzler MJ, Gordon T, Hardin JA, Kalden JR, Lahita RG, Maini RN, Rothfield NF, Smeenk RJ, Takasaki Y, Van Venrooij WJ, Wiik A, Wilson MR, Koziol JA. A critical evaluation of enzyme immunoassays for the detection of antinuclear antibodies of defined specificities. I. Precision, sensitivity and specificity. Arthritis Rheum. 1999;42:455–464. doi: 10.1002/1529-0131(199904)42:3<455::AID-ANR10>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Bayer PM, Bauerfeind S, Bienvenu J, Fabien N, Frei PC, Gilburd B, Heide KG, Hoier-Madsen M, Meroni PL, Monier JC, Monneret G, Panzeri P, Shoenfeld Y, Spertini F, Wiik A. Multicenter evaluation study on a new HEp2 ANA screening enzyme immune assay. J Autoimmun. 1999;13:89–93. doi: 10.1006/jaut.1999.0298. [DOI] [PubMed] [Google Scholar]
- Bayer PM, Fabian B, Hubl W. Immunofluorescence assays (IFA) and enzyme-linked immunosorbent assays (ELISA) in autoimmune disease diagnostics – technique, benefits, limitations and applications. Scand J Clin Lab Invest Suppl. 2001;235:68–76. doi: 10.1080/003655101753352077. [DOI] [PubMed] [Google Scholar]
- Wiik A. Testing for ANA and ANCA – diagnsotic value and pitfalls. In Rheumatology. London: Mosby 2003.
- von Muhlen CA, Tan EM. Autoantibodies in the diagnosis of systemic rheumatic disease. Semin Arthritis Rheum. 1995;24:323–358. doi: 10.1016/s0049-0172(95)80004-2. [DOI] [PubMed] [Google Scholar]
- Rondell JMM, Van Gelder M, Van der Leeden H, Dinkelaar RB. Different strategies in the laboratory diagnosis of autoimmune disease: immunofluorescence, enzyme-linked immunosorbent assay or both? Ann Clin Biochem. 1999;36:189–195. doi: 10.1177/000456329903600209. [DOI] [PubMed] [Google Scholar]
- Tan EM, Feltkamp TEW, Smolen JS, Butcher B, Dawkins R, Frit-zler MJ, Gordon T, Hardin JA, Kalden JR, Lahita RG, Maini RN, McDougal JS, Rothfield NF, Smeenk RJ, Takasaki Y, Wiik A, Koziol JA. Range of antinuclear antibodies in 'healthy' individuals. Arthritis Rheum. 1997;40:1601–1611. doi: 10.1002/art.1780400909. [DOI] [PubMed] [Google Scholar]
- Targoff IN. Laboratory testing in the diagnosis and management of idiopathic inflammatory myopathies. Rheum Dis Clin North Am. 2002;28:859–890. doi: 10.1016/s0889-857x(02)00032-7. viii. [DOI] [PubMed] [Google Scholar]
- Ho KT, Reveille JD. The clinical relevance of autoantibodies in scleroderma. Arthritis Res Ther. 2003;5:80–93. doi: 10.1186/ar628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cervera R, Khamashta MA, Font J, Sebastiani GD, Gil A, Lavilla P, Domenech I, Aydintug AO, Jedryka-Goral A, de Ramon E, Galeazzi M, Haga HJ, Mathieu A, Houssiau F, Ingelmo M, Hughes GR. Systemic lupus erythematosus: clinical and immunologic patterns of disease expression in a cohort of 1,000 patients. Medicine. 1993;72:113–124. [PubMed] [Google Scholar]
- Wiik A. Clinical use of serological tests for antineutrophil cytoplasmic antibodies. What do the studies say? Rheum Dis Clin North Am. 2003;27:799–813. doi: 10.1016/s0889-857x(05)70236-2. [DOI] [PubMed] [Google Scholar]
- Permin H, Hørbov S, Wiik A, Knudsen JV. Antinuclear antibodies in juvenile chronic arthritis. Acta Paediatr Scand. 1978;67:181–185. doi: 10.1111/j.1651-2227.1978.tb16300.x. [DOI] [PubMed] [Google Scholar]
- Rosenberg AM. The clinical associations of antinuclear antibodies in juvenile rheumatoid arthritis. Clin Immunol Immunopathol. 1988;49:19–27. doi: 10.1016/0090-1229(88)90091-8. [DOI] [PubMed] [Google Scholar]
- Chugani DC, Rome LH, Kedersha NL. Evidence that vault ribonucleoprotein particles localize to the nuclear pore complex. J Cell Sci. 1993;106:23–29. doi: 10.1242/jcs.106.1.23. [DOI] [PubMed] [Google Scholar]
- Solomon DH, Kavanaugh AJ, Schur PH. Evidence-based guidelines for the use of immunologic tests: antinuclear antibody testing. Arthritis Rheum. 2002;47:434–444. doi: 10.1002/art.10561. [DOI] [PubMed] [Google Scholar]
- Kavanaugh AF, Solomon DH. Guidelines for immunologic laboratory testing in the rheumatic diseases: anti-DNA antibody tests. Arthritis Rheum. 2002;47:546–555. doi: 10.1002/art.10558. [DOI] [PubMed] [Google Scholar]
- Kavanaugh A. The utility of immunologic laboratory tests in patients with rheumatic diseases. Arthritis Rheum. 2001;44:2221–2223. doi: 10.1002/1529-0131(200110)44:10<2221::aid-art383>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- Smolen JS, Steiner G, Tan EM. Standards of care: the value and importance of standardization. Arthritis Rheum. 1997;40:410–412. doi: 10.1002/art.1780400303. [DOI] [PubMed] [Google Scholar]
- Tozzoli R, Bizzaro N, Tonutti E, Villalta D, Bassetti D, Manoni F, Piazza A, Pradella M, Rizzotti P. Guidelines for the laboratory use of autoantibody tests in the diagnosis and monitoring of autoimmune rheumatic diseases. Am J Clin Pathol. 2002;117:316–324. doi: 10.1309/Y5VF-C3DM-L8XV-U053. [DOI] [PubMed] [Google Scholar]
- Keren DF, Nakamura RM. Progress and controversies in autoimmune disease testing. Clin Lab Med. 1997;17:483–497. [Google Scholar]
- Van Eenennaam H, Vogelzangs JHP, Bisschops L, Te Boome LCJ, Seelig HP, Renz M, De Rooij DJ, Brouwer R, Pluk H, Pruijn GJM, Van Venrooij WJ, van den Hoogen FHJ. Autoantibodies against small nucleolar ribonucleoprotein complexes and their clinical associations. Clin Exp Immunol. 2002;130:532–540. doi: 10.1046/j.1365-2249.2002.01991.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blass S, Engel J-M, Burmester GR. The immunologic homunculus in rheumatoid arthritis. Arthritis Rheum. 1999;42:2499–2506. doi: 10.1002/1529-0131(199912)42:12<2499::AID-ANR1>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- Deng JS, Rubin RL, Lipscomb MF, Sontheimer RD, Gilliam JN. Reappraisal of the specificity of the Crithidia luciliae assay for nDNA antibodies: evidence for histone antibody kinetoplast binding. Am J Clin Pathol. 1984;82:448–452. doi: 10.1093/ajcp/82.4.448. [DOI] [PubMed] [Google Scholar]
- Kumar V, Krasny S, Beutner EH. Specificity of the Crithidia luciliae method for detecting anti-DNA antibodies. Effect of absorption for lipoproteins. Immunol Invest. 1985;14:199–210. doi: 10.3109/08820138509076144. [DOI] [PubMed] [Google Scholar]
- Vogel JC, Roberts JL, Lewis EJ. A non-anti-DNA antibody detected with the Crithidia luciliae anti-DNA assay. N Engl J Med. 1980;303:458–459. doi: 10.1056/NEJM198008213030813. [DOI] [PubMed] [Google Scholar]
- Smolen JS, Butcher B, Fritzler MJ, Gordon T, Hardin J, Kalden JR, Lahita R, Maini RN, Reeves W, Reichlin M, Rothfield N, Takasaki Y, Van Venrooij WJ, Tan EM. Reference sera for antinuclear antibodies. Arthritis Rheum. 1997;40:413–418. doi: 10.1002/art.1780400304. [DOI] [PubMed] [Google Scholar]
- Smolen JS, Butcher B, Fritzler MJ, Gordon T, Hardin J, Kalden JR, Lahita R, Maini RN, Reeves W, Reichlin M, Rothfield N, Takasaki Y, Van Venrooij WJ, Tan EM. Reference sera for antinuclear antibodies. 2. Further definition of antibody specificities in international antinuclear antibody reference sera by immunofluorescence and Western blotting. Arthritis Rheum. 1999;40:413–418. doi: 10.1002/art.1780400304. [DOI] [PubMed] [Google Scholar]