

Abstract
Controlled damage by light energy has been a valuable tool in studies of cell function. Here, we show that the Ti:Sapphire laser in a multiphoton microscope can be used to cause localized damage within unlabeled cells or tissues at greater depths than previously possible. We show that the damage is due to a multiphoton process and made wounds as small as 1 μm in diameter 20 μm from the surface. A characteristic fluorescent scar allows monitoring of the damage and identifies the wound site in later observations. We were able to lesion a single axon within a bundle of nerves, locally interrupt organelle transport within one axon, cut dendrites in a zebrafish embryo, ablate a mitotic pole in a sea urchin egg, and wound the plasma membrane and nuclear envelope in starfish oocytes. The starfish nucleus collapsed ∼1 h after wounding, indicating that loss of compartmentation barrier makes the structure unstable; surprisingly, the oocyte still completed meiotic divisions when exposed to maturation hormone, indicating that the compartmentalization and translocation of cdk1 and its regulators is not required for this process. Multiphoton excitation provides a new means for producing controlled damage deep within tissues or living organisms.
INTRODUCTION
Light energy is an effective means for producing a localized cellular disruption (Berns et al., 1998). It is possible to photodynamically destroy individual structures or cells labeled with fluorescent dyes (e.g., Tamm, 1978), or specific proteins that have been targeted with fluorescent antibodies (CALI; Buchstaller and Jay, 2000). With the appropriate fluorescent label, the plasma membrane can be disrupted using a laser in a confocal microscope (Bi et al., 1995), and it has been shown that neurons loaded with specific dyes are susceptible to ablation (Liu and Fetcho, 1999). It is also possible to damage unlabeled structures. UV light from a mercury arc lamp has been used to cut microtubules (Walker et al., 1989), whereas individual Caenorhabditis elegans cells can be killed with energy from a dye laser (Bargmann and Avery, 1995), and centrosomes in cultured cells have been ablated with pulses from an Nd-YAG laser (Khodjakov et al., 1997).
We investigated the use of multiphoton excitation as a potential new way of producing a highly localized disruption at greater depths than has been possible. Under conventional continuous illumination conditions, a single photon causes the transition of an electron to a higher energy state within a molecule. A mode-locked Ti-Sapphire laser can provide high-density photon pulses such that two or more photons combine to cause transitions, and because the probability of excitation depends on the square of photon density, excitation occurs only in the focal plane rather than all along the light path (Denk et al., 1990, 1995). This small excitation volume has proven effective at reducing photodamage during live cell imaging (e.g., Jontes et al., 2000). Additional benefits of multiphoton microscopes are the longer wavelengths that provide deeper specimen penetration and the ability of commercial multiphoton microscopes to precisely vary the intensity and x-y position of this light. By taking advantage of the high degree of spatial localization, depth penetration, and the ability to precisely vary the laser's intensity and position, we have produced site-specific controlled disruption in unstained cells and tissues at greater depths than previously possible.
MATERIALS AND METHODS
Biological Preparations
Sea urchins (Lytechinus variegatus) were obtained from Duke University Marine Lab, Beaufort, NC, and starfish (Asterina miniata) were obtained from Bodega Marine Laboratory, Bodega Bay, CA. Methods for obtaining gametes and for making injection chambers are as described previously (Terasaki et al., 1997). At the two-cell stage, the chamber was brought to the multiphoton microscope for wounding. To monitor development after wounding, a Bio-Rad MRC 600 (Cambridge, MA) coupled to a Zeiss Axioskop was used, with a Zeiss 20× N.A. 0.5 lens (Carl Zeiss Inc., Thornwood, NY). Centrifugation of starfish oocytes was done as previously described (Kishimoto et al., 1977). Oocytes in the maturation hormone 1-methyladenine (1 μM) were used as soon as a majority had undergone nuclear envelope breakdown (∼20 min); they were layered on 23% 70 kDa ficoll (Sigma Chemical Co., St. Louis, MO) in sea water (50 μl oocytes on 50 μl ficoll in a 1.5-ml Eppendorf tube) and centrifuged at 14,000 × g for 15 min at 4°C.
Squid (Loligo pealeii) were obtained through the Marine Resources Center at the Marine Biological Laboratory (Woods Hole, MA). The hindmost stellar nerve or giant axon (380–450-μm diameter) and the adjoining fin nerve bundle were dissected under running seawater and maintained in artificial seawater (Galbraith et al., 1999). For wounding experiments both the squid giant axon and the fin nerve bundle were mounted between a pair of number 0 coverslips with parallel lines of silicone grease acting as spacers. The chamber was periodically perfused with artificial seawater. In experiments that fluorescently labeled the mitochondria the axon was incubated in 10 μg/ml Rhodamine 123 for 30 min before imaging.
Zebrafish embryos (Brachydanio rerio) were obtained from the zebrafish facility at the Marine Biological Laboratory. Embryos were injected at the one-cell stage with a transfection vector containing a HuC promoter driving expression of GFP (Park et al., 2000); this results in mosaic expression in embryonic neurons. At 1.5 d, embryos were embedded in 1.5% agarose at 45°C with MS 222 (0.15 mg/ml) to inhibit movement and phenylthiocarbamide (25 μg/ml) to block development of pigment cells. Embryos were held within a well cut in a culture dish with a coverslip bottom and were kept at room temperature (∼22°C). Wounded embryos were imaged using the Bio-Rad confocal microscope with a Zeiss 40× numerical aperture (NA) 0.75 water immersion objective lens.
Multiphoton Microscopy
Experiments were done with a Zeiss LSM 510 multiphoton microscope using a Coherent Mira 900 Ti:Sapphire laser pumped with a either 5 W or 10 W Verdi solid state laser (Coherent Inc., Santa Clara, CA) using software that allows for line scan or spot mode and a Meta attachment for spectrum analysis. Emission spectra were corrected for background autofluorescence. The laser was coupled to a Zeiss Axiovert 200 (Carl Zeiss Inc.) inverted microscope. A Zeiss 25× multi-immersion lens (NA 0.8) was used for most experiments. We were also able produce multiphoton damage with Bio-Rad and Leica multiphoton microscope systems.
Most experiments were performed using 800-nm light; for the Ti:Sapphire laser pumped with the 5 W Verdi laser, the maximum laser output was ∼600 mW as measured with a Coherent Fieldmaster energy level meter. After passing through the intervening optics between the laser and the back aperture plane of the objective lens this energy level was reduced to ∼100 mW. With the 25× multi-immersion lens, the energy at the specimen was ∼60 mW; to make this measurement, a pair of back to back matched objectives were used to eliminate errors associated with internal reflection (Misawa et al., 1991; Liang et al., 1996). The Ti:Sapphire laser pulse frequency was 76 MHz, so that at full laser power, each pulse was ∼0.8 nJ at the specimen; the pulse duration was ∼100 femtoseconds. We found that the best way to cause controlled damage was to use the line scan mode of the 510 LSM microscope with the laser at full power (lowest attenuation setting) and slow scan speed (72 μs per pixel, or ∼5000 pulses per pixel). In many cases a single line scan was sufficient to cause damage. A single scan of 100 pixels in length provided an exposure of 7.2 ms and 430 μJ.
It is interesting to compare the exposure settings used for wounding to those used for live cell imaging. A typical setting for obtaining images in a long-term time-lapse sequence is ∼10% laser power and ∼1-μs dwell time. To create deliberate damage, each pulse has 10× as many photons (100% laser power), and each pixel is exposed to ∼70× as many pulses (72-μs pixel dwell time), so that there is ∼700× difference in exposures between live cell imaging and creating this type of damage.
RESULTS
Current methods for light induced ablation in unlabeled specimens are limited to within a few microns of the coverslip (Bargmann and Avery, 1995; Cole et al., 1995; Khodjakov et al., 1997). We set out to make damage at greater depths using the Ti:Sapphire laser in a multiphoton microscope.
We used echinoderm eggs as a test specimen and 800 nm light from the Ti:Sapphire laser. We also used a water immersion lens because these are less subject to spherical aberration at large distances from the coverslip than oil immersion lenses (e.g., Keller, 1995). Laser power levels that would be used for long-term live cell observations caused no detectable damage. When the laser power and the pixel dwell time were increased to the maximum settings on the microscope (see MATERIALS AND METHODS), a distinct, condensed “scar” was produced that is visible by both transmitted light and fluorescence microscopy (Figure 1A). The same exposure created a scar in A6 cells (a frog epithelial cell line) and human cheek epithelial cells. In these cells, as well as all other specimens we used, the transmitted light scar and fluorescent scar appeared together; we saw no instances of a transmitted light scar without fluorescence, or a fluorescent scar without a transmitted light scar.
Figure 1.

(A) Relation between wounding and autofluorescence. The cytoplasm of an immature oocyte is more autofluroescent than the nucleus (∼70-μm diameter dark region in the image). Five scars were made in the cytoplasm by single line scans of ∼10-μm length (the light, focused to a spot, was scanned along one dimension only, and in this case, just once). Several 10-line scans (i.e., the spot was scanned along one dimension 10 times over the same 10-μm path) were made in the nucleus without any evidence for damage by fluorescence or by transmitted light imaging. Scale bar, 50 μm. (B) Emission spectrum of the fluorescent scar with 800 nm excitation. (C) Energy dependence of wounding. The total energy deposited in a line scan wound is proportional to the product of laser attenuation and pixel dwell time (i.e., pulse energy and number of pulses). The graph shows the threshold pixel dwell time for making a scar as a function of laser power. The data is fit by a 2.5 power dependence (line) indicating that the damage is due to a multiphoton process. The progressively smaller scars (clockwise starting from top) on the sea urchin egg (inset) were obtained by decreasing laser power at constant pixel dwell time.
Scars could be created with wavelengths between ∼720 nm and ∼850 nm, but it was more difficult to produce damage with longer wavelengths. This wavelength dependence is similar to that of multiphoton autofluorescence, which also decreases at longer wavelengths. In starfish oocytes, cytoplasm is more autofluorescent at 800 nm than the nuclear interior and also was easier to damage (Figure 1A); similarly, in the squid fin nerve, variations in autofluorescence corresponded with the ease of making wounds. However, autofluorescence itself is not sufficient, because the autofluorescent oil cap of centrifuged oocytes (see Figure 5) was not easier to damage. Also, oocytes injected with fluorescent dextran were not easier to damage than uninjected oocytes, so that the damage is not simply due to fluorescence excitation.
Figure 5.
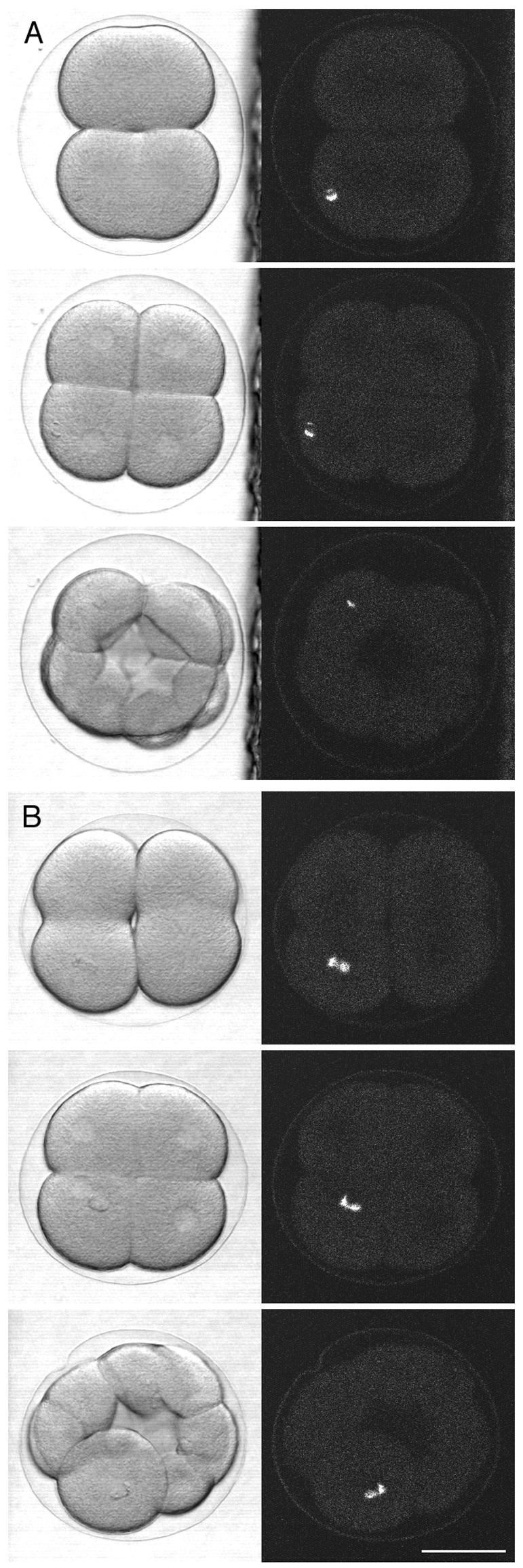
Damage of a mitotic pole. Multiphoton damage was made in two-cell stage sea urchin embryos; subsequent development was observed using a conventional confocal microscope. (A) Control embryo, in which the damage was deliberately made away from the mitotic pole. Scanning transmission images (left column) were obtained simultaneously with fluorescence images (right column; 488-nm excitation) that show the fluorescent scar. The first image pair was taken ∼3 min after the damage was made, with the timing of the later images shown on the figure. The daughter cell that inherited the damage divided at the same time as the undamaged daughter cell. (B) Damage was made at the center of the mitotic aster as located by bright field microscopy. The first image was taken ∼3 min afterward; this was taken at lower magnification so the mitotic aster is not easily visible. The daughter cell that inherited the damage did not divide, whereas the undamaged daughter cell divided. This indicates that sea urchin blastomeres cannot continue normal development without a centrosome. Scale bar, 50 μm.
The bright fluorescence from the scar is visible by multiphoton excitation or by conventional fluorescence with 488- or 568-nm excitation. The emission spectrum (Figure 1B) has a broad peak between 475 and 525 nm. The fluorescence develops immediately and can be used to monitor the degree of damage during the scanning process. The scar persisted in the specimens that we tried and was useful for identifying the damage site during long-term experiments.
The Ti:Sapphire laser can provide photons either continuously (continuous wave mode) or as brief, intense, repetitive pulses (mode lock). For these two cases, the same amount of photons (or equivalently, energy) is delivered when averaged over time. The normal operating condition is mode lock, because the pulses provide the high intensities required for multiphoton excitation of fluorescence. Likewise, damage occurred with the laser in mode lock, but not in continuous wave mode, where the same total amount of energy is delivered to the specimen at a steady, low rate.
We examined the dependence of wounding on pulse energy. Within the multiphoton microscope, an acousto-optic device attenuates pulse energy, whereas the number of pulses delivered to the specimen is controlled by the scan speed of the galvanometer mirror through the pixel dwell time setting. For each pixel dwell time setting, we determined the minimum energy required to create a fluorescent scar (Figure 1C). The data was fit by E ∝ (1/T)2.5 where E = energy, T = pixel dwell time. This means, for instance, that pulses of twice the energy require ∼1/6, instead of 1/2, the number of pulses to cause the same damage. The nonlinear dependence indicates that the damage is due to a multiphoton process.
We investigated the size of the damage caused by multiphoton wounding. For high exposures to light energy, there must be some spreading of damage from the region that is exposed to light; this is evident from Figure 1C, where all of the wounds were created by a line scan that delivers light to a thin strip of cytoplasm. In wounds larger than ∼25 μm, the shape of the wound was often very irregular, and a small air bubble often appeared at the damage site, possibly because of plasma formation from decomposition of water (Noack and Vogel, 1999). We found that we could make much smaller wounds by carefully controlling the exposure levels. We used 0.8 and 1.2 NA water immersion lenses (25× and 63× magnification, respectively) to make wounds in the squid fin nerve (Figure 2, A–F). The x, y, and z full width half-maximal values for the smallest wound produced by the 25× lens were 2.69, 1.88, and 4.14 μm (middle wound; Figure 2, A–C) and for the 63× lens were 0.79, 0.61, and 1.0 μm (bottom wound; Figure 2, D–F). This shows that it is possible to create small, precise wounds by using appropriate energy levels and optical conditions.
Figure 2.

Small localized wounds in the squid fin nerve and giant axon. Different size wounds were made in a fin nerve of the squid with either a 25× 0.8 NA multi-immersion lens (A–C) or a 63× 1.2 NA (D–F) water immersion lens. Arrowheads point to the wound in the transmitted light images A and D. The corresponding fluorescent scar is shown in the x-y (C and F) and y-z (B and E) planes. The corresponding volumes for these fluorescent scars are 76, 10.9, and 376.1 μm3 (B and C) and 10.9 and 0.253 μm3 (E and F). (G) Spot wound in the squid giant axon using a 63× 1.2 NA water immersion lens. To confirm that damage was confined to the wound plane axonal transport was monitored by taking time series of Rhodamine 123 labeled mitochondria at the wound plane, 4 μm above and 4 μm below the wound plane (see online movies: above.mov and below.mov). The volume of the wound was 13 μm3. Arrowheads indicate the wound site in the out of plane images. Scale bar for all images, 10 μm.
It is possible that the region of structural damage extends beyond the fluorescent scar or that diffusible toxins can damage regions beyond the fluorescent scar. To investigate this, a wound was made in a squid axon with fluorescently stained mitochondria. In time lapse sequences, mitochondria were observed to move through regions just beyond the edges of the fluorescent scar (Figure 2G), which had x and y dimensions of 2.3 μm and a depth of 4.7 μm. This suggests that the damage is restricted to the region of the fluorescent scar.
Biological Applications: Damage of Axons
Individual axons from the fin nerve bundle of the squid >50 μm beneath the surface were damaged, demonstrating the ability of multiphoton illumination to produce a small volume wound deep within a tissue. The damage shown in Figure 3A was generated using a line scan across a single axon and a 25× 0.8 NA lens water immersion lens. These axons were typically 10 μm in diameter, and Z-series images confirmed that disruption was limited to one axon. In addition, transmitted light images showed an accumulation of organelles on the proximal side of the scar, indicating that transport was blocked at that location. This transport block was observed only in the damaged axon and not in any of the surrounding axons.
Figure 3.

Multiphoton damage in axons created by a line scan mode with a 25× 0.8 NA water immersion lens. (A) The squid fin nerve contains a bundle of ∼10-μm diameter axons. The multiphoton microscope was focused to a plane ∼50 μm from the surface of the axon bundle, and a line scan at full power created a wound within an axon; axons above and below were undamaged. The transmitted light image shows an accumulation of organelles on the proximal side of the wound after 10 min, indicating blockage of axonal transport. Scale bar, 50 μm. (B) The squid giant axon is ∼500 μm in diameter. A line scan wound was made within the axoplasm ∼25 μm from the surface of the axon. The axon had been preincubated with rhodamine 123 to label mitochondria. After 10 min, an accumulation of anterograde and retrograde moving mitochondria was seen at the scar as demonstrated by the difference in intensity profiles (bottom) of a band the full width of the scar (denoted by bar with arrows). Scale bar, 50 μm.
A more localized interruption of axonal transport was also accomplished by disrupting the axoplasm of the giant axon again with the 25× lens and a line scan ∼ 25 μm from the axon surface (Figure 3B). Fluorescently labeled mitochondria were observed by timelapse confocal microscopy to move linearly in a saltatory manner through the axoplasm before damage. However, after damage, mitochondria moving along a line that intersected the scar tended to stop and accumulate at the scar, whereas mitochondria moving along lines that did not intersect the scar were not affected (Figure 3B). Like other large scars (>25 μm) some variation is seen in the fluorescent intensity along the length, but transport was blocked at all points along the band.
Severing of a Dendrite
We cut single neuronal processes within living zebrafish embryos. A transfection vector with a neuronal promoter driving GFP expression was injected into one cell stage zebrafish embryos, resulting in mosaic expression of GFP in a small fraction of embryonic neurons. GFP-labeled Rohon-Beard cells were used as targets. These cells are mechanosensory neurons that are easily identifiable in 2-d-old embryos. The cell bodies, which are located in the dorsal spinal cord, send out dendritic processes that innervate the skin (Figure 4).
Figure 4.

Severing of a neuronal dendrite. Injection of a transfection vector into fertilized zebrafish eggs resulted in mosaic labeling of neurons by GFP in 1.5-d-old embryos. A Rohon-Beard cell dendrite was severed by a single multiphoton line scan. Z series images were taken at 30–45-min intervals by confocal microscopy, with the time after wounding indicated on the figure. The severed dendrite remained intact for several hours. At 5 h, the beginning of degeneration was evident by the beaded appearance of the dendrite; the apparent connection at another point has not occurred and instead is due to the superposition of z series images. By 7 h, the remnants of the dendrite had disappeared. Scale bar, 10 μm.
GFP-labeled Rohon-Beard cells were located by conventional confocal microscopy. The dendritic branches are much thinner (2–3 μm) than the squid axons described in the previous section, so that multiphoton line scan wounds resulted in severing rather than creation of damage within the dendrite. The presence of GFP did not seem to reduce the amount of energy required for wounding. Over the next 6 h Z-series images of the neuron were obtained by conventional confocal microscopy at ∼30–45-min intervals. The severed dendritic branch remained intact for ∼5 h and then began to vesiculate and eventually disappeared (Figure 4). Multiphoton excitation was also effective at severing axons within the spinal cord (unpublished data).
Ablation of a Mitotic Pole
We used sea urchin embryos, a classic model for mitosis. At about the time of anaphase onset, microtubules grow from the two mitotic poles away from the spindle and are easily seen in bright field microscopy as prominent star-like patterns (mitotic asters). The microtubules of the aster are involved in setting up the position of the cleavage furrow, and the cell becomes committed to divide sometime before the cell begins to constrict (Rapoport, 1981). Multiphoton damage was made to the mitotic pole at the center of the aster during the ∼3-min period between the first appearance of the asters and the beginning of constriction.
Two cell stage embryos were used. As control, a wound was made deliberately away from the mitotic pole (Figure 5A). The blastomere that inherited the fluorescent scar divided normally and in synchrony with the undamaged blastomere (2/2 embryos). In embryos where the damage was made at the center of a mitotic aster, the damaged cell completed the first division normally. However, of the four resulting daughter cells, the cell that inherited the fluorescent scar failed to divide in the next cycle (Figure 5B; 3/3 embryos). In the case of the first division, the cell was probably able to divide because the commitment to form a cleavage furrow had already occurred at the time of the wounding. The failure of the second division indicates that sea urchin blastomeres cannot continue normal development without a centrosome.
Plasma Membrane Wounding
We used the starfish oocyte for wounding of the plasma membrane and the nuclear envelope (next section). Repair of plasma membrane wounds requires fusion of intracellular organelles at the injury site (Steinhardt et al., 1994; McNeil and Steinhardt, 1997; Reddy et al., 2001). In echinoderm eggs, the primary organelle involved in repair of large wounds appears to be the yolk platelets (Terasaki et al., 1997; McNeil et al., 2000).
When echinoderm eggs are centrifuged, the yolk platelets are displaced to the centrifugal end, leaving the other half optically clear. Heilbrunn (1958) noted that surface wounds become repaired on the yolk containing end but not on the clear end; not believing in the existence of the plasma membrane, he advocated protein coagulation for repair. We confirmed Heilbrunn's observation using microneedle wounds, then used multiphoton excitation to make wounds. No cytoplasmic changes occurred when a line scan wound was made at the yolk containing end (3/3 eggs), whereas a wound at the clear end resulted in degeneration of the clear cytoplasm and the establishment of an apparent new cellular boundary between the clear and yolk containing cytoplasm (6/6 eggs) (Figure 6). Multiphoton damage within the clear zone cytoplasm did not cause any changes in the oocyte (6/6 eggs; Figure 6). The simplest interpretation of these results is that the plasma membrane of the clear end does not become sealed because of the lack of yolk platelets and that the raised intracellular Ca in the clear end causes fusion of the bordering yolk platelets to create a new boundary (McNeil et al., 2000).
Figure 6.

Wounding of the plasma membrane. Starfish eggs were stratified by centrifugation. The yolk platelets were redistributed to the centrifugal end, leaving most of the centripetal half clear with an autofluorescent “oil cap.” Heilbrunn (1958) noted that stratified eggs heal better on the yolk-containing end. We tested this by multiphoton line scanning across the cell surface. Simultaneous scanning transmission and fluorescence images were taken ∼3 min after the wounds were made. (A) A cell surface wound on the yolk side (9 line scans; arrowhead) caused no apparent change, indicating successful healing. (B) A cell surface wound on the clear end (5 line scans, arrowhead) resulted in degeneration of the clear cytoplasm and formation of a new boundary between the yolk end and the former clear end. (C) A wound made within clear cytoplasm (5 line scans) caused no change. These results provide further evidence that the yolk platelets are the primary organelle involved in repair of large plasma membrane wounds in echinoderm eggs. Scale bar, 50 μm.
Nuclear Envelope Wounding
The starfish oocyte has a 60–70-μm diameter nucleus (germinal vesicle) positioned close to the cell surface at the animal pole. The oocyte is arrested in prophase of meiosis I, which is the stage just before nuclear envelope breakdown. Meiotic maturation is initiated by the hormone 1-methyladenine, whereupon nuclear envelope breakdown occurs at ∼20–30 min, followed by polar body extrusion and formation of the female pronucleus by ∼3 h.
The nuclear envelope is a double membrane boundary associated with an underlying lamina. As in other cells, the starfish nucleus can be injected, indicating that a rupture in the nuclear envelope can be sealed. However, the nuclear envelope does not reseal multiphoton excitation wounds. A line scan wound at the nuclear envelope resulted in slow leakage of cytoplasmic molecules into the nucleus; fluorescent 70 kDa dextran came to equilibrium by ∼40–50 min (Figure 7, A and B).
Figure 7.

Wounding of the nuclear envelope of starfish oocytes. (A) A multiphoton line scan was done across the nuclear envelope. This fluorescence image was obtained by multiphoton excitation at 800 nm. The nuclear interior has less autofluorescence than cytoplasm and is visible as the dark oval. The fluorescent scar is present at the site of the wound. The surface of the oocyte contains autofluorescent organelles. Scale bar, 20 μm. (B) Oocytes were injected in the cytoplasm with either rhodamine 70 kDa dextran or fluorescein 500 kDa dextran, and then the nuclear envelope was wounded by multiphoton line scan as above. The entry of fluorescence dextran in the nucleus was monitored. The fluorescence level at the start was set to 0 and was normalized to twice the starting cytoplasmic fluorescence because the nuclear space for soluble markers is about twice that in cytoplasm due to volume exclusion by yolk platelets (Terasaki, 1994). (C) Collapse of the nucleus after a line scan wound at the nuclear envelope. Images were taken every 30 s (see online move: collapse.mov). The first image, shown on the left, was taken 13 min after the wound. The image on the right was taken at 50 min, after the nucleus had collapsed. Scale bar, 20 μm. (D) Oocytes with a collapsed nucleus are still able to undergo meiotic divisions. The same oocyte shown in the previous panel was exposed to the maturation hormone 2.0 h after the nuclear envelope wound. Images were taken every 30 s beginning 2 min after application of the hormone (see online movie: division.mov). The left panel was taken at 91 min, soon after the first polar body had formed (black arrowhead; inset shows polar body at twice magnification). The small circle within the egg cytoplasm is the oil drop from the injection of 70 kDa rhodamine dextran, which helps in visualizing the nuclear envelope for wounding. The right panel was taken at 4.0 h and shows the egg pronucleus (white arrowhead). Scale bars, 20 μm.
The outline of wounded nuclei remained unchanged until ∼45–90 min when the nuclear envelope outline collapsed (Figure 6C). Small remnants of the nuclear envelope remained visible, and the collapse differed from the breakdown of the nucleus that normally occurs during meiotic maturation because polar bodies and the female pronuclei never formed. The oocyte was stable for ∼12 h then underwent blebbing and deterioration. Oocytes that were wounded in the cytoplasm instead of at the nuclear envelope survived and were as healthy as unwounded oocytes for at least 2 d.
During meiotic and mitotic cell cycles, nuclear envelope breakdown is preceded by the translocation of several cell cycle regulatory proteins between nucleus and cytoplasm (Pines, 1999; further details in DISCUSSION). By wounding the starfish nuclear envelope, the normal nucleocytoplasmic compartmentalization is disrupted so that the normal interactions and translocations that involve compartmentalization will not be able to occur. We tested whether this disruption would impair the normal meiotic cell cycle by adding maturation hormone to oocytes 2 h after the nuclear envelope had been wounded. Surprisingly, the oocytes completed the meiotic divisions on a normal time schedule as evidenced by formation of polar bodies and the female pronucleus (14/15 oocytes from three animals; Figure 7D).
DISCUSSION
We were able to use the Ti:Sapphire laser that is available on commercial multiphoton microscopes to make controlled damage in thick specimens. The wounds were produced at larger depths than with previously used laser techniques. The lack of toxicity of the wounds for the surrounding cells or cytoplasm was evidenced by continuing transport in neighboring axons within a bundle of axons or in adjacent axoplasm of a single axon, continued development in the zebrafish embryo, division of the neighboring cells in the sea urchin embryo, and healing of the starfish oocyte surface. We have thus shown that it is feasible to selectively damage axons, cut dendrites, ablate mitotic poles, and lesion the cell surface and nuclear envelope in thick specimens.
Two recent studies have investigated damage in multiphoton microscopes, primarily with the purpose of minimizing photodamage during imaging of living cells, rather than the deliberate creation of damage. Damage was assessed by nonphysiological rises in intracellular calcium in neurons and chromaffin cells or degranulation in the chromaffin cells (Koester et al., 1999; Hopt and Neher, 2001). Both studies found a 2.5 power dependence on energy for damage, indicating a multiphoton process (a one photon process is expected to produce a linear power dependence). Hopt and Neher speculated that damage involves a significant amount of three photon absorption. This seems to correspond with our observation that multiphoton damage is easiest with wavelengths < ∼850 nm, because 800/3 = 266 nm is a highly absorbing region of the spectrum for the aromatic amino acids and nucleotides, while there is less absorption at 900/3 = 300 nm. We found a similar ∼2.5 power dependence for deliberately induced multiphoton damage as assayed by fluorescent scar formation. This indicates that the wounds should be restricted to the focal plane region (Denk et al., 1995) and is consistent with our experimental observations that wounds with dimensions of 1 μm or less can be made under appropriate conditions.
As with other forms of laser induced damage, the physical/chemical mechanisms in multiphoton damage are not well understood and are likely to be complex. There may be interesting differences, because the Ti:Sapphire laser makes fluorescent scars and has no effect on glass at the power levels used for wounding, whereas the Nd-YAG and dye lasers make nonfluorescent scars and damage glass (Bargmann and Avery, 1995; Khodjakov et al., 1997).
A related issue is the spatial extent of multiphoton damage. Because high numerical aperture objective lenses can focus light to a volume comparable to the wavelength of light, it may be feasible to restrict damage to a similar volume. With a Nd-YAG laser focused to a stationary spot by an oil immersion lens (NA 1.4), it is possible to make very precise wounds that are ∼0.4 μm in diameter at a depth of a few micrometers into a tissue culture cell (Khodjakov et al., 1997). Using a water immersion lens (NA 1.2), we were able to make and detect a fluorescent scar as small as ∼1 μm at a depth of 20 μm in the squid fin nerve. It is possible that nonfluorescent structural damage extends beyond the observed fluorescent scar or that diffusible toxins may be generated. However, in squid axons, we observed that mitochondria are able to move in the region just adjacent to the fluorescent scar; this is evidence that damage is restricted to the region of the fluorescent scar.
Two features of multiphoton wounding are advantageous for the practical use of this technique. The fluorescent scar is very useful for monitoring the damage as it is occurring and also for locating the damage site later in long-term observations. It is also very convenient that the Ti:Sapphire laser is integrated within a scanning microscope. The location and intensity of the laser illumination are controlled precisely with high time resolution, resulting in much spatial and temporal flexibility in making damage. The normal imaging mode of the multiphoton microscope is also convenient for characterizing the damage or wound response afterward.
We used multiphoton wounding on four different biological systems. In the first, axons within the squid fin nerve as well as the giant axon were selectively damaged. This shows that it is possible to make wounds deep within tissues or cytoplasm without damaging surrounding regions, offering the possibility of laser microsurgery with the multiphoton microscope. In the second system, a dendritic branch of Rohon-Beard neurons within zebrafish embryos was selectively cut; because of the thinness of the dendrites, multiphoton wounding caused severing rather than damage within the dendrite. This ability to sever neuronal processes at locations that have previously been difficult to reach provides a better opportunity to investigate mechanisms of axonal degeneration, regeneration, and pathfinding.
In the third system, a mitotic pole in sea urchin embryos was damaged. In previous studies where cultured mammalian cell centrosomes were ablated with a Nd-YAG laser, the centrosomes could be ablated early in the cell cycle because they were labeled by GFP-gamma tubulin; it was found that the cell can assemble a mitotic spindle and divide, but the daughter cell that inherits a damaged centrosome becomes arrested in G1 (Khodjakov and Rieder, 2001). Multiphoton damage should provide the opportunity to pursue these findings in other systems such as embryonic cells or cells within tissues.
Starfish oocytes were the fourth system used. Previous work had indicated that yolk platelets are the primary intracellular organelle involved in repair of large wounds in echinoderm eggs (Terasaki et al., 1997; McNeil et al., 2000). We tested this by examining the ability of the plasma membrane to repair wounds in stratified oocytes, and the results were consistent this idea. Other work shows that fluorescently labeled plasma membranes can be specifically damaged by multiphoton excitation (McNeil et al., 2001), whereas our studies involve unstained membranes.
Multiphoton wounds in the starfish oocyte nuclear envelope were not healed, as shown by the slow leak of large cytoplasmic fluorescent dextrans into the nucleus. After 45–90 min, the nucleus underwent collapse, indicating that this disruption somehow makes the nuclear structure unstable. Dissipation of the nuclear Ran GTP gradient (Kalab et al., 2002) would disrupt active nucleocytoplasmic transport and might lead to destabilization of the normal nuclear structure. The collapse of nucleus was not immediately detrimental to the oocyte, which survived for ∼12 h and then degenerated.
We used multiphoton disruption of the nucleus to investigate requirements for nuclear compartmentation and translocation during meiosis. The presence of the nucleus is not always required for cell cycle–related events. For instance, in enucleated fertilized eggs, a system in which the rapid embryonic division cycle has already been initiated, centrosome duplication and surface contraction cycles continue (Hara et al., 1980; Sluder et al., 1986). The presence of the nucleus is required though, during meiotic maturation in starfish oocytes. The full level of maturation promoting factor appears in the cytoplasm only after nuclear envelope breakdown, and removal of the nucleus prevents full development of maturation promoting factor activity (reviewed in Kishimoto, 1999); these effects are not well understood.
There are also complex movements across the nucleus before breakdown occurs (e.g., Pines, 1999; Takizawa and Morgan, 2000). Nuclear envelope breakdown is preceded by the entry into the nucleus of cdc2 kinase (Pines and Hunter, 1991; Ookata et al., 1992). Depending on the species and cell type, the regulators of cdc2 are compartmentalized differently and undergo translocations between nucleus and cytoplasm. In starfish, the inhibitory myt1 and the activating phosphatase cdc25 are both cytoplasmic, with cdc25 translocating into the nucleus before nuclear envelope breakdown (Okano-Uchida et al., 1998; Kishimoto, 1999), whereas in mammalian cells, wee1 is nuclear and cdc25C is cytoplasmic and undergoes translocation to the nucleus (Heald et al., 1993). In addition, cyclin B apparently must be phosphorylated for cdc2 to enter the nucleus (Toyoshima-Morimoto et al., 2001). The reasons for the localizations and movements in the activation and function of cdc2 are not well understood.
The collapse of the starfish oocyte nucleus caused by multiphoton wounding was not followed by meiotic divisions. This shows that disruption of the nucleocytoplasmic compartmentalization by itself is not sufficient to start meiosis; in these oocytes, for instance, cdc2 and cdc25 presumably can mix with the former nuclear contents. We tested whether the loss of nucleo-cytoplasmic compartmentalization would affect the ability of the oocyte to undergo meiotic maturation. In the oocytes with a collapsed nucleus, many of the normal interactions or translocations that involve compartmentalization will not be able to occur. Surprisingly, these oocytes were still able to complete meiotic divisions when exposed to maturation hormone. This seems to show that the localization of the cdc2 and its regulatory molecules and their translocations are not required for initiation and completion of starfish meiotic maturation. It is possible that the compartmentalizations and translocations are only involved in nuclear envelope breakdown and are not required for initiating any of the following events in meiosis. Another possibility is that they are involved in monitoring whether nuclear envelope has occurred, and if inactivated, will simply allow meiotic divisions to occur.
In conclusion, we have used multiphoton excitation to make controlled damage in several biological systems and have shown that it is possible to make precise wounds within thick specimens. This significantly expands the capabilities for controlled damage within cells and tissues. We foresee that controlled damage by multiphoton excitation will become a useful tool for experimental cell biology.
Supplementary Material
Acknowledgments
Much of this work was done during the MBL Neurobiology course in 2000–2002 and was begun as a project with Jean Livet, a student in the 2000 course. We also thank other members of the course, especially Winfried Denk and Jeff Lichtman for discussions and advice as well as Tom Reese for continuing support. We also thank Zeiss, Bio-Rad, and Leica for providing the multiphoton microscopes to the Neurobiology Course that were used in these studies as well as Louis Kerr and the Central Microscope Facility at the MBL and the Center for Biomedical Imaging Technology (University of Connecticut Health Center), where some experiments were performed. We thank Brian Link of the Grass Fellows for providing zebrafish expressing GFP neurons and Mary Dickinson for help in obtaining the emission spectrum. We also thank Paul Campagnola, Alexei Khodjakov, Cathy Galbraith, Andrew Millard, Tom Reese, Carolyn Smith, Laurinda Jaffe, Volodya Rodionov, Paul McNeil, and John Jordan for comments and/or reading the manuscript. This work was partially supported by R01-GM60389 and a grant from the Human Frontiers Foundation to M.T.
Article published online ahead of print. Mol. Biol. Cell 10.1091/mbc.E02-03-0163. Article and publication date are at www.molbiolcell.org/cgi/doi/10.1091/mbc.E02-03-0163.
Online version of this article contains video materials. Online version is available at www.molbiolcell.org.
References
- Bargmann, C.I., and Avery, L. (1995). Laser killing of cells in Caenorhabditis elegans. Methods Cell Biol. 48, 225–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berns, M.W., Tadir, Y., Liang, H., and Tromberg, B. (1998). Laser scissors and tweezers. Methods Cell Biol. 55, 71–98. [DOI] [PubMed] [Google Scholar]
- Bi, G.Q., Alderton, J.M., and Steinhardt, R.A. (1995). Calcium-regulated exocytosis is required for cell membrane resealing. J. Cell Biol. 131, 1747–1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchstaller, A., and Jay, D.G. (2000). Micro-scale chromophore-assisted laser inactivation of nerve growth cone proteins. Microsc. Res. Tech. 48, 97–106. [DOI] [PubMed] [Google Scholar]
- Cole, R.W., Khodjakov, A., Wright, W.H., and Rieder, C.L. (1995). A differential interference contrast-based light microscopic system for laser microsurgery and optical trapping of selected chromosomes during mitosis in vivo. J. Microsc. Soc. Am. 1(5), 203–215. [Google Scholar]
- Denk, W., Strickler, J.H., and Webb, W.W. (1990). Two-photon laser scanning fluorescence microscopy. Science 248, 73–76. [DOI] [PubMed] [Google Scholar]
- Denk, W., Piston, D.W., and Webb, W.W. (1995). Two-photon excitation in laser-scanning microscopy. In: Handbook of Biological Confocal Microscopy, 2nd ed., ed. J.B. Pawley, New York: Plenum Press, 445–458.
- Galbraith, J.A., Reese, T.S., Schlief, M.L., and Gallant, P.E. (1999). Slow transport of unpolymerized tubulin and polymerized neurofilament in the squid giant axon. Proc. Natl. Acad. Sci. USA 96, 11589–11594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara, K., Tydeman, P., and Kirschner, M. (1980). A cytoplasmic clock with the same period as the division cycle in Xenopus eggs. Proc. Natl. Acad. Sci. USA 77: 462–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heald, R., McLoughlin, M., and McKeon, F. (1993). Human wee1 maintains mitotic timing by protecting the nucleus from cytoplasmically activated Cdc2 kinase. Cell 74, 463–474. [DOI] [PubMed] [Google Scholar]
- Heilbrunn, L.V. (1958). The Dynamics of Living Protoplasm. New York: Academic Press. 634 pp.
- Hopt, A., and Neher, E. (2001). Highly nonlinear photodamage in two-photon fluorescence microscopy. Biophys. J. 80, 2029–2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jontes, J.D., Buchanan, J., and Smith, S.J. (2000). Growth cone and dendrite dynamics in zebrafish embryos: early events in synapto-genesis imaged in vivo. Nat. Neurosci. 3, 231–237. [DOI] [PubMed] [Google Scholar]
- Kalab, P., Weis, K., and Heald, R. (2002). Visualization of a Ran-GTP gradient in interphase and mitotic Xenopus egg extracts. Science 295, 2452–2456. [DOI] [PubMed] [Google Scholar]
- Keller, H.E. (1995). Objective lenses for confocal microscopy. In: Handbook of Biological Confocal Microscopy, 2nd ed., ed. J.B. Pawley, New York: Plenum Press, 111–126.
- Khodjakov, A., and Rieder, C.L. (2001). Centrosomes enhance the fidelity of cytokinesis in vertebrates and are required for cell cycle progression. J. Cell Biol. 153, 237–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khodjakov, A., Cole, R.W., and Rieder, C.L. (1997). A synergy of technologies: combining laser microsurgery with green fluorescent protein tagging. Cell Motil. Cytoskeleton 38, 311–317. [DOI] [PubMed] [Google Scholar]
- Kishimoto, T. (1999). Activation of MPF at meiosis reinitiation in starfish oocytes. Dev. Biol. 214, 1–8. [DOI] [PubMed] [Google Scholar]
- Kishimoto, T., Kubota, J., and Kanatani, H. (1977). Distribution of maturation-promoting factor in starfish oocyte stratified by centrifugation. Dev. Growth Diff. 19, 283–288. [DOI] [PubMed] [Google Scholar]
- Koester, H.J., Baur, D., Uhl, R., and Hell, S.W. (1999). Ca2+ fluorescence imaging with pico- and femtosecond two-photon excitation: signal and photodamage. Biophys. J. 77, 2226–2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang, H., Vu, K.T., Krishnan, P., Trang, T.C., Shin, D., Kimel, S., and Berns, M.W. (1996). Wavelength dependence of cell cloning efficiency after optical trapping. Biophys. J. 70, 1529–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, K.S., and Fetcho, J.R. (1999). Laser ablations reveal functional relationships of segmental hindbrain neurons in zebrafish. Neuron 23, 325–335. [DOI] [PubMed] [Google Scholar]
- McNeil, P.L., and Steinhardt, R.A. (1997). Loss, restoration, and maintenance of plasma membrane integrity. J. Cell Biol. 137, 1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeil, P.L., Vogel, S.S., Miyake, K., and Terasaki, M. (2000). Patching plasma membrane disruptions with cytoplasmic membrane. J. Cell Sci. 113, 1891–1902. [DOI] [PubMed] [Google Scholar]
- McNeil, P.L., Miyake, K., and Vogel, S.S. (2001). Rapid resealing requirement for internal membrane. Mol. Biol. Cell 12s, 75a. [Google Scholar]
- Misawa, H., Koshioka, M., Sasaki, K., Kitamura, N., and Masuhara, H. (1991). Three-dimensional optical trapping and laser ablation of a single polymer latex particle in water. J. Appl. Phys. 70, 3829–3836. [Google Scholar]
- Noack, J., and Vogel, A. (1999). Laser-induced plasma formation in water at nanosecond to femtosecond time scales: calculation of thresholds, absorption coefficients and energy density. IEEE J. Quantum Electron. 35, 1156–1167. [Google Scholar]
- Okano-Uchida, T., Sekiai, T., Lee, K.-s., Okumura, E., Tachibana, T., and Kishimoto, T. (1998). In vivo regulation of cyclin A/cdc2 and cyclin B/cdc2 through meiotic and early cleavage cycles in starfish. Dev. Biol. 197, 39–53. [DOI] [PubMed] [Google Scholar]
- Ookata, K., Hisanaga, S.-I., Okano, T., Tachibana, K., and Kishimoto, T. (1992). Relocation and distinct subcellular localization of p34cdc2–cyclin B complex at meiosis reinitiation in starfish oocytes. EMBO J. 11, 1763–1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, H.C. et al. (2000). Analysis of upstream elements in the HuC promoter leads to the establishment of transgenic zebrafish with fluorescent neurons. Dev. Biol. 227, 279–293. [DOI] [PubMed] [Google Scholar]
- Pines, J. (1999). Checkpoint on the nuclear frontier. Nature 397, 104–105. [DOI] [PubMed] [Google Scholar]
- Pines, J., and Hunter, T. (1991). Human cyclins A and B1 are differentially located in the cell and undergo cell cycle-dependent nuclear transport. J. Cell Biol. 115, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapoport, R. (1981). Cytokinesis: cleavage furrow establishment in cylindrical sand dollar eggs. J. Exp. Zool. 221, 399–403. [Google Scholar]
- Reddy, A., Caler, E.V., and Andrews, N.W. (2001). Plasma membrane repair is mediated by Ca2+-regulated exocytosis of lysosomes. Cell 106, 157–169. [DOI] [PubMed] [Google Scholar]
- Sluder, G., Miller, F.J., and Rieder, C.L. (1986). The reproduction of centrosomes: nuclear versus cytoplasmic controls. J. Cell Biol. 103, 1873–1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinhardt, R.A., Bi, G., and Alderton, J.M. (1994). Cell membrane resealing by a vesicular mechanism similar to neurotransmitter release. Science 263, 390–393. [DOI] [PubMed] [Google Scholar]
- Takizawa, C.G., and Morgan, D.O. (2000). Control of mitosis by changes in the subcellular location of cyclin-B1-Cdk1 and Cdc25C. Curr. Opin. Cell Biol. 12, 658–6. [DOI] [PubMed] [Google Scholar]
- Tamm, S.L. (1978). Laser microbeam study of a rotary motor in termite flagellates. Evidence that the axostyle complex generates torque. J. Cell Biol. 78, 76–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terasaki, M. (1994). Redistribution of cytoplasmic components during germinal vesicle breakdown in starfish oocytes. J. Cell Sci. 107, 1797–1805. [DOI] [PubMed] [Google Scholar]
- Terasaki, M., Miyake, K., and McNeil, P.L. (1997). Large plasma membrane disruptions are rapidly resealed by Ca2+-dependent vesicle-vesicle fusion events. J. Cell Biol. 139, 63–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyoshima-Morimoto, F., Taniguchi, E., Shinya, N., Iwamatsu, A., and Nishida, E. (2001). Polo-like kinase 1 phosphorylates cyclin B1 and targets it to the nucleus during prophase. Nature 410, 215–220. [DOI] [PubMed] [Google Scholar]
- Walker, R.A., Inoue, S., and Salmon, E.D. (1989). Asymmetric behavior of severed microtubule ends after ultraviolet-microbeam irradiation of individual microtubules in vitro. J. Cell Biol. 108, 931–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}