

Abstract
Erythritol biosynthesis is catalyzed by erythrose reductase, which converts erythrose to erythritol. Erythrose reductase, however, has never been characterized in terms of amino acid sequence and kinetics. In this study, NAD(P)H-dependent erythrose reductase was purified to homogeneity from Candida magnoliae KFCC 11023 by ion exchange, gel filtration, affinity chromatography, and preparative electrophoresis. The molecular weights of erythrose reductase determined by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and gel filtration chromatography were 38,800 and 79,000, respectively, suggesting that the enzyme is homodimeric. Partial amino acid sequence analysis indicates that the enzyme is closely related to other yeast aldose reductases. C. magnoliae erythrose reductase catalyzes the reduction of various aldehydes. Among aldoses, erythrose was the preferred substrate (Km = 7.9 mM; kcat/Km = 0.73 mM−1 s−1). This enzyme had a dual coenzyme specificity with greater catalytic efficiency with NADH (kcat/Km = 450 mM−1 s−1) than with NADPH (kcat/Km = 5.5 mM−1 s−1), unlike previously characterized aldose reductases, and is specific for transferring the 4-pro-R hydrogen of NADH, which is typical of members of the aldo/keto reductase superfamily. Initial velocity and product inhibition studies are consistent with the hypothesis that the reduction proceeds via a sequential ordered mechanism. The enzyme required sulfhydryl compounds for optimal activity and was strongly inhibited by Cu2+ and quercetin, a strong aldose reductase inhibitor, but was not inhibited by aldehyde reductase inhibitors and did not catalyze the reduction of the substrates for carbonyl reductase. These data indicate that the C. magnoliae erythrose reductase is an NAD(P)H-dependent homodimeric aldose reductase with an unusual dual coenzyme specificity.
Erythritol, a four-carbon polyol, is widely distributed in nature (16). Like most other polyols, erythritol is a metabolite or storage compound and is found in seaweed, mushrooms, and fruits. It is a noncaloric, noncariogenic sweetener that is safe for diabetics (35). Erythritol has about 70% of the sweetness of sucrose in a 10% (wt/vol) solution and a very high negative heat capacity, providing a strong cooling effect when dissolved (16). Erythritol can be synthesized from dialdehyde starch by a high-temperature chemical reaction in the presence of a nickel catalyst (39). This process has not been industrialized because of its low efficiency. Erythritol also can be produced by microbial methods that utilize osmophilic yeasts and some bacteria (42, 48), and it has been produced commercially by using a mutant of Aureobasidium that produces erythritol in high yield (44% [wt/wt] glucose) (20). Recently, a high-erythritol-producing yeast strain was isolated from honeycombs and the new isolate was identified as Candida magnoliae KFCC 11023 (44). This strain can produce erythritol in high yield (43% [wt/wt] glucose) when cultured appropriately (42, 44).
There are many reports of enzymes that catalyze the formation of glycerol, mannitol, and xylitol, e.g., glyceraldehyde dehydrogenase, mannitol dehydrogenase, and xylose reductase (14, 21), respectively, but there are few reports of enzymes that catalyze the formation of erythritol from erythrose. Erythritol is synthesized from erythrose-4-phosphate, an intermediate in the pentose-phosphate pathway, by dephosphorylation and the subsequent reduction of erythrose. Erythrose reductase (ER), which catalyzes the final step in this pathway, is a key enzyme in the biosynthesis of erythritol (19, 27, 46). Braun and Niederpruem studied erythritol metabolism in Schizophyllum commune and observed high erythrose-reducing activity in the cell extract (8), with the highest activity toward d-erythrose. Ishizuka et al. (19) and Lee et al. (27) purified ERs from an Aureobasidium sp. mutant and Torula corallina, respectively. Although there have been three reports of the purification of ER from microbial sources (8, 19, 27, 46), to our knowledge, ER has never been purified from C. magnoliae, and this enzyme has not been characterized in terms of amino acid sequence homology and kinetic mechanism.
In this study, we purified the enzyme catalyzing the conversion of erythrose to erythritol from C. magnoliae KFCC 11023 to homogeneity and characterized its physical and kinetic properties, identifying it as an aldose reductase (ADR), a member of the aldo-keto reductase (AKR) superfamily, with properties similar to those of a wide range of ADRs found in other microorganisms and animals.
MATERIALS AND METHODS
Materials.
d-Erythrose, d-fructose, d-galactose, d-glucose, d-ribose, d-arabinose, d-xylose, d-glyceraldehyde, meso-erythritol, p-nitrobenzaldehyde, p-carboxybenzaldehyde, menadione, sodium valproate, barbiturate, quercetin, indomethacin, pyrazole, ethacrynic acid, Coomassie brilliant blue R-250, and the column chromatographic support media, including Cibacron Blue 3GA affinity resin, were obtained from Sigma Chemical Co. (St. Louis, Mo.). Enzyme cofactors (NAD, NADH, NADP, and NADPH) were purchased from Boehringer Mannheim (Indianapolis, Ind.), and the polyvinylidene difluoride membrane was purchased from Bio-Rad Laboratories (Hercules, Calif.). Proteins used for calibration and assay runs were obtained from Pharmacia Fine Chemicals (Uppsala, Sweden). Diaflo YM 10 (cutoff, 10 kDa) ultrafiltration membranes were purchased from Amicon, Inc. (Danvers, Mass.). All other chemicals were of analytical grade or higher.
Microorganism and culture conditions.
C. magnoliae was isolated from honeycombs in this laboratory (44) and deposited in the Korean Culture Center for Microorganisms (KFCC 11023). The fermentation medium for erythritol production was: 10% (wt/vol) glucose, 0.5% yeast extract, 0.5% KH2PO4, 0.2% (NH4)2SO4, and 0.04% MgSO4 · 7H2O. The strain was cultured as described previously (42).
Purification of ER.
All procedures were performed at 4°C, and 50 mM potassium phosphate buffer (pH 7.0) containing 1 mM dithiothreitol (DTT) was used in the purification procedures unless otherwise stated. Protein was measured by the method of Lowry et al. (29), with bovine serum albumin as a standard. Protein in the column effluents was monitored by measuring the absorbance at 280 nm. All chromatographic separations were performed with a BioLogic LP system (Bio-Rad).
Step 1: preparation of cell extract.
Cells from the culture broth were harvested by centrifugation at 10,000 × g for 30 min. After washing with 50 mM potassium phosphate buffer (pH 7.0), harvested cells were resuspended in homogenization buffer containing 50 mM potassium phosphate buffer (pH 7.0), 10 mM MgCl2, and 1 mM phenylmethylsulfonyl fluoride. The cell suspension was incubated for 1 h at room temperature and then homogenized by grinding with 0.5-mm-diameter glass beads (Sigma). Cell extracts were obtained by pelleting cell debris by centrifugation at 10,000 × g for 30 min. The supernatants were combined, concentrated, and desalted by ultrafiltration through a YM10 membrane in a stirred cell (Amicon).
Step 2: ammonium sulfate fractionation.
Cell extracts were fractionated by ammonium sulfate precipitation. The fraction precipitated between 3.0 and 5.3 M ammonium sulfate was collected by centrifugation and redissolved in 50 mM potassium phosphate buffer (pH 7.0). Insoluble material was removed by centrifugation at 10,000 × g for 1 h, and the enzyme solution was dialyzed against the same buffer at 4°C for 24 h.
Step 3: DEAE-cellulose chromatography.
The dialyzed enzyme solution was loaded onto a DEAE-cellulose column (1.4 by 20 cm) equilibrated with 50 mM potassium phosphate buffer at pH 7.0, and protein was eluted with a 180-min linear gradient of 0 to 0.5 M NaCl in the same buffer at a flow rate of 0.5 ml/min. Active fractions were pooled, dialyzed against the same buffer, and concentrated by ultrafiltration.
Step 4: Sephadex G-100 chromatography.
The concentrated enzyme solution was loaded onto a Sephadex G-100 column (0.7 by 40 cm) equilibrated with 50 mM potassium phosphate buffer at pH 7.0, and protein was eluted with the same buffer at a flow rate of 0.3 ml/min. Active fractions were pooled, dialyzed against the same buffer, and concentrated by ultrafiltration.
Step 5: Cibacron Blue 3GA affinity chromatography.
The enzyme was further purified with an affinity column (1.4 by 20 cm) of Cibacron Blue 3GA previously equilibrated with 10 mM phosphate (pH 7.0). The enzyme was eluted with a 180-min linear gradient of 0 to 1.0 M NaCl in 10 mM potassium phosphate buffer (pH 7.0) at a flow rate of 0.5 ml/min. The combined active fractions were pooled, concentrated, dialyzed against the same buffer, and concentrated by ultrafiltration.
Step 6: preparative electrophoresis.
The enzyme was mixed with native polyacrylamide gel electrophoresis (PAGE) sample buffer and applied to the preparative electrophoresis system (Bio-Rad) for purification. The gel composition was 9% T-2.6% C, with a height of 10 cm and a gel tube size of 28 mm, where the following formulas apply: % T = [{acrylamide (in grams) + bis (in grams)}/volume (milliliters)] × 100%, % C = [bis (in grams)/{acrylamide (in grams) + bis (in grams)}] × 100%. The stacking gel composition was 5% T-2.6% C, with a height of 2.0 cm. Running conditions were 30 mA of constant current for 8 to 10 h. After elution of bromophenol blue tracking dye, 2-ml fractions were collected. The combined active fractions were pooled, concentrated, dialyzed against the same buffer, concentrated with a Centricon (Millipore Corp., Bedford, Mass.) ultrafiltration device with a molecular mass cutoff of 10 kDa, and then used as a purified enzyme in the following experiments.
ER activity assay.
The activity of ER was determined spectrophotometrically by monitoring the change in A340 upon oxidation or reduction of NADP(H) at 37°C (45). Unless otherwise indicated, the ER assay mixture (1.0 ml) for reduction contained 50 mM potassium phosphate buffer (pH 7.0), 50 μM NADH, 20 mM erythrose, and enzyme solution (0.1 ml). The ER assay mixture for oxidation contained 50 mM potassium phosphate buffer (pH 8.5), 50 μM NAD, 50 mM erythritol, and enzyme solution. One unit of enzyme activity represents 1 μmol of NADH consumed or produced per min. Activities were expressed as units/milligram of protein, and the results presented are the means of triplicate assays. The amount of variation observed was <10%.
pH profiles of the kinetic parameters.
For assays at different pH values, the reactions were performed at 37°C in the following buffers (50 mM) and pH values: sodium citrate, pH 4.5 to 6.0; potassium phosphate, pH 6.0 to 8.0; Tris-HCl, pH 8.0 to 9.0; and glycine-NaOH, pH 9.0 to 10.0. In all experiments, d-erythrose was the variable substrate and NADH remained at a fixed concentration of 50 μM. To construct the pH profiles, the kinetic parameters kcat and kcat/Km were determined for d-erythrose between pH 5.0 and 10.0, and the pH dependence of Y (kcat/Km) was fitted to a bell-shaped curve described by the following equation:
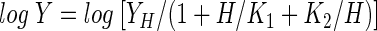 |
where H is the proton concentration, K1 and K2 are the dissociation constants for the groups that ionize at low and high pHs, respectively, and YH is the pH-independent plateau value of Y at an intermediate pH. The pH profile of kcat was constructed in a point-to-point manner.
PAGE and molecular mass determination.
For the determination of subunit molecular mass, sodium dodecyl sulfate (SDS)-PAGE was performed as described by Laemmli (25) with 10% gels. Protein bands were visualized with Coomassie brilliant blue R-250 (Sigma). Native PAGE was performed with 10% polyacrylamide gels without SDS. ER activity was stained on the polyacrylamide gel by using the method of Birken and Pisano (4). Isoelectric focusing was performed with 7.5% polyacrylamide gels (0.5 by 10 cm) supplemented with ampholytes (0.4% pH 3 to 10) as described by O'Farrel (37). The molecular mass of the purified enzyme was determined by size exclusion chromatography with a Superose 12 column (Amersham Pharmacia Biotech) attached to a BioLogic LP system (Bio-Rad). The enzyme was eluted with 10 mM Tris-HCl buffer (pH 7.0) at a flow rate of 0.1 ml/min.
Amino acid composition and partial amino acid sequencing.
Approximately 500 μg of enzyme was precipitated with 30% (wt/vol) trichloroacetic acid, and the precipitate was washed with ice-cold acid acetone (0.1% [vol/vol] concentrated HCl in acetone). The amino acid composition of the dried acetone powder was analyzed by ion-exchange chromatography on an amino acid high-performance liquid chromatography (HPLC) system (Waters, Milford, Mass.). The compositional relatedness between C. magnoliae ER and AKRs from other sources was assessed by the method of Metzger et al. (31). The purified protein was resolved by SDS-PAGE and then electroblotted onto a polyvinylidene trifluoride membrane (Bio-Rad). Protein cleavage for peptide mapping was carried out at 37°C for 4 h with 100 ng of endoproteinase Glu-C (Promega, Madison, Wis.) to digest 20 μg of purified enzyme in 50 μl of 100 mM (NH4)2CO3 (pH 8.5). The resulting peptide fragments were separated by SDS-PAGE (15% polyacrylamide), and the separated peptides were transferred to a polyvinylidene trifluoride membrane by electroblotting. Peptide bands were visualized by 0.1% Coomassie brilliant blue R-250 staining in 40% methanol. The partial amino acid sequence was determined by Edman degradation with an automatic protein sequencer (model 491A; Applied Biosystems, Division of Perkin-Elmer) at The National Instrumentation Center for Environmental Management (Suwon, Korea). The partial amino acid sequence was used to identify analogous proteins through a BLAST search of the nonredundant protein database.
Stereospecificity of hydride transfer from NADH.
We used 1H nuclear magnetic resonance (NMR), performed with a Bruker ARX Fourier transform spectrometer (Bruker Instruments, Inc., Billerica, Mass.) operating at 400 MHz in the pulsed Fourier transform mode. Spectra were recorded at 27°C, with D2O (99.9%) and 3-(trimethylsilyl)-1-propanesulfonic acid (sodium salt) as references. The (R) and (S) isomers of [4-D]NADH were prepared as previously described (35). The deuterium content of (4R)-[4-D]NADD and (4S)-[4-D]NADD was determined to be greater than 98%. A reaction mixture contained 2 mg of (4R)-[4-D]NADD or (4S)-[4-D]NADD, 1 U of ER, and 20 mM d-erythrose dissolved in 50 mM phosphate buffer (pD 7.0). When the oxidation of NADD was complete (monitored spectrophotometrically at 340 nm), the sample was used for NMR analysis without further treatment. The stereospecificity for the hydride transfer for ER was determined by comparing the integrated peak area of C4H (δ, 8.75 ppm) for the reaction mixtures containing B- or A-side-labeled NADH as the coenzyme.
CD spectroscopy.
Circular dichroism (CD) spectra of C. magnoliae ER were recorded by using a Jasco 715 CD spectrophotometer (Easton, Md.) with a cell with a path length of 0.1 cm and a protein concentration of 0.05 mg/ml in 20 mM potassium phosphate buffer (pH 7.0) at 22°C. The mean of 115 scans between 200 and 250 nm was calculated and corrected by subtraction of the buffer spectra. The percentage of secondary structure elements was calculated by using the Jasco secondary structure estimation program.
RESULTS
Purification of ER.
Crude extracts of C. magnoliae were obtained from cells on the third day of culture, when ER activity was high, and the enzyme was purified (Table 1). One peak of enzyme activity was eluted after the extract was subjected to a DEAE-cellulose step, followed by Sephadex G-100 gel filtration and a Cibacron Blue 3GA affinity step. The pooled, active fractions were then applied to a preparative electrophoresis system. This method resulted in an 850-fold purification of ER with a recovery of 5.8%. The yield of the purified enzyme (specific activity, 20 U/mg) was 0.28 mg starting from 270 g (wet weight) of C. magnoliae cells. PAGE of the enzyme after the preparative electrophoresis step yielded a single protein band in both the absence (Fig. 1A) and presence (Fig. 1B) of SDS when the gels were stained with Coomassie brilliant blue. Activity staining of the nondenaturing gel resulted in a single band with the same mobility as that of the protein band (Fig. 1A-a). These results indicate that the purified enzyme is homogeneous. The UV-visible spectrum of the purified enzyme had one absorption maximum at 280 nm. No absorbance was detectable above 320 nm, indicating that the enzyme does not contain flavin, which is the coenzyme in most quinone reductases (15).
TABLE 1.
Purification of ER from the cell extract of C. magnoliae
| Procedure | Total protein (mg) | Total activity (U) | Specific activity (U/mg of protein) | Yield (%) | Purification (fold) |
|---|---|---|---|---|---|
| Cell extract obtainment | 4,100 | 95 | 0.023 | 100 | 1.0 |
| Ammonium sulfate fractionation | 850 | 52 | 0.061 | 55 | 2.7 |
| DEAE-cellulose ion-exchange chromatography | 72 | 48 | 0.662 | 50 | 29 |
| Sephadex G-100 gel filtration chromatography | 20 | 35 | 1.71 | 36 | 74 |
| Cibacron Blue 3GA affinity chromatography | 2.7 | 21 | 7.83 | 23 | 340 |
| Preparative electrophoresis | 0.3 | 5.5 | 19.6 | 5.8 | 850 |
FIG. 1.

PAGE of ER purified from the newly isolated C. magnoliae. (A) Native PAGE. Lane 1, cell extract; lane 2, DEAE ion-exchange fraction; lane 3, Sephadex G-100 fraction; lane 4, Cibacron Blue 3GA affinity fraction; lane 5, preparative electrophoresis fraction. (A-a) Activity staining after native PAGE. (B) SDS-PAGE. The enzyme solution was run on a 10% (wt/vol) polyacrylamide slab gel as described in Materials and Methods. The standard proteins (Bio-Rad) used for the estimation of molecular mass were phosphorylase B (113 kDa), bovine serum albumin (92 kDa), ovalbumin (52.3 kDa), carbonic anhydrase (39.2 kDa), soybean trypsin inhibitor (28.9 kDa), and lysozyme (21 kDa).
HPLC analysis of the reaction products formed after the enzyme was incubated at 37°C in a mixture of buffer and erythrose confirmed the identity of the enzyme under examination, with and without NADH. Each mixture was cooled to 4°C, diluted, and chromatographed. The HPLC chromatogram showed a significant decrease in erythrose content for the sample containing NADH accompanied by the formation of a new peak that chromatographed with and had the same mass profile as authentic erythritol (Fig. 2); this substance was absent when the enzyme extract and NADH were incubated without erythrose.
FIG. 2.

HPLC (BioLC; Dionex, Sunnyvale, Calif.) analysis of the reaction products of C. magnoliae ER. Erythrose (5 mM) was incubated with C. magnoliae ER (1 U) at 37°C for 2 h. The enzyme was incubated in a mixture of buffer and erythrose with 1.0 mM NADPH. The sample was eluted isocratically on a CarboPac MA1 column with 500 mM NaOH at a flow rate of 0.4 ml/min and detected with a BioLC ED50A electrochemical detector. Panels A and B show the respective HPLC profile and mass spectrum of authentic erythrose while panels C and D show those of authentic erythritol. Panel E shows the HPLC profile of the reaction products. The reaction mixture was diluted, filtered through a 0.2-μm-pore-size membrane, and subjected to HPLC analysis. The results for the residual substrate erythrose (34.6 min), the product erythritol (11.2 min), and the contaminants (22.4 and 24.5 min) are illustrated. The peaks were assigned based on the retention times of the standard samples. Panel F shows the mass spectrum of erythritol enzymatically formed by the C. magnoliae ER.
Molecular weight and CD spectra of erythrose reductase.
One band with an Mr of 38,800 ± 1,000 (n = 3) was revealed by gel electrophoresis in the presence of SDS (Fig. 1B). Size exclusion chromatography on Superose 12 resulted in elution of the enzyme activity as a symmetrical peak corresponding to an Mr of approximately 79,000. This result, combined with those from SDS-PAGE separations, indicates that the C. magnoliae ER is a homodimeric enzyme. The yeast ADRs that have been purified and characterized in some detail occur chiefly as homodimers with a subunit molecular mass of 35 to 40 kDa (21, 49). The C. magnoliae ER was further characterized by CD spectroscopy (data not shown). The CD spectrum of C. magnoliae ER is very similar in shape and intensity to that of human ADR (38), indicating that both proteins have similar folding patterns. The calculated α-helix content of the C. magnoliae ER was 38%.
Partial amino acid sequencing and amino acid composition.
Pure enzyme (1.5 μg) was separated by SDS-PAGE and blotted onto a polyvinylidene trifluoride membrane. Automated Edman degradation of the enzyme protein was unsuccessful, implying that the N terminus of the enzyme was blocked. Two small molecular fragments (peptide 1 molecular weight, 11,000; peptide 2 molecular weight, 16,000) were analyzed on a protein sequencer. Eight amino acid residues (LVDYQPAR) of peptide 1 and 18 residues (GKVVIGFGPGCYVAAIKH) of peptide 2 were determined by automated Edman degradation. Peptide 1 has no similarity to known sequences, but the 18 residues of peptide 2 are similar to several ADRs (Fig. 3). The sequence of each reductase contains a highly conserved region (positions 1 to 18, according to the C. magnoliae ER numbering) and possesses a unique 7-amino-acid-long peptide (GXXXGXG) that is similar to the Wierenga coenzyme-binding motif of other dehydrogenases (23). The first two Gly residues of this sequence are strictly conserved among the yeast enzymes while the third Gly can be replaced by other residues (26): Asp in the Saccharomyces cerevisiae enzyme, Glu in the Kluyveromyces lactis enzyme, and Tyr in C. magnoliae ER.
FIG. 3.

Partial amino acid sequence alignment of ADRs from C. magnoliae (this study), S. cerevisiae (22), K. lactis (3), P. stipitis (1), P. tannophilus (6), and C. tropicalis (54). Identical residues are given against a black background, and similar residues are boxed.
The similarity between the C. magnoliae ER and AKRs from different species also is evident from their amino acid compositions. All AKRs have a high proline content, but ADRs are clearly distinguished from aldehyde reductases (ALRs) by their higher cysteine contents (9 to 10 versus 3 to 4 Cys residues) (55). The amino acid composition of C. magnoliae ER is high in proline (6.1%), which is typical of AKRs (7, 32). The C. magnoliae ER also had a high cysteine content (2.2% mol content), which is typical of ADRs (26). Indeed, the overall amino acid composition of the C. magnoliae ER is quite similar to that of ADRs from human muscle (33), pig lens (7), and Candida tropicalis (55).
Optimum pH and thermal stability.
The optimum pH for the reduction of d-erythrose by purified C. magnoliae ER was 7.0, with 72 and 58% of maximum activity at pH 6.0 and 8.0, respectively. The optimum pH for oxidation was 9.0, with 62 and 91% of the maximum activity at pH 8.0 and 10.0, respectively. Maximal reductase activity at pH 7.0 and an alkaline pH optimum for erythritol oxidation are common features of similar enzymes isolated from diverse microbial systems (28). In addition, the rate of the reverse reaction with 50 mM erythritol was insignificant (<5% of the forward reaction rate with 20 mM erythrose), as is typical of an AKR. The isoelectric point of the ER from C. magnoliae was pH 5.1 as determined by isoelectric focusing.
The stability of ER was tested at pH 7.0 in standard buffer containing 1 mM DTT. Preparations were incubated at 4, 20, 30, 45, and 50°C and retained 50% of their initial activities after 60 days, 21 days, 5 days, 15 h, and 8 min, respectively.
Kinetic parameters.
Initial-velocity studies were performed with erythrose as a variable substrate in the presence of fixed concentrations of NADH. Plots of the reciprocal of the initial velocity against the reciprocal of the erythrose concentration gave a family of straight lines that intersected in the left quadrant (Fig. 4). When NADH was used as a variable substrate, similar straight lines intersecting in the left quadrant were obtained. These results indicate that the reaction proceeds via the formation of a ternary complex of the enzyme with NADH and d-erythrose and rule out the possibility of a ping-pong mechanism (11). The Kms for NADH and d-erythrose were calculated to be 12.8 μM and 7.9 mM, respectively, from the secondary plots of the intercepts versus the reciprocal concentrations of the other substrate (Fig. 4, inset). The Vmax was determined to be 20.1 μmol/min · mg of protein.
FIG. 4.

Initial-velocity double-reciprocal plot with d-erythrose as the varied substrate at NADH concentrations of 5 (♦), 10 (▾), 25 (▴), 50 (▪), and 100 (•) μM. The initial velocities were measured by the standard assay at pH 7.0. Other conditions are described in Materials and Methods. The inset shows secondary plots of the intercepts versus the fixed NADH concentrations.
Under nonsaturating conditions, erythritol inhibition was noncompetitive for NADH and d-erythrose (Fig. 5), and erythritol binds to C. magnoliae ER with a Ki of 276 mM. The inhibition by NAD+ was competitive with NADH and was noncompetitive with d-erythrose (data not shown). The slopes versus the NAD+ concentrations were plotted, and the product inhibition data are shown in Table 2.
FIG. 5.

Graphical analysis of the inhibition of C. magnoliae ER by erythritol. The effects of increasing erythritol (product) concentration on the apparent Km and Vmax values for erythrose and NADH were examined. Analysis of these data by double-reciprocal plots indicated that erythritol inhibited ER noncompetitively with respect to erythrose (A) and NADH (B). In panel C, the secondary plots for noncompetitive inhibition with erythrose and NADH are shown. The erythritol product binds to ER with a Ki of 276 mM.
TABLE 2.
Product inhibition pattern of C. magnoliae ER
| Product | Substrate | Inhibition | Ki |
|---|---|---|---|
| Erythritol | Erythrose | Noncompetitive | 280 mM |
| Erythritol | NADH | Noncompetitive | 280 mM |
| NAD+ | Erythrose | Noncompetitive | 150 μM |
| NAD+ | NADH | Competitive | 48 μM |
pH profiles of the kinetic parameters.
The pH profiles of the kinetic parameters with NADH and d-erythrose are shown in Fig. 6. The effect of pH on the ionization of free C. magnoliae ER was visualized by the plot of log kcat/Km (per molarity per second) versus pH. The pH profile of log kcat/Km for d-erythrose reduction is dependent on two ionizing groups, indicating that a group with a pK of 5.9 has to be unprotonated and a group with a pK of 9.0 has to be protonated for activity. In the log kcat (per second) versus pH plot, the log kcat increased steadily up to pH 7.0 and then decreased with a slope of less than 0.3.
FIG. 6.

pH dependence of log kcat (•) and log kcat/Km (○) values for C. magnoliae ER at 37°C with d-erythrose. The assay and plotting were performed as described in Materials and Methods. Each value represents the mean of triplicate measurements and varied from the mean by not more than 10%.
Substrate and coenzyme specificity.
Purified C. magnoliae ER has broad substrate specificity (Table 3). The C. magnoliae ER is active with various aldose substrates and its high affinity for aromatic aldehydes, such as p-nitrobenzaldehyde, is typical of AKRs (47). Aromatic aldehydes are followed in catalytic effectiveness by aliphatic aldehydes and then sugar aldehydes. This order is similar to those reported for AKRs from various mammals (2, 7) and the yeasts Pichia stipitis (49), Pachysolen tannophilus (14), and S. cerevisiae (24).
TABLE 3.
Substrate and coenzyme specificity of C. magnoliae ERa
| Substrate | Km (mM) | kcat (s−1) | kcat/Km (mM−1 s−1) |
|---|---|---|---|
| p-Nitrobenzaldehyde | 0.13 | 1.8 | 14 |
| p-Carboxybenzaldehyde | 0.53 | 1.1 | 2.1 |
| Benzaldehyde | 0.59 | 1.2 | 2.1 |
| d-Glyceraldehyde | 3.8 | 6.6 | 1.7 |
| d-Erythrose | 7.9 | 5.7 | 0.73 |
| d-Xylose | 36 | 2.3 | 0.064 |
| d-Arabinose | 42 | 1.4 | 0.034 |
| d-Ribose | 61 | 1.3 | 0.022 |
| d-Galactose | 70 | 0.70 | 0.010 |
| d-Glucose | 76 | 1.3 | 0.017 |
| NADH | 0.013 | 5.8 | 450 |
| NADPH | 0.12 | 0.66 | 5.5 |
The purified enzymes were assayed under standard assay conditions with various substrates. Each value represents the mean of triplicate measurements and varied from the mean by not more than 10%.
Most of the polyol-oxidizing and -reducing enzymes described to date are pyridine nucleotide linked, requiring either NADH or NADPH as a coenzyme. C. magnoliae ER showed dual coenzyme specificity for both NADPH and NADH. Although the ADRs generally have a greater affinity for NADPH than NADH in mammalian tissues (53) and yeasts (49), ADRs often utilize both cofactors (36, 49). C. magnoliae ER had a higher affinity for NADH (Km = 12.8 μM) than for NADPH (Km = 116 μM) in the presence of 20 mM erythrose, which makes this enzyme very interesting.
Stereospecificity of hydride transfer.
The stereospecificity of the hydride transfer step was examined by using stereospecifically labeled NADH, and the oxidized NAD+ generated during the reaction was analyzed by 1H NMR (34). When (4R)-[4-D]NADD was used as a coenzyme for the reduction of d-erythrose, the H-4 signal at δ 8.75 was retained in the NAD+ species, indicating the transfer of the pro-R deuterium at the C-4 position of the nicotinamide ring. By contrast, incubation with (4S)-[4-D]NADD resulted in the absence of the δ 8.75 signal, due to the depletion of (4R)-hydrogen on C. magnoliae ER-catalyzed oxidation of NADD.
Effects of metal ions and other compounds.
Both divalent and monovalent cations affect ADRs (13, 18). ER activity was measured in the presence of metal ions (1 mM) and other compounds with erythrose as the substrate. Ag+, Zn2+, Cu2+, and Al3+ inhibited C. magnoliae ER. At 1 mM concentrations, Mg2+, Mn2+, Zn2+, Ag+, and Ba2+ slightly inhibited (10 to 20% inhibition), Al3+ significantly inhibited (56% inhibition), and Cu2+ completely inhibited (100% inhibition) C. magnoliae ER with a Ki value of 12 μM. Pretreatment of C. magnoliae ER with the same concentration of EDTA protected against the inhibitory effect of Cu2+, and the ER activity previously inactivated with Cu2+ was completely restored by the addition of EDTA. The protection and reactivation of the enzyme with EDTA suggest the effect of Cu2+ is reversible. The effects of Cu2+ on AKRs and the enzymatic production of erythritol by ER have also been reported previously (27, 51).
Dependence of the enzyme activity on sulfhydryl compounds has been reported for the ER from Aureobasidium sp. (19) and several ADRs purified from Candida tenuis (36), pig lens (7), and rabbit muscle (12). The addition of 1 mM 2-mercaptoethanol, glutathione, cysteine, or DTT to the reaction mixture increased the enzyme activity by 21, 32, 40, and 55%, respectively. These results suggest that sulfhydryl compounds, including DTT, the best reductant, keep the active enzyme in a reduced state. N-Ethylmaleimide is often used to inactivate enzymes, presumably by reacting with the thiol group of cysteine residues. Incubation of purified C. magnoliae ER with 5 mM N-ethylmaleimide resulted in about 70% inhibition of ER activity, suggesting that cysteine is, at least in part, responsible for ER activity, and the time required to reach half-maximal inhibition (t1/2) was approximately 3.5 min.
The effects on C. magnoliae ER of several drugs that are commonly used to inhibit ADRs and to discriminate between ALRs and ADRs were also examined. Drugs used to inhibit ADRs, such as quercetin and sorbinil, were the most potent inhibitors of C. magnoliae ER, with complete inhibition at 0.1 mM. Sodium valproate and barbiturates, which inhibit ALRs significantly (12, 47), were ineffective against the purified C. magnoliae ER up to 1 mM. Characteristic inhibitors of carbonyl reductase, e.g., indomethacin, pyrazole, and ethacrynic acid, had no effect up to 1 mM.
DISCUSSION
In previous studies, the isolation of a C. magnoliae strain that could produce high levels of erythritol was reported (42). ER catalyzes erythritol biosynthesis in C. magnoliae fermentation of glucose. AKRs, such as ALR and carbonyl reductase, from C. magnoliae have been reported (50, 51), and there are reports on the purification and properties of ER from three strains (19, 27, 46). However, this is the first report on the purification and characterization of ER, an ADR, from C. magnoliae, an organism with the potential for use in industrial erythritol production. Since the rate of erythritol oxidation never exceeded 10% of that of erythrose reduction (with 20 mM erythrose) at pH 7, its optimal pH, the enzyme is a reductase. Since the oxidative reaction of C. magnoliae ER is much slower than the reductive one and C. magnoliae ER is oxidatively active at alkaline pH, the differences in the pH activity profile and substrate specificity in the oxidative reaction have no practical meaning in living cells, where the pH is weakly acidic. Therefore, this enzyme is thought to catalyze exclusively the reduction of d-erythrose (formation of erythritol) in C. magnoliae.
The Km for d-erythrose (7.9 mM) is comparable to the values for the ERs from Aureobasidium sp. (46), T. corallina (27), and S. commune (8), which have Kms of 8.0, 7.1, and 5.0 mM, respectively. The Km of the C. magnoliae ER was also similar to the values reported for ADRs from the yeasts P. tannophilus (14) and P. stipitis (49).
The results of kinetic analyses suggest that NADH and NAD+ bind to the free form of the enzyme and rule out the possibility of a random mechanism (11). The pattern of product inhibition observed in a two-substrate reaction, under nonsaturating conditions, can be diagnostic for the mechanism of catalysis by that enzyme (41). For Theorell-Chance and ping-pong mechanisms, each product is a competitive inhibitor of one substrate and a noncompetitive inhibitor of the other. In random mechanisms, each product can competitively inhibit each substrate. Only an ordered Bi Bi reaction mechanism exhibits a pattern of inhibition in which one product is a noncompetitive inhibitor of both substrates and the other product is competitive and noncompetitive for the respective substrates. The product inhibition data (Table 2) and the results of the initial-velocity product inhibition studies suggest that the enzyme reaction proceeds via a sequential ordered Bi Bi mechanism, in which NADH binds first to the enzyme and is followed by d-erythrose, and that erythritol leaves the enzyme before the NAD+ is released (11). This type of reaction mechanism is typical of ADRs (5). The C. magnoliae ER specifically transfers the 4-pro-R hydrogen from the C-4 of the nicotinamide ring to the re face of the carbonyl carbon of the substrate, which also is typical of all studied members of the AKR superfamily. The stereochemistry of the enzymatic reaction is expected to be a constant characteristic of members of an enzyme family. Short-chain dehydrogenases and reductases show B-type stereospecificity, which transfers the 4-pro-S hydrogen in a si side attack to the carbonyl group (23). The C. magnoliae ER had no activity towards d-glucuronate, a typical substrate used to characterize ALRs of the AKR superfamily (17). ADRs are generally less active with uronic acids than are ALRs. Unlike ADR, ALR prefers substrates with a negatively charged carboxyl group, such as glucuronate and succinic semialdehyde (52). No activity was observed with menadione or benzoquinone, which are good substrates for carbonyl reductase (47). These results further confirm that the C. magnoliae ER is an ADR of the AKR superfamily.
Although the C. magnoliae ER is similar to the homologous ADRs described in other microorganisms and mammalian tissues, it has novel properties. Most members of the AKR superfamily appear to have a strong preference for NADPH over NADH (30). However, a few members of the family can use both cofactors. For example, xylose reductases from the yeasts P. tannophilus (6) and P. stipitis (49), an ADR from the yeast C. tenuis (36), a 3α-hydroxysteroid dehydrogenase from rat liver (40), and a 3-dehydroecdysone 3β-reductase cloned from Spodoptera littoralis (9, 10) are thought to have dual cofactor specificity. As far as we are aware, no members of the ADR family are specific for NADH only or show higher affinity for NADH than for NADPH. The dual nucleotide specificity of the C. magnoliae ER makes this enzyme interesting, especially compared to an NADPH-specific enzyme. Moreover, the C. magnoliae ER showed much higher catalytic efficiency with NADH (kcat/Km = 450 mM−1 s−1) than with NADPH (kcat/Km = 5.5 mM−1 s−1), which makes this enzyme even more interesting. NADH is less costly, more prevalent in the cell, and more stable than NADPH; therefore, process improvements could be obtained if the natural cofactor specificity of the enzyme could be broadened to enable the use of NADH. Many researchers are attempting to improve the activity of ADRs with NADH as a cofactor, using rational protein design (43, 49). Glutathione reductase, an NADP-dependent enzyme, was converted to an NAD-dependent enzyme by a point mutation, and this mutant enzyme had an 8-fold-higher preference for NAD than did the wild-type glutathione reductase (43). However, this mutant had only 3% of the activity of the wild type. P. stipitis xylose reductase also was mutagenized to modify cofactor specificity. However, replacement of Lys270 by Met resulted in an 80 to 90% loss in activity (49). Therefore, the C. magnoliae ER, an ADR with a high preference for NADH, is a good candidate for studying cofactor specificity and for use in the industrial production of sugar alcohols, including erythritol, by fermentation and other techniques.
Our results improve the understanding of erythritol biosynthesis in C. magnoliae and should contribute to better industrial production of erythritol by biological processes. However, definitive proof for the characteristics of the C. magnoliae ER requires complete primary structure and further crystallographic analysis of the enzyme or enzyme-coenzyme complex. We believe that further studies of the molecular determinants of coenzyme and substrate binding will facilitate engineering of ADRs, including the C. magnoliae ER, with greater NADH specificity and catalytic activity for use in the efficient production of a useful product.
Acknowledgments
We thank Ji-Hye Kil (Korea Basic Science Institute, Seoul) for help with mass spectrometry and CD analysis, Ji-Hyun Yoo for proofreading, and Su-Jin Kim and Keun-Sup Lee for technical assistance.
This work was supported by grants from the Ministry of Science and Technology of Korea (M1011100007-01A160000510) and from the Ministry of Information and Communication of Korea (IMT 2000 AIT-143).
REFERENCES
- 1.Amore, R., P. Kotter, C. Kuster, M. Ciriacy, and C. P. Hollenberg. 1991. Cloning and expression in Saccharomyces cerevisiae of the NAD(P)H-dependent xylose reductase-encoding gene (XYL1) from the xylose-assimilating yeast Pichia stipitis. Gene 109:89-97. [DOI] [PubMed] [Google Scholar]
- 2.Ansari, N. H., A. Bhatnagar, S. Lui, and S. K. Srivastava. 1991. Purification and characterization of aldose reductase and aldehyde reductase from human kidney. Biochem. Int. 25:755-765. [PubMed] [Google Scholar]
- 3.Billard, P., S. Menart, R. Fleer, and M. Bolotin-Fukuhara. 1995. Isolation and characterization of the gene encoding xylose reductase from Kluyveromyces lactis. Gene 162:93-97. [DOI] [PubMed] [Google Scholar]
- 4.Birken, S., and M. A. Pisano. 1976. Purification and properties of a polyol dehydrogenase from Cephalosporium chrysogenus. J. Bacteriol. 125:225-232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boghosian, R. A., and E. T. McGuinness. 1981. Quick brain aldose reductase: a kinetic study using the centrifugal fast analyzer. Int. J. Biochem. 13:909-914. [DOI] [PubMed] [Google Scholar]
- 6.Bolen, P. L., G. T. Hayman, and H. S. Shepherd. 1996. Sequence and analysis of an aldose (xylose) reductase gene from the xylose-fermenting yeast Pachysolen tannophilus. Yeast 12:1367-1375. [DOI] [PubMed] [Google Scholar]
- 7.Branlant, G. 1982. Properties of an aldose reductase from pig lens. Comparative studies of an aldehyde reductase from pig lens. Eur. J. Biochem. 129:99-104. [DOI] [PubMed] [Google Scholar]
- 8.Braun, M. L., and D. J. Niederpruem. 1969. Erythritol metabolism in wild-type and mutant strains of Schizophyllum commune. J. Bacteriol. 100:625-634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen, J., P. C. Turner, and H. H. Rees. 1999. Molecular cloning and characterization of hemolymph 3-dehydroecdysone 3β-reductase from the cotton leafworm, Spodoptera littoralis. A new member of the third superfamily of oxidoreductases. J. Biol. Chem. 274:10551-10556. [DOI] [PubMed] [Google Scholar]
- 10.Chen, J., T. J. Webb, R. Powls, and H. H. Rees. 1996. Purification and characterization of haemolymph 3-dehydroecdysone 3β-reductase in relation to ecdysteroid biosynthesis in the cotton leafworm Spodoptera littoralis. Eur. J. Biochem. 242:394-401. [DOI] [PubMed] [Google Scholar]
- 11.Cleland, W. W. 1970. Steady-state kinetics, p. 1-65. In P. D. Boyer (ed.), The enzymes, 3rd ed., vol. 2. Academic Press, Inc., New York, N.Y.
- 12.Cromlish, J. A., and T. G. Flynn. 1983. Purification and characterization of two aldose reductase isoenzymes from rabbit muscle. J. Biol. Chem. 258:3416-3424. [PubMed] [Google Scholar]
- 13.Das, B., and S. K. Srivastava. 1985. Purification and properties of aldose reductase and aldehyde reductase II from human erythrocyte. Arch. Biochem. Biophys. 238:670-679. [DOI] [PubMed] [Google Scholar]
- 14.Ditzelmuller, G., C. P. Kubicek, W. Wohrer, and M. Rohr. 1984. Xylose metabolism in Pachysolen tannophilus: purification and properties of xylose reductase. Can. J. Microbiol. 30:1330-1336. [Google Scholar]
- 15.Ernster, L. 1967. DT diaphorase, p. 309-317. In R. W. Estabrook and M. E. Pullman (ed.), Methods in enzymology, vol. 10. Academic Press, Inc., New York, N.Y.
- 16.Goossen, J., and H. Röper. 1994. Erythritol, a new sweetener. Confect. Prod. 24:182-188. [Google Scholar]
- 17.Griffin, B. W. 1992. Functional and structural relationships among aldose reductase, l-hexonate dehydrogenase (aldehyde reductase), and recently identified homologous proteins. Enzyme Microb. Technol. 14:690-695. [DOI] [PubMed] [Google Scholar]
- 18.Hermann, R. K., P. F. Kador, and J. H. Kinoshita. 1983. Rat lens aldose reductase: rapid purification and comparison with human placental aldose reductase. Exp. Eye Res. 37:467-474. [DOI] [PubMed] [Google Scholar]
- 19.Ishizuka, H., K. Tokuoka, T. Sasaki, and H. Taniguchi. 1992. Purification and some properties of an erythrose reductase from an Aureobasidium sp. mutant. Biosci. Biotechnol. Biochem. 56:941-945. [DOI] [PubMed] [Google Scholar]
- 20.Ishizuka, H., H. Wako, T. Kasumi, and T. Sasaki. 1989. Breeding of a mutant of Aureobasidium sp. with high erythritol production. J. Ferment. Bioeng. 68:310-314. [Google Scholar]
- 21.Jez, J. M., M. J. Bennett, B. P. Schlegel, M. Lewis, and T. M. Penning. 1997. Comparative anatomy of the aldo-keto reductase superfamily. Biochem. J. 326:625-636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Johnston, M., S. Andrews, R. Brinkman, J. Cooper, H. Ding, J. Dover, Z. Du, A. Favello, L. Fulton, and S. Gattung. 1994. Complete nucleotide sequence of Saccharomyces cerevisiae chromosome VIII. Science 265:2077-2082. [DOI] [PubMed] [Google Scholar]
- 23.Jornvall, H., B. Persson, M. Krook, S. Atrian, R. Gonzalez-Duarte, J. Jeffery, and D. Ghosh. 1995. Short-chain dehydrogenases/reductases (SDR). Biochemistry 34:6003-6013. [DOI] [PubMed] [Google Scholar]
- 24.Kuhn, A., C. van Zyl, A. van Tonder, and B. A. Prior. 1995. Purification and partial characterization of an aldo-keto reductase from Saccharomyces cerevisiae. Appl. Environ. Microbiol. 61:1580-1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680-685. [DOI] [PubMed] [Google Scholar]
- 26.Lee, H. 1998. The structure and function of yeast xylose (aldose) reductases. Yeast 14:977-984. [DOI] [PubMed] [Google Scholar]
- 27.Lee, J. K., B. S. Koo, and S. Y. Kim. 2002. Fumarate-mediated inhibition of erythrose reductase, a key enzyme for erythritol production by Torula corallina. Appl. Environ. Microbiol. 68:4534-4538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lewis, D. H., and D. C. Smith. 1967. Sugar alcohols (polyols) in fungi and green plants. I. Distribution, physiology and metabolism. New Phytol. 66:143-184. [Google Scholar]
- 29.Lowry, O. H., N. J. Rosebrough, A. L. Farr, and R. J. Randall. 1951. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 193:265-275. [PubMed] [Google Scholar]
- 30.Ma, H., K. Ratnam, and T. M. Penning. 2000. Mutation of nicotinamide pocket residues in rat liver 3α-hydroxysteroid dehydrogenase reveals different modes of cofactor binding. Biochemistry 39:102-109. [DOI] [PubMed] [Google Scholar]
- 31.Metzger, H., M. B. Shapiro, J. E. Mosimann, and J. E. Vinton. 1968. Assessment of compositional relatedness between proteins. Nature 219:1166-1168. [DOI] [PubMed] [Google Scholar]
- 32.Morjana, N. A., and T. G. Flynn. 1989. Aldose reductase from human psoas muscle. Purification, substrate specificity, immunological characterization, and effect of drugs and inhibitors. J. Biol. Chem. 264:2906-2911. [PubMed] [Google Scholar]
- 33.Morjana, N. A., C. Lyons, and T. G. Flynn. 1989. Aldose reductase from human psoas muscle. Affinity labeling of an active site lysine by pyridoxal 5′-diphospho-5′-adenosine. J. Biol. Chem. 264:2912-2919. [PubMed] [Google Scholar]
- 34.Mostad, S. B., and A. Glasfeld. 1993. Using high field NMR to determine dehydrogenase stereospecificity with respect to NADH. J. Chem. Educ. 70:504-506. [Google Scholar]
- 35.Munro, I. C., W. O. Bernt, J. F. Borzelleca, G. Flamm, B. S. Lynch, E. Kennepohl, E. A. Bar, and J. Modderman. 1998. Erythritol: an interpretive summary of biochemical, metabolic, toxicological and clinical data. Food Chem. Toxicol. 36:1139-1174. [DOI] [PubMed] [Google Scholar]
- 36.Neuhauser, W., D. Haltrich, K. D. Kulbe, and B. Nidetzky. 1997. NAD(P)H-dependent aldose reductase from the xylose-assimilating yeast Candida tenuis. Biochem. J. 326:683-692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.O'Farrel, P. H. 1975. High resolution 2-dimensional electrophoresis of proteins. J. Biol. Chem. 250:4007-4021. [PMC free article] [PubMed] [Google Scholar]
- 38.Petrash, J. M., T. Harter, I. Tarle, and D. Borhani. 1993. Kinetic alteration of human aldose reductase by mutagenesis of cysteine residues, p. 289-300. In H. Weiner, D. W. Crabb, and T. G. Flynn (ed.), Enzymology and molecular biology of carbonyl metabolism 4. Plenum Press, New York, N.Y. [DOI] [PubMed]
- 39.Pfeifer, V. F., V. E. Sohns, H. E. Conway, E. B. Lancaster, S. Dabic, Jr., and E. L. Griffin. 1960. Two-stage process for dialdehyde starch using electrolytic regeneration of periodic acid. Ind. Eng. Chem. 52:201-205. [Google Scholar]
- 40.Ratnam, K., H. Ma, and T. M. Penning. 1999. The arginine 276 anchor for NADP(H) dictates fluorescence kinetic transients in 3α-hydroxysteroid dehydrogenase, a representative aldo-keto reductase. Biochemistry 38:7856-7864. [DOI] [PubMed] [Google Scholar]
- 41.Rudolph, F. B. 1979. Product inhibition and abortive complex formation. Methods Enzymol. 63:411-436. [DOI] [PubMed] [Google Scholar]
- 42.Ryu, Y. W., C. Y. Park, J. B. Park, S. Y. Kim, and J. H. Seo. 2000. Optimization of erythritol production by Candida magnoliae in fed-batch culture. J. Ind. Microbiol. Biotechnol. 25:100-103. [Google Scholar]
- 43.Scrutton, N. S., A. Berry, and R. N. Perham. 1990. Redesign of the coenzyme specificity of a dehydrogenase by protein engineering. Nature 343:38-43. [DOI] [PubMed] [Google Scholar]
- 44.Seo, J. H., Y. W. Ryu, S. R. Jung, and S. Y. Kim. September 2001. Fermentation processes for preparing erythritol by a high salt tolerant mutant of Candida sp. U.S. patent 6,287,830 B1.
- 45.Stephen, B., and J. S. Robert. 1991. The enzyme determination of d-mannitol with mannitol dehydrogenase from Agaricus bisporus. Carbohydr. Res. 216:505-509. [Google Scholar]
- 46.Tokuoka, K., H. Ishizuka, K. Wako, and H. Taniguchi. 1992. Comparison of three forms of erythrose reductase from an Aureobasidium sp. mutant. J. Gen. Appl. Microbiol. 38:145-155. [DOI] [PubMed] [Google Scholar]
- 47.Turner, A. J., and T. G. Flynn. 1982. The nomenclature of aldehyde reductases, p. 401-402. In H. Weiner and B. Wermuth (ed.), Enzymology of carbonyl metabolism: aldehyde dehydrogenase and aldo-keto reductase. Alan R. Liss, Inc., New York, N.Y.
- 48.Veiga-Da-Cunha, M., P. Firme, M. V. San Romao, and H. Santosm. 1992. Application of 13C nuclear magnetic resonance to elucidate the unexpected biosynthesis of erythritol by Leuconostoc oenos. Appl. Environ. Microbiol. 58:2271-2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Verduyn, C., R. Van Kleef, J. Frank, H. Schreuder, J. P. van Dijken, and W. A. Scheffers. 1985. Properties of the NAD(P)H-dependent xylose reductase from the xylose-fermenting yeast Pichia stipiti. Biochem. J. 226:669-677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wada, M., M. Kataoka, H. Kawabata, Y. Yasohara, N. Kizaki, J. Hasegawa, and S. Shimizu. 1998. Purification and characterization of an aldehyde reductase from Candida magnoliae. Biosci. Biotechnol. Biochem. 62:280-285. [DOI] [PubMed] [Google Scholar]
- 51.Wada, M., H. Kawabata, M. Kataoka, Y. Yasohara, N. Kizaki, J. Hasegawa, and S. Shimizu. 1999. Purification and characterization of an aldehyde reductase from Candida magnoliae. J. Mol. Catal. B 6:333-339. [Google Scholar]
- 52.Wermuth, B. 1985. Aldo-keto reductases, p. 209-230. In T. G. Flynn and H. Weiner (ed.), Enzymology of carbonyl metabolism 2: aldehyde dehydrogenase, aldo-keto reductase and alcohol dehydrogenase. Alan R. Liss, Inc., New York, N.Y.
- 53.Wermuth, B., H. Burgisser, K. Bohren, G. Heinemann, and J. P. Von Wartburg. 1982. Purification and characterization of human-brain aldose reductase. Eur. J. Biochem. 127:279-284. [DOI] [PubMed] [Google Scholar]
- 54.Yokoyama, S., Y. Kinoshita, T. Suzuki, K. Kawai, H. Horitsu, and K. Takamizawa. 1995. Cloning and sequencing of two d-xylose reductase genes (xyrA and xyrB) from Candida tropicalis. J. Ferment. Bioeng. 80:603-605. [Google Scholar]
- 55.Yokoyama, S., T. Suzuki, K. Kawai, H. Horitsu, and K. Takamizawa. 1995. Purification, characterization and structure analysis of NADPH dependent d-xylose reductases from Candida tropicalis. J. Ferment. Bioeng. 79:217-223. [Google Scholar]