

Abstract
Formation of a specific contact between two residues of a polypeptide chain is an important elementary process in protein folding. Here we describe a method for studying contact formation between tryptophan and cysteine based on measurements of the lifetime of the tryptophan triplet state. With tryptophan at one end of a flexible peptide and cysteine at the other, the triplet decay rate is identical to the rate of quenching by cysteine. We show that this rate is also close to the diffusion-limited rate of contact formation. The length dependence of this end-to-end contact rate was studied in a series of Cys-(Ala-Gly-Gln)k-Trp peptides, with k varying from 1 to 6. The rate decreases from ∼1/(40 ns) for k = 1 to ∼1/(140 ns) for k = 6, approaching the length dependence expected for a random coil (n−3/2) for the longest peptides.
To understand how proteins fold, it is essential to know the time scales and mechanisms of its elementary processes (1). The formation of a contact between two residues in an unfolded polypeptide chain is one of the most fundamental of these processes and is central to many aspects of protein-folding mechanisms. Most helical segments in proteins are unstable in isolation (2, 3) and require interactions with residues in other parts of the chain to form stable folded structures. Similarly, interaction between side chains on opposite strands of an antiparallel β sheet are required for stability (4). Initiation of a parallel β sheet requires a contact between residues distant in sequence. Because of its central role in protein folding, it is important to develop kinetic methods to investigate contact formation. Here we describe a technique with nanosecond time resolution and apply it to the investigation of the end-to-end contact formation rate in a flexible peptide.
An initial glimpse of the kinetics of contact formation was obtained in a study of unfolded cytochrome c (5–7). In these experiments, photodissociation of carbon monoxide from the heme with a nanosecond laser pulse was used to initiate an intramolecular ligand-binding process. CO dissociation opens up a binding site at the heme iron, permitting binding kinetics to be measured by time-resolved absorption spectroscopy. This study showed that intramolecular methionine binding is very close to diffusion limited. The measured binding rate of ∼1/(40 μs) is therefore also the rate of forming a contact, in this case between positions separated by ∼50 residues in the sequence. An obvious limitation of this technique, however, is that it is restricted to very few proteins (a subset of heme proteins). A more generic method for measuring contact formation was clearly needed.
One such method has been introduced recently by Bieri et al. (8), who attached thioxanthone at one end of a flexible peptide and naphthalene near the other. On optical excitation, the triplet state of thioxanthone forms and transfers its excitation energy to naphthalene on close contact in an apparently diffusion-limited process. The rate at which the thioxanthone triplet state decays is therefore the rate of contact formation. This method has two important limitations. First, with the current probes, positioning of the triplet states for efficient energy transfer requires the use of ethanol/water mixtures. Second, extrinsic probes are used. A method that takes advantage of properties of naturally occurring amino acids would be more useful. It would simplify investigations of contact formation in protein folding as well as allow exploitation of the power of site-directed mutagenesis.
We have developed a method to measure the rate of contact formation that utilizes unique but relatively unfamiliar properties of two amino acids—the long lifetime of the triplet state of tryptophan and its quenching by cysteine. The basic idea of the experiment is that cysteine is such an efficient quencher that the rate of forming a contact between the side chains of tryptophan and cysteine in a polypeptide is very close to the measured decay rate of the tryptophan triplet state (Fig. 1). In the absence of any quenchers, this triplet state lives for more than ∼40 μs (9–11), providing considerable dynamic range for investigating contact formation rates. The unique quenching property of cysteine was demonstrated in an important series of experiments by Gonnelli and Strambini (12), who used phosphorescence lifetime measurements to determine the bimolecular quenching rates of the tryptophan triplet by all the amino acids, except glycine and alanine. They showed that at neutral pH, cysteine is by far the most efficient quencher, with a rate of 5 × 108 M−1s−1. Except for tryptophan itself, all other amino acids quench with rates that are more than ∼400-fold smaller (the tyrosine rate).
Figure 1.

Determination of the rate of contact formation between tryptophan and cysteine at the ends of a flexible peptide. Pulsed optical excitation leads to population of the lowest excited triplet state of tryptophan. Tryptophan can then contact cysteine in a diffusion-limited process with rate kD+uni. During the lifetime of this contact pair, quenching of the triplet occurs with a probability [φ = q/(q + kD−uni)], determined by the quenching rate q and rate of the diffusive dissociation of the contact pair kD−uni. The observed quenching rate is given by kD+uniφ. Therefore, if the probability of quenching at contact, φ, can be determined, the contact formation rate, kD+uni, can be determined from the observed rate.
To demonstrate the utility of this method, we have investigated one of the simplest dynamical properties of a polypeptide chain—the rate of end-to-end contact formation. We have used optical (triplet→triplet) absorption (9, 11, 13, 14) to measure the lifetime of the excited triplet state of tryptophan at the C terminus of a flexible peptide with either cysteine, cystine, or lipoate at the N terminus as quencher. Lipoate is a cyclic disulfide and is known to be a near diffusion-limited quencher of the tryptophan triplet state (11). The peptide separating tryptophan and quencher consists of multiples of the sequence Ala-Gly-Gln, which was chosen because it does not contain charged side chains, and peptides with this composition form no secondary structure. The experiments show that quenching by cysteine is much closer to the diffusion limit for the intramolecular (unimolecular) process than for the bimolecular process. A simple kinetic model explains this as resulting from a smaller rate for diffusive dissociation of the tryptophan/quencher contact pair when they are attached to the same molecule compared to this dissociation rate when tryptophan and quencher are on different molecules. The length dependence of the end-to-end contact rate was also studied in a series of Cys-(Ala-Gly-Gln)k-Trp peptides, with k varying from 1 to 6. We compare these results to a simple statistical mechanical theory of Szabo, Schulten, and Schulten (SSS) (15) on the end-to-end contact rates for idealized polymer chains.
Materials and Methods
N-acetyl tryptophan amide (NATA), l-tryptophan, and lipoic acid (also known as thioctic acid) were obtained from Sigma. Peptides were synthesized by California Peptide Research (Napa, CA) and were greater than 95% pure. The peptides are listed in Table 1. CD measurements showed no evidence for any secondary structure formation. All experiments were carried out at room temperature on aqueous solutions containing 100 mM potassium phosphate buffer at pH 7.0. Samples were deoxygenated to prevent quenching by O2 (16), to minimize photodamage (17), and to prevent disulfide crosslinking of the peptides by oxidation of the cysteine thiol groups. N2O was used for deoxygenation because it is an efficient scavenger of hydrated electrons produced by tryptophan excitation (11). Measurements of tryptophan decay kinetics were found to be the same with and without the addition of an oxygen-consuming enzyme system (16), indicating that deoxygenation was sufficient. Also, disulfide formation was minimal (<5%), as measured by optical absorption in the 220- to 300-nm region of the cysteine undecapeptide (peptide c of Table 1) in the absence and presence of dithiothreotol (18).
Table 1.
Peptide sequences
| Name | Sequence* |
|---|---|
| w† | Gln-(Ala-Gly-Gln)3-Trp |
| c | Cys-(Ala-Gly-Gln)3 |
| cc | Cys(Cys)-(Ala-Gly-Gln) |
| l | Lipoate-(Ala-Gly-Gln)3 |
| cw | Cys-(Ala-Gly-Gln)3-Trp |
| ccw | Cys(Cys)-(Ala-Gly-Gln)3-Trp |
| lw | Lipoate-(Ala-Gly-Gln)3-Trp |
| p | Lipoate-(Pro)9-Trp |
| a | Lipoate-(Ala)2-Arg-(Ala)4-Arg-Ala-Trp |
All peptides were amidated at the C terminus.
Peptide w was acetylated at the N terminus.
The decay of the tryptophan triplet state was monitored by triplet→triplet absorption measurements after pulsed excitation. Absorption was chosen over phosphorescence because a single instrument could be constructed to measure the decay over a wide temporal range. The instrument was designed to have a collinear geometry. This geometry permits the use of cuvettes with pathlengths that vary from 1 mm to 10 cm, which are necessary for determining the concentration dependence of the quenching rate. To excite tryptophan, the 266-nm fourth harmonic of a Continuum (Santa Clara, CA) Surelite II Nd∶YAG laser was used to pump a Lambda Physik (Acton, MA) UV Star Ce∶LiCAF laser, producing a ∼1-mJ, ∼8-ns pulse at 290 nm [initial experiments with 266 nm Nd∶YAG excitation confirmed earlier results that the rate of tryptophan photodestruction increases considerably at shorter excitation wavelengths (19)]. A continuous wave probe beam was produced by a Lexel (Fremont, CA) Model 95 argon ion laser, which could be tuned to any of 8 wavelengths between 457 nm and 514 nm. The probe beam was split to generate a reference beam and a sample beam that passed collinearly through the cuvette with the pump beam. The intensity of each beam was measured with a photodiode (NewFocus, Santa Clara, CA, Model 1621), recorded with a digital oscilloscope (Hewlett–Packard 54420A or Tektronix TDS 3032), and stored on a computer. To eliminate high-frequency cable noise and leakage of the pump beam, background signals measured in the absence of the probe beam were subtracted from the measured sample and reference signals (in the presence of the probe beam). An apparent optical density was calculated as the logarithmic ratio of the corrected sample and reference intensities, thereby removing low-frequency intensity drift in the probe laser. Finally, the change in optical density was calculated by subtracting the apparent optical density at negative time.
To reduce the uncertainty in the determination of the decay rate, the signal was averaged for 256 laser shots. In a typical experiment, between three and five averages were fitted. The scatter in the rates from these fits was typically larger than the uncertainty from each fit, so the quoted uncertainty is the standard deviation from the mean. No more than five sets were collected to limit the exposure of the sample to UV radiation, which causes irreversible photodamage. At the end of a typical experiment, the absorption spectrum indicated that less than 10–20% had been photochemically transformed.
Results
Identification of Triplet Decay Kinetics.
Optical excitation of tryptophan transiently populates both singlet and triplet states. The singlet state decays in a few nanoseconds and does not influence measurement of triplet state lifetimes, which are much longer. However, excitation also generates additional photoproducts that can be problematic (11). The production of hydrated electrons and neutral radicals interferes with absorption measurements of triplet-state lifetimes. The radical absorbs at wavelengths that overlap the triplet→triplet absorption (11). Combination of the radical with the hydrated electrons is presumably responsible for a significant spectral amplitude at ∼3 μs, as this amplitude was almost completely eliminated by degassing the solutions with the electron scavenger, N2O (20). Radical/radical recombination also occurs, but its rate is much slower (∼3 × 108 M−1 s−1) (21). At the estimated concentrations of radicals in our experiments (1 μm–10 μm), this process occurs with a relaxation rate of 300–3,000 s−1, much slower than the tryptophan decay rates (see below).
Fig. 2 shows a representative trace of the optical density change at 457 nm after 290 nm excitation of peptide w (Table 1), which contains no quencher. Only two processes are observed—an exponential process with a decay time of ∼40 μs followed by a slower process with a much smaller amplitude. The 40-μs process was identified as the triplet-state decay by measuring its amplitude as a function of the eight wavelengths available with the argon ion laser. A comparison of these amplitudes with the triplet→triplet absorption spectrum reported by Bent and Hayon (11) for tryptophan (Fig. 2 Inset) confirms that the species decaying at 40 μs is indeed the triplet state. A similar transient spectrum has been reported by Sudhakar et al. (9, 22). The amplitude of the slow process was too small for accurate wavelength-dependent measurements, and we tentatively attribute it to recombination of the neutral radicals.
Figure 2.

Decay of tryptophan triplet→triplet absorption after nanosecond excitation. Optical density measured at 457 nm is plotted vs. the logarithm of time after excitation of the tryptophan to the triplet state. Data are shown for peptides with and without cysteine as a quencher (peptides w and cw, Table 1). The smooth curves are fits to the data with an exponential followed by a single second-order decay for peptide w [y = a + b exp(−ct) + d/(1 + d*e*t)] and an exponential followed by two second-order decays for peptide cw (y = a + b exp(−ct) + d/(1 + d*e*t) + f/(1 + f*g*t)]. (Inset): Comparison of triplet→triplet absorption spectrum (scaled) reported by Bent and Hayon (dashed line) for tryptophan with the amplitudes of the exponential decays for peptide w (circles).
The measured lifetime of 40 μs in the absence of quencher is very close to that found for NATA at the same concentration (33 μs at 15 μM). For tryptophan at 70 μM, a 20-μs lifetime was observed. These values are very similar to those previously reported from triplet→triplet absorption measurements, i.e., 20 μs for tryptophan at 150 μM (11) and 30 μs for NATA at 50 μM (9). A considerably longer lifetime was obtained by Gonnelli and Strambini [∼1 ms for 6 μM NATA (10)] from the decay of phosphorescence. They argued that the longer lifetime results from removal of (unidentified) quenching impurities in commercial samples.
Quenching Rates.
Our results on quenching rates are summarized in Table 2. To interpret intramolecular quenching experiments in terms of contact formation, it is necessary to perform bimolecular quenching experiments for two reasons. One is to establish how the rate of intramolecular quenching is related to the rate of contact formation. The second is to determine the contribution from the bimolecular process of a quencher on one peptide quenching tryptophan on another. Bent and Hayon showed that the cyclic disulfide, lipoate, is an extremely efficient quencher of the tryptophan triplet (11). We measured the rate of the triplet decay of free tryptophan as a function of (excess) lipoate concentration, yielding a bimolecular quenching rate of 3.0 (±0.1) × 109 M−1 s−1, very close to the value of 3.6 (±0.4) × 109 M−1 s−1 reported by Bent and Hayon (11).* Lipoate also quenches with a very high rate when attached to a peptide. The bimolecular rate for quenching of tryptophan on peptide w by lipoate on peptide l is 1.4 × 109 M−1 s−1 (Tables 1 and 2).
Table 2.
Observed quenching rates*
| Bimolecular rates, M−1 s−1 | |
| w + w (tryptophan) | 2.1 (±0.2) × 108 |
| w + c (cysteine) | 1.3 (±0.1) × 108 |
| w + cc (cystine) | 1.9 (±0.1) × 108 |
| w + l (lipoate) | 1.4 (±0.1) × 109 |
| Unimolecular rates, s−1 | |
| w | 2.3 (±0.1) × 104 |
| cw (cysteine) | 9.1 (±0.1) × 106 |
| ccw (cystine) | 1.1 (±0.1) × 107 |
| lw (lipoate) | 1.7 (±0.1) × 107 |
| p (lipoate) | 6.2 (±0.5) × 104 |
| a (lipoate) | 2.4 (±0.1) × 107 |
See Table 1 for peptide sequences. Quencher indicated in parentheses.
Experiments on quenching peptide w by the cysteine-containing peptide c (Table 1) yield a bimolecular quenching rate ∼10-fold smaller. Quenching by the cystine-containing peptide cc was also studied. Finally, the bimolecular rate of tryptophan triplet quenching by (ground state) tryptophan itself was studied, yielding a bimolecular rate slightly higher than that for either cysteine or cystine (Table 2). This rate is ∼10-fold higher than the value reported by Gonnelli and Strambini (10, 12) and may result from impurity quenching. Fortunately, however, even the higher bimolecular rate does not significantly influence our intramolecular quenching experiments.
Intramolecular quenching rates were measured for peptides with tryptophan at the C terminus and either cysteine, cystine, or lipoate at the N terminus, i.e., peptides cw, ccw, and lw (Table 1). Fig. 2 shows the decay kinetics for the cysteine-containing peptide cw. There is a dramatic increase in the triplet decay rate, from ∼104 s−1 for the peptide without quencher (peptide w) to ∼107 s−1 for peptides containing any of the three quenchers. Because these rates are so high, there is very little contribution from bimolecular quenching at the concentrations of the measurements (10–100 μM). At the highest concentration of 100 μM, for example, bimolecular quenching increases the rate only by about 1% compared to the value measured at 10 μM. In Table 2, we therefore report a simple average of the unimolecular rates obtained at all concentrations.
The intramolecular quenching rate as a function of chain length was studied in the series of peptides, Cys-(Gln-Gly-Ala)k-Trp, with k = 1–6. Fig. 3 shows that the rate decreases with increasing chain length. Finally, to begin to explore the effect of sequence on contact formation, we measured the decay rates for two other sequences of the same length (Table 1). In the alanine peptide a contact formation is slightly faster than in peptide lw. In contrast, the decay rate for the proline peptide p is much slower (see Table 2).
Figure 3.

Dependence of the intramolecular quenching rate on peptide length. The plotted values were obtained by dividing the observed rate of tryptophan triplet quenching by cysteine by the probability factor φ = q/(q + kD−uni) = 0.5 (Eq. 3). The solid line is the function k ∝ n−3/2 scaled to the observed rate for the longest peptide, n = 19.
Discussion
Our experiments demonstrate that there is a dramatically decreased lifetime of the triplet state of tryptophan at the C terminus of a flexible peptide when cysteine is attached at the N terminus. Without cysteine, the lifetime is ∼40 μs (peptide w). For the peptide with tryptophan and cysteine separated by 10 peptide bonds, the lifetime is reduced ∼400-fold to ∼100 ns (peptide cw). We interpret the markedly reduced lifetime as resulting from quenching on formation of a contact between cysteine and tryptophan at the ends of the peptide. This interpretation is supported by a number of previously published results on the short-range nature of tryptophan triplet state quenching (16, 12, 23, 24) and is also supported by our finding on the proline peptide. When all of the amino acids between tryptophan and the quencher are replaced by proline, the lifetime is reduced less than 3-fold (peptide p, Table 2). Polyproline is known to be helical (25, 26), providing a rod-like spacer of ∼3 nm between tryptophan and quencher. Our results are also consistent with those of Bieri et al. (8), who measured the rate of triplet energy transfer between thioxanthone and naphthalene in peptides containing multiples of Ser-Gly pairs.†
Determination of Contact Formation Rate from Triplet Decay Rate.
The quantity of interest in this study is the rate of contact formation between tryptophan and cysteine at the ends of the peptide. To establish this rate, we studied both the bimolecular and unimolecular quenching rates of the tryptophan triplet state. The disulfide lipoate was used in this study because it was known from previous work that its bimolecular quenching rate of 3 × 109 M−1 s−1 is near the expected diffusion limit (11). In the simplest picture of a diffusion-limited process, due to Smoluchowski, reaction occurs only when the reacting pair come to within a center-to-center distance a. For distances less than or equal to a, reaction is instantaneous and irreversible, whereas at distances larger than a, the reaction rate is zero. The Smoluchowski rate is given by kD+bi = 4πDbia, where Dbi is the sum of the diffusion coefficients of the reactants. In real systems, there is a distance dependence to this rate, and a is therefore a less well-defined quantity. Zhou and Szabo have developed a comprehensive theory for diffusion-influenced reactions, which requires knowing the diffusion coefficients of the reactants, the distance dependence of the reaction rate, and the absolute reaction rate at some specified distance (27). The latter two quantities are not yet known for quenching of tryptophan. The mechanism of quenching, moreover, is not completely understood, but in the case of disulfides is thought to occur via electron transfer from the tryptophan triplet state to the sulfur atoms of the quencher (11, 12, 23, 24, 28).‡ Also, the quencher and tryptophan at the ends of a peptide cannot access each other at all angles because of excluded volume effects, so that the factor of 4 in the Smoluchowski equation represents a maximum value. Given these uncertainties, we must adopt the admittedly naive picture of assuming isotropic quenching at a contact distance a and no quenching at longer distances.
Assessing whether a bimolecular reaction is purely diffusion limited in this simple picture therefore requires knowing only the diffusion constants of the reactants and the distance a. The simplest assumption is that a is the sum of the van der Waals radii, which is ∼0.4 nm. This choice may compensate for a finite quenching (e.g., electron-transfer) rate at distances larger than a, which would make the rate higher than the Smoluchowski kD+bi and excluded volume effects, which would produce a rate lower than kD+bi. To obtain diffusion constants, we use the empirical formula obtained by fitting to the data compiled by Creighton (29), D0bi = 10−7 exp(α − β ln m) cm2 s−1, where m is the molecular weight, α = 6.42 ± 0.06, and β = 0.42 ± 0.01. The diffusion coefficients are estimated to be 6.6 × 10−6 cm2 s−1 for both lipoate and tryptophan and 3.3 × 10−6 cm2 s−1 for the peptides. With these values, the Smoluchowski equation yields a rate of 4.0 × 109 M−1 s−1 for free tryptophan and lipoate and 2.0 × 109 M−1 s−1 for the peptides (w + l), both very close to the measured values of 3.0 × 109 M−1 s−1 and 1.4 × 109 M−1 s−1, respectively. A second indication that the quenching by lipoate is near the diffusion limit is that the 2-fold slower rate for the peptides can be attributed entirely to the 2-fold smaller diffusion coefficient.
When lipoate is replaced by cysteine or cystine (peptides c and cc, Table 1), the bimolecular rates are about an order of magnitude smaller (Table 2). In contrast, the unimolecular rates for quencher and tryptophan on the same peptide (peptides cw, lw, and ccw) are all the same to within a factor of 2. To explain this result, we use a simple kinetic model that shows how the observed unimolecular and bimolecular rates can be combined to yield the intramolecular contact formation and dissociation rates.
The observed triplet decay rate, kobs, can be written in general as:
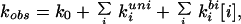 |
1 |
where k0 is the rate in the absence of quencher, kiuni and kibi are unimolecular and bimolecular quenching rates for quencher i, and [i] is the quencher concentration. For peptides lw, cw, and ccw, there is effectively only a single quencher, i (lipoate, cysteine, or cystine). Also, kobs ∼ kiuni, because kiuni is almost three orders of magnitude faster than the rate in the absence of quencher (k0), and because the bimolecular rates do not contribute significantly at the low concentrations used in these experiments (Table 2). For the overall quenching reaction, we assume the simple mechanism analogous to that described by Hagen et al. (6, 7) and used earlier by Wang and Davidson for cyclization of DNA (30, 31), in which both unimolecular and bimolecular reactions are treated as two-step processes (Fig. 1). The first step is a diffusional process that brings the quencher within a distance a of the excited tryptophan to form what is often called the “encounter complex” or “contact pair.” In the second step, the quencher can either remove the tryptophan triplet excitation energy (e.g., by electron transfer) or diffuse away. The overall rates of unimolecular and bimolecular reaction then are the product of the diffusion-limited rate of contact formation (kD+) and the probability that quenching occurs during the lifetime of the contact pair, i.e.:
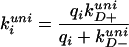 |
2 |
and
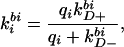 |
3 |
where kD− is the rate at which the tryptophan/quencher contact pair dissociates, and qi is its quenching rate at contact. Eqs. 2 and 3 show that for a diffusion-limited reaction, kD− ≪ qi. If we assume that qi is the same in the unimolecular and bimolecular processes, then Eqs. 2 and 3 can be combined to give:
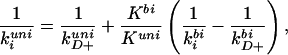 |
4 |
where K ≡ kD+/kD− is the equilibrium constant for forming the contact pair. In the bimolecular case, the equilibrium constant for forming the contact pair is simply the volume defined by the contact distance a, i.e., Kbi ≡ kD+bi/kD−bi = (4/3)πa3. Kuni will be discussed below.
Eq. 4 shows that by assuming the same value for the contact distance a, of 0.4 nm for all three quenchers, Kbi/Kuni and kD+uni can be obtained as the slope and intercept of a plot of the measured unimolecular and bimolecular rates. The plot shown in Fig. 4 is indeed linear and gives Kbi/Kuni = 7 ± 1 (M−1) and 1/kD+uni = 60 ± 1 ns. A value of kD−uni ≈7 × 108 s−1 is then obtained from (4/3)πa3/(kD+uni/kD−uni) = 7. The corresponding bimolecular rate is given by kD−bi = kD+bi/Kbi = 3Dbi/a2 ≈1 × 1010 s−1. The values of qi for lipoate and cysteine are then calculated from Eqs. 2 or 3 to be ≳3 × 1010 s−1 and ∼8 × 108 s−1.
Figure 4.

Unimolecular vs. bimolecular decay rates plotted according to Eq. 4. The observed triplet lifetimes (1/kobs = 1/kiuni) for peptides cw, ccw, and lw (Table 1) are plotted vs. the difference between the reciprocal of the observed bimolecular rates (1/kibi) and the reciprocal diffusion-limited bimolecular rate (1/kD+bi = 1/(2.0 × 109 M−1 s−1), assumed to be the same for all three quenchers. A least-squares fit yields a slope of Kbi/Kuni = 7 ± 1 (M−1) and an intercept of 1/kD+uni = 60 ± 1 ns (Eq. 5).
The net result of this analysis is that intramolecular quenching by lipoate is diffusion limited (qlipoate/(qlipoate + kD−uni) ≈1), and the observed rate is the rate of forming the tryptophan/lipoate contact pair. Intramolecular quenching by cysteine is near diffusion limited, and the contact formation rate can be obtained from the observed rate by dividing by the probability factor: qcysteine/(qcysteine + kD−uni) ≈0.5. Even though qcysteine, the quenching rate at contact for cysteine, is more than ∼35-fold smaller than qlipoate, the dissociation of the contact pair (kD−uni) is sufficiently slow that the observed rate of intramolecular quenching by cysteine in peptide cw is less than a factor of 2 smaller than the rate of contact formation. In the bimolecular case, there is a much larger difference between the overall quenching rates of lipoate and cysteine, because the dissociation rate (kD−bi) is much faster. It is possible that a stabilizing van der Waals interaction between cysteine and tryptophan could slow the dissociation of the intramolecular contact pair. However, there is no obvious reason why a barrier to dissociation of the intramolecular pair would be larger than for an intermolecular pair. We therefore assume that the slower dissociation rate of the intramolecular contact pair represents a dynamical property of the polypeptide chain.
As a framework for explaining this result, we use the first passage time theory of SSS (15), which is the simplest description of end-to-end polymer contact dynamics. In SSS theory, the contact rate is obtained by solving a diffusion equation for an idealized chain having a Gaussian end-to-end distribution, viewed as radial diffusion of one end relative to the other in a purely entropic harmonic potential. For a contact distance a, which is small compared to the length of the chain segment, the SSS rate is given by (15):
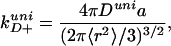 |
5 |
where 〈r2〉 is the equilibrium mean-squared end-to-end distance, and Duni is the relative diffusion coefficient between the ends of the chain. Because the unimolecular equilibrium constant Kuni = v/(2π〈r2〉/3)3/2 (31, 32), where v = (4/3)πa3, the dissociation rate kD−uni has the same form as the corresponding rate in the bimolecular case (kD−bi), that is kD−uni = 3Duni/a2. According to this theory, the ∼16-fold difference between the two dissociation rates therefore comes entirely from the difference in the diffusion constants Duni (≈4 × 10−7 cm2 s−1), and Dbi (= 6.6 × 10−6 cm2 s−1). Similar values have been reported previously. Hagen et al. used SSS theory to estimate Duni = 4–7 × 10−7 cm2 s−1 for cytochrome c in 5.6 M guanidinium chloride at 40°C (6, 7). In a fluorescence resonance energy transfer study on ribonuclease in 6.7 M GdnHCl at 22°C, Buckler et al. obtained a relative diffusion constant of 2–8 × 10−7 cm2 s−1 between donor and acceptor separated by 21-residues (33).
Why is Duni less than Dbi? SSS theory ignores intrachain interactions, excluded volume, chain stiffness, and hydrodynamic interaction (15, 34). For an idealized chain, Duni is the sum of the diffusion coefficients of the monomers in the polymer–tryptophan and cysteine in our case (34). However, in a real polymer, intrachain interactions, e.g., transient hydrogen bonds between peptide groups, could produce kinetic barriers. This effect may have little or no influence on the bimolecular process but would yield a lower value for the diffusion coefficient Duni calculated from SSS theory. Intrachain interactions can be viewed as introducing “roughness” to the entropic harmonic potential of SSS theory. Zwanzig has shown that for diffusion in one dimension, roughness reduces the diffusion coefficient by a factor: exp[−(ɛ/kBT)2], where ɛ is the root-mean-squared height of the energy barriers having a Gaussian distribution (35). A reduction of a factor of 16 could therefore result from ɛ of only 1.7 kBT.
Dependence of End-to-End Contact Rate on Peptide Length.
An important part of the physical description of any polymer property is how it depends on the chain length. We therefore investigated the quenching rate of tryptophan by cysteine as a function of the number of intervening residues. For this study, we used the same Ala-Gly-Gln triplet in the series: Cys-(Ala-Gly-Gln)k-Trp, with k varying from 1 to 6, corresponding to 4 to 19 peptide bonds. If we make the SSS assumption that the contact dissociation rate is independent of chain length, the contact formation rate at all chain lengths can be obtained by simply dividing the observed rate by the probability factor: qcysteine/(qcysteine + kD−uni) = 0.5. These rates are shown in Fig. 3. As expected, the rates decrease with increasing chain length from 2.7 × 107 to 7.2 × 106 s−1. Note that the slope of the log–log plot is not constant but increases with increasing chain length. Such curvature was not observed by Bieri et al. (8), who varied the number of peptide bonds from 3 to 9 by changing the number of serine/glycine pairs between thioxanthone and naphthalene.
In SSS theory, the rate for a Gaussian chain depends on the mean-squared end-to-end distance, 〈r2〉. For a chain of n peptide bonds, 〈r2〉 = Cnn2vl2, where l is the distance between consecutive Cα atoms of the peptide (0.38 nm), Cn is the Flory characteristic ratio (36), and v is an exponent that is determined by the relative strengths of chain-solvent interactions and intrachain interactions. [For n = 10, we have determined 〈r2〉 = 2.4 nm2 from Kbi/Kuni = (2π〈r2〉/3)3/2), known as the Jacobsen–Stockmayer factor (31)]. For a “good solvent” in which chain-solvent interactions are favored over intrachain interactions, the polymer is more expanded, and v ≈ 3/5. In a so-called “θ solvent,” intrachain interactions and chain-solvent interactions are balanced, and v = 1/2 (37). SSS theory (Eq. 5) predicts that in this case the end-to-end contact formation rate, kD+uni, scales as n−3v = n−3/2. The results in Fig. 3 show that at the longest chain lengths, the dependence approaches k ∝ n−3/2 but depends less on n for shorter peptides. One possible explanation is that for shorter chains, the persistence length becomes comparable to the length of the chain, and the Gaussian chain approximation breaks down. If this is true, using Eq. 5 to calculate the unimolecular diffusion coefficient is not strictly correct. However, because the intramolecular quenching rate for n = 10 (peptide cw) does not deviate significantly from the extrapolated value for the longest peptide, we expect Duni to be approximately correct. Thirumalai and coworkers have considered the effect of stiffness in a closely related problem—the rate of interior loop formation—and have derived an expression that predicts that for a generic peptide the rate will exhibit a sharp maximum at 10 peptide bonds (38–40). The absence of such a maximum for our peptide presumably reflects a greater flexibility because of the high density of glycines.
Finally, we should compare our results with those of Hagen et al. on heme–methionine binding in unfolded cytochrome c (6, 7). They determined a contact rate of ∼2.5 × 104 s−1 between positions separated by ∼50 peptide bonds. These experiments were performed at denaturant concentrations producing θ solvent conditions (v = 1/2). Hagen et al. therefore used a length scaling of n−3/2 in their estimate of the rate of ∼3 × 105 s−1 for contacting ends separated by 10 peptide bonds (6, 7) compared to our rate of 1.7 × 107 s−1 and the rate of Bieri et al. of 1.4 × 107 for 9 peptide bonds. Several factors account for this difference. This is most easily shown by rescaling our rate using Eq. 5 and 〈r2〉 = Cnn2vl2, with the Hagen et al. values for Duni ≈ 5 × 10−7 cm2 s−1, a = 0.15 nm, Cn = 8, v = ½ (Cn ≈ 1.7 for our peptide), and a factor of two introduced because methionine can access only one side of the heme. All of these factors slow the rate. The rescaled rate of ∼4 × 105 s−1 is essentially identical to the value predicted by Hagen et al. These factors were not taken into account by Bieri et al. (8) when they suggested that their measured rates were inconsistent with the rate estimated by Hagen et al. (6).
Conclusion
Our results and analysis show that measurements of the decay rate of the triplet state of tryptophan by excited-state optical absorption measurements can accurately determine the rate of forming an intermolecular contact with cysteine. Because cysteine quenches at a rate that is so much faster than that of any other amino acid, measuring the triplet decay rate should provide a useful method for investigating the kinetics of forming a specific intermolecular contact in an unfolded or folding protein. We have assumed that quenching occurs only at van der Waals contact, and a remaining issue is a precise determination of the mean contact distance associated with the measured rate.
Our study also shows that the technique can be used to address fundamental issues in polypeptide dynamics. It will be important in future studies to determine how the mean-squared end-to-end distance, contact rate, and effective end-to-end diffusion constant depend on peptide length, composition, sequence, and solvent conditions. We hope that such experimental studies will stimulate further theoretical development in this area. The present work, moreover, shows that end-to-end contact formation in flexible peptides occurs on a time scale accessible to all-atom molecular dynamics calculations and should therefore provide an experimental benchmark for simulations on polypeptides.
Acknowledgments
We thank Attila Szabo, Robert Zwanzig, John Louis, Victor Muñoz, and Jane Vanderkooi for helpful discussions.
Abbreviations
- SSS
Szabo, Schulten, and Schulten
- NATA
N-acetyl tryptophan amide
Footnotes
Free tryptophan also exhibits a 30-ns relaxation of comparable amplitude to the 40-μs triplet decay, which was also observed by Bent and Hayon. There is, however, no evidence of this relaxation in NATA or in the peptides. This is consistent with the observation of Bent and Hayon that the 30-ns relaxation is not present unless an amino group is present in its protonated form (11). There is, as yet, no convincing explanation for these observations.
Volk et al. (41) measured the recombination of thiyl radicals at the ends of a helix-forming alanine peptide, which were produced by subpicosecond photolysis of an aryl disulfide. They found recombination occurring from picoseconds to microseconds with a power law time course. However, in these experiments the initial condition is a contact pair, in contrast to the equilibrium end-to-end distribution in our experiments and in those of Bieri et al.
References
- 1.Eaton W A, Muñoz V, Hagen S J, Jas G S, Lapidus L J, Henry E R, Hofrichter J. Annu Rev Biophys Biomol Struct. 2000;29:327–359. doi: 10.1146/annurev.biophys.29.1.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chakrabartty A, Baldwin R L. Adv Protein Chem. 1995;46:141–176. [PubMed] [Google Scholar]
- 3.Muñoz V, Serrano L. Curr Opin Biotechnol. 1995;6:382–386. doi: 10.1016/0958-1669(95)80066-2. [DOI] [PubMed] [Google Scholar]
- 4.Gellman S H. Curr Opin Struct Biol. 1998;2:717–725. doi: 10.1016/s1367-5931(98)80109-9. [DOI] [PubMed] [Google Scholar]
- 5.Jones C M, Henry E R, Hu Y, Chan C-K, Luck S D, Bhuyan A, Roder H, Hofrichter J, Eaton W A. Proc Natl Acad Sci USA. 1993;90:11860–11864. doi: 10.1073/pnas.90.24.11860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hagen S J, Hofrichter J, Szabo A, Eaton W A. Proc Natl Acad Sci USA. 1996;93:11615–11617. doi: 10.1073/pnas.93.21.11615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hagen S J, Hofrichter J, Eaton W A. J Phys Chem. 1996;101:2352–2365. [Google Scholar]
- 8.Bieri O, Wirz J, Hellrung B, Schutkowski M, Drewello M, Kiefhaber T. Proc Natl Acad Sci USA. 1999;96:9597–9601. doi: 10.1073/pnas.96.17.9597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sudhakar K, Phillips C M, Owen C S, Vanderkooi J M. Biochemistry. 1995;34:1355–1363. doi: 10.1021/bi00004a030. [DOI] [PubMed] [Google Scholar]
- 10.Strambini G B, Gonnelli M. J Am Chem Soc. 1995;117:7646–7651. [Google Scholar]
- 11.Bent D V, Hayon E. J Am Chem Soc. 1975;97:2612–2619. doi: 10.1021/ja00843a004. [DOI] [PubMed] [Google Scholar]
- 12.Gonnelli M, Strambini G B. Biochemistry. 1995;34:13847–13857. doi: 10.1021/bi00042a017. [DOI] [PubMed] [Google Scholar]
- 13.Volkert W A, Kuntz R R, Ghiron C A, Evans R F, Santus R, Bazin M. Photochem Photobiol. 1977;26:3–9. [Google Scholar]
- 14.Gershenson A, Gafni A, Steel D. Photochem Photobiol. 1998;67:391–398. [PubMed] [Google Scholar]
- 15.Szabo A, Schulten K, Schulten Z. J Chem Phys. 1980;72:4350–4357. [Google Scholar]
- 16.Calhoun D B, Englander W S, Wright W W, Vanderkooi J M. Biochemistry. 1988;27:8466–8474. doi: 10.1021/bi00422a026. [DOI] [PubMed] [Google Scholar]
- 17.Davidson R S. J Photochem Photobiol B. 1996;33:3–25. [Google Scholar]
- 18.Cleland W W. Biochemistry. 1964;3:480–482. doi: 10.1021/bi00892a002. [DOI] [PubMed] [Google Scholar]
- 19.Amouyal E, Bernas A, Grand D. Photochem Photobiol. 1979;29:1071–1077. [Google Scholar]
- 20.Janata E, Schuler R H. J Phys Chem. 1982;86:2078–2084. [Google Scholar]
- 21.Santus R, Patterson L K, Bazin M, Maziere J C, Morliere P. Free Rad Res. 1998;29:409–419. doi: 10.1080/10715769800300451. [DOI] [PubMed] [Google Scholar]
- 22.Sudhakar K, Phillips C M, Williams S A, Vanderkooi J M. Biophys J. 1993;64:1503–1511. doi: 10.1016/S0006-3495(93)81519-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li Z, Lee W E, Galley W C. Biophys J. 1989;56:361–367. doi: 10.1016/S0006-3495(89)82682-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li Z, Bruce A, Galley W C. Biophys J. 1992;61:1364–1371. doi: 10.1016/S0006-3495(92)81943-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cowen P M, McGavin S. Nature (London) 1955;176:470–478. [Google Scholar]
- 26.Harrington W F, Sela M. Biochim Biophys Acta. 1958;27:24–40. doi: 10.1016/0006-3002(58)90289-0. [DOI] [PubMed] [Google Scholar]
- 27.Zhou H-X, Szabo A. Biophys J. 1996;71:2440–2457. doi: 10.1016/S0006-3495(96)79437-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bonifačić M, Asmus K-D. J Chem Soc Perkin Trans II. 1986;11:1805–1809. [Google Scholar]
- 29.Creighton T E. Proteins: Structures and Molecular Properties. New York: Freeman; 1993. p. 266. and 269. [Google Scholar]
- 30.Wang J C, Davidson N. J Mol Biol. 1966;15:111–123. doi: 10.1016/s0022-2836(66)80213-9. [DOI] [PubMed] [Google Scholar]
- 31.Wang J C, Davidson N. J Mol Biol. 1966;19:469–482. doi: 10.1016/s0022-2836(66)80017-7. [DOI] [PubMed] [Google Scholar]
- 32.Jacobson H, Stockmayer W H. J Chem Phys. 1950;18:1600–1606. [Google Scholar]
- 33.Buckler D R, Haas E, Scheraga H A. Biochemistry. 1995;34:15965–15978. doi: 10.1021/bi00049a011. [DOI] [PubMed] [Google Scholar]
- 34.Pastor R W, Zwanzig R, Szabo A. J Chem Phys. 1996;105:3878–3882. [Google Scholar]
- 35.Zwanzig R. Proc Natl Acad Sci USA. 1988;85:2029–2030. doi: 10.1073/pnas.85.7.2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Flory P J. Statistical Mechanics of Chain Molecules. New York: Interscience; 1969. p. 11. [Google Scholar]
- 37.Tanford C. Physical Chemistry of Macromolecules. New York: Wiley; 1961. p. 164. [Google Scholar]
- 38.Guo Z, Thirumalai D. Biopolymers. 1995;36:83–102. [Google Scholar]
- 39.Camacho J, Thirumalai D. Proc Natl Acad Sci USA. 1995;92:1277–1281. doi: 10.1073/pnas.92.5.1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thirumalai D. J Phys Chem B. 1999;103:608–610. [Google Scholar]
- 41.Volk M, Kholodenko Y, Lu H S M, Gooding E A, DeGrado W F, Hochstrasser R M. J Phys Chem B. 1997;101:8607–8616. [Google Scholar]
- 42.Moser C C, Keske J M, Warncke K, Farid R S, Dutton P L. Nature (London) 1992;355:796–802. doi: 10.1038/355796a0. [DOI] [PubMed] [Google Scholar]