

Abstract
DHDPS (dihydrodipicolinate synthase) catalyses the branch point in lysine biosynthesis in bacteria and plants and is feedback inhibited by lysine. DHDPS from the thermophilic bacterium Thermotoga maritima shows a high level of heat and chemical stability. When incubated at 90 °C or in 8 M urea, the enzyme showed little or no loss of activity, unlike the Escherichia coli enzyme. The active site is very similar to that of the E. coli enzyme, and at mesophilic temperatures the two enzymes have similar kinetic constants. Like other forms of the enzyme, T. maritima DHDPS is a tetramer in solution, with a sedimentation coefficient of 7.2 S and molar mass of 133 kDa. However, the residues involved in the interface between different subunits in the tetramer differ from those of E. coli and include two cysteine residues poised to form a disulfide bond. Thus the increased heat and chemical stability of the T. maritima DHDPS enzyme is, at least in part, explained by an increased number of inter-subunit contacts. Unlike the plant or E. coli enzyme, the thermophilic DHDPS enzyme is not inhibited by (S)-lysine, suggesting that feedback control of the lysine biosynthetic pathway evolved later in the bacterial lineage.
Keywords: dihydrodipicolinate synthase, (S)-lysine inhibition, (S)-lysine biosynthesis, thermophilic enzyme, Thermotoga maritima
Abbreviations: (S)-ASA, (S)-aspartate-semialdehyde; DHDPR, dihydrodipicolinate reductase; DHDPS, dihydrodipicolinate synthase; HTPA, (4S)-4-hydroxy-2,3,4,5-tetrahydro-(2S)-dipicolinic acid; rmsd, root mean square deviation
INTRODUCTION
Thermotoga maritima is a thermophilic, Gram-negative, rod-shaped bacterium that has an optimum growth temperature of 80 °C [1]. Small-subunit ribosomal RNA phylogeny has placed this bacterium as one of the deepest and most slowly evolving lineages in the bacteria [2], suggesting that knowledge of its structural biology will provide important insights into protein evolution. This has been strengthened by recent work on the structural genomics of T. maritima [3]. Thermophilic enzymes are also of interest because of the differences in structural dynamics that are required to maintain their activity at high temperatures. Experiments looking at enzyme dynamics have shown that at a given temperature, thermostable enzymes are less flexible than thermolabile ones [4], and that at lower temperatures, enzymes from extreme thermophiles are often less active than those from mesophiles [5,6]. At the optimum growth temperature for an organism, the flexibilities of enzymes from mesophiles and extreme thermophiles are similar [7].
Various theories have been put forward to explain the stability of thermophilic enzymes, which may be associated with more salt bridges and buried hydrophobic regions. For example, triose phosphate isomerase from T. maritima [8] and Pyrococcus woesei [9] achieves an increased number of buried hydrophobic residues by existing as a tetramer instead of the homodimeric protein found in other organisms. Disulfide bonds have also been proposed to stabilize hyperthermic proteins [10], an example being methylthioadenosine phosphorylase from Pyrococcus furiosus [11]. Additional enzyme stability has also been engineered by the incorporation of disulfide bonds [12]. Some thermophilic organisms, including T. maritima, have been shown to contain disulfide oxidoreductase protein, which is thought to have a role in intracellular disulfide bond formation [13]. However, recent comprehensive studies of a large number of thermophilic proteins, compared with their mesophilic counterparts, have suggested that the main features responsible for thermostability are increased compactness and an increase in electrostatic interactions, with oligomerization order, hydrogen bonds and secondary structure playing minor roles [14,15].
DHDPS (dihydrodipicolinate synthase; EC 4.2.1.52) is a key enzyme in the (S)-lysine biosynthesis pathway and an important antibiotic target [16]. It catalyses the condensation of (S)-ASA [(S)-aspartate semialdehyde] and pyruvate to form a product, currently thought to be HTPA [(4S)-4-hydroxy-2,3,4,5-tetrahydro-(2S)-dipicolinic acid] [17]. This is the first reaction that is unique to (S)-lysine biosynthesis, and DHDPS is generally feedback regulated by (S)-lysine [18,19]. Another control point in the synthesis of (S)-lysine is the aspartate kinase enzyme, regulation of which controls the synthesis of all of the aspartate family of amino acids. Isozymes of DHDPS can be grouped according to their regulatory properties with respect to (S)-lysine. Plant enzymes are strongly inhibited by (S)-lysine (IC50=0.01−0.05 mM) [20–25] and DHDPS appears to be an important metabolic step in (S)-lysine biosynthesis [18,19,26]. DHDPSs from Gram-negative bacteria are only weakly inhibited (IC50=0.25–1.0 mM) [27–29], with (S)-lysine acting as a mixed partial inhibitor of DHDPS with respect to pyruvate and a partial non-competitive inhibitor with respect to (S)-ASA [30]. Conversely, DHDPS enzymes from Gram-positive bacteria show little or no feedback inhibition by (S)-lysine [31–36]. The feedback inhibition properties of lysine in the T. maritima DHDPS were therefore of interest.
DHDPS from Escherichia coli has been rigorously characterized in terms of its kinetic and reaction mechanism. In the first step of the reaction, pyruvate forms a Schiff base with a lysine residue in the active site. This is followed by the binding of the second substrate, (S)-ASA, and subsequent dehydration and cyclization to form HTPA [37]. Structural features of E. coli DHDPS have also been well characterized, with the enzyme consisting of a homotetramer that is made up of a dimer of ‘tight-dimers’ (see Figure 1B). There are many interactions between monomers A and B but few between the two tight dimers [38,39]. The active site is located in the centre of a (β/α)8-barrel in each monomer, while the lysine-binding site is situated in the cleft at the tight dimer interface, with one lysine binding per monomer, but each lysine molecule being co-ordinated by residues from each monomer within the tight dimer [40].
Figure 1. X-ray crystal structures of DHDPS from (A) T. maritima, (B) E. coli and (C) N. sylvestris.

Each enzyme is a homotetramer composed of two tight-dimer units (A–B and C–D), but the arrangement of the two dimeric units is different. The structures in (B) and (C) were drawn using the co-ordinates described in [38] and [17] respectively. The structures were drawn using Pymol (http://www.pymol.org).
The X-ray crystal structure for DHDPS has also been solved for the plant Nicotiana sylvestris [41]. Comparison of the bacterial and plant structures reveals some intriguing differences (see Figure 1). While each enzyme is a homotetramer, the quaternary architecture is different in the bacterial and plant enzymes. Specifically, each enzyme exists as a dimer of tight dimers, with a common tight-dimer structure. Each tight-dimer interface has a binding site for lysine. The reason for the enzyme adopting a dimeric structure is thus clear, especially in the light of findings that the key catalytic motif, a triad of amino acid residues (Tyr133, Thr44 and Tyr107) spans the tight-dimer interface [39]. Curiously, although both the E. coli and N. sylvestris enzymes have adopted a dimer of dimers as their functional unit, the arrangement of these dimers is entirely different. This led us to hypothesize that each protein had evolved from an ancestral dimeric protein, and the quaternary structure of T. maritima DHDPS was of particular interest.
In the present paper, we describe the X-ray crystal structure of T. maritima DHDPS, the co-ordinates of which have been deposited by Joint Center for Structural Genomics (Genomics Institute, San Diego, CA, U.S.A.) in the Protein Data Bank (PDB ID number 1O5K), and characterize its biochemical and biophysical properties, with a particular focus on the feedback inhibition properties by lysine and the quaternary structure of the enzyme in solution.
EXPERIMENTAL
Materials
Unless otherwise stated, all chemicals were obtained from Sigma, GE Biosciences or Invitrogen. Protein concentration was measured by the method of Bradford [42]. Unless otherwise stated, enzymes were manipulated at 4 °C or on ice. (S)-ASA was synthesized using the method of Roberts et al. [43], and was of high quality (>95%) as judged by 1H-NMR and the coupled assay with DHDPR (dihydrodipicolinate reductase) [29,37]. DHDPR from E. coli was purified by methods reported previously [30,44].
Cloning, overexpression and purification
Primer pairs encoding the predicted 5′- and 3′-ends of the TM1521 open reading frame [45] were used to amplify the dapA gene T. maritima strain MSB8 genomic DNA. The PCR product included a purification tag (MGSDKIHHHHHH) at the N-terminus of the full-length protein, and was cloned into the pMH1 plasmid [3], which was a donation from Scott Lesley and Heath Klock (Joint Center for Structural Genomics, Genomics Institute of the Novartis Research Foundation) and introduced to the Epicurian Coli® XL-1 Blue strain. Protein expression from Epicurian Coli® XL-1 Blue cells was performed in LB (Luria–Bertani) medium, and expression was induced by the addition of 0.15% arabinose for 3 h. The cells were harvested by centrifugation (10 min and 13000 g) and resuspended in 2 vol. of extraction buffer (50 mM NaH2PO4, pH 8.0, 20 mM imidazole and 300 mM NaCl). After lysis by sonication, cell debris was pelleted by centrifugation at 13000 g for 10 min, and the supernatant was applied to a His-Trap column (GE Biosciences). The column was washed with extraction buffer for 3 column volumes and then protein was eluted with elution buffer (50 mM NaH2PO4, pH 8.0, 300 mM imidazole and 300 mM NaCl). Fractions containing DHDPS activity were pooled, dialysed against storage buffer (20 mM Tris/HCl, pH 8.0) and stored at −20 °C.
Analytical ultracentrifugation
Sedimentation experiments were performed in a Beckman Coulter Model XL-A analytical ultracentrifuge equipped with UV–Vis scanning optics and an An-60 Ti 4-hole rotor. Protein sample and reference (20 mM Tris/HCl and 150 mM NaCl, pH 8.0) solutions were loaded into 12 mm double sector cells with quartz windows. For sedimentation velocity experiments, samples at an initial protein concentration of 0.5 mg·ml−1 (380 μl) and reference (400 μl) were centrifuged at 40000 rev.·min−1 at 20 °C and data were collected in continuous mode at 235 nm every 8 min without averaging. Data were fitted to a continuous size-distribution model [46] using the program SEDFIT (which is available from http://www.analyticalultracentrifugation.com). The partial specific volume (v) of the sample (0.743 ml·g−1), buffer density (1.005 g·ml−1) and buffer viscosity (1.021 cp) were computed using the program SEDNTRP [47]. For sedimentation equilibrium experiments, samples (100 μl) and reference (120 μl) solutions were centrifuged at 10000 and 16000 rev.·min−1 for at least 24 h until sedimentation equilibrium was attained. This was determined by overlaying scans taken at 2 h intervals. At equilibrium, the final absorbance compared with the radial position profile was collected at 280 nm and 20 °C, with a step size of 0.001 cm and ten averages. Sedimentation equilibrium data obtained at rotor speeds of 10000 and 16000 rev.·min−1 were globally fitted to a single species with mass conservation constraints using the program SEDPHAT [48] (also available from http://www.analyticalultracentrifugation.com) to determine the equivalent molar mass (Meq) according to eqn (1) below.
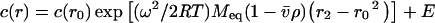 |
(1) |
where c(r) is the concentration at radius r, c(r0) is the concentration at the reference radius r0, ω is the rotor angular velocity, R is the gas constant, T is temperature, v is the partial specific volume of the solute, ρ is the solvent density and E is the baseline offset.
Kinetics
DHDPS activity was measured using a coupled assay with DHDPR, as previously described [37]. The assays were initiated by the addition of DHDPS, and the temperature was kept constant at 30 °C by the use of a circulating water bath. Stock solutions of (S)-ASA, NADPH, pyruvate and (S)-lysine were prepared fresh for each experiment, and care was taken to ensure that an excess of DHDPR was present in the assays. The amount of DHDPS in the assays was 0.2–1 μg·ml−1. Initial rate data were analysed using non-linear regression software (OriginLab, Northampton, MA, U.S.A.), and fitted to the Ping Pong model, also known as the double displacement enzyme mechanism [49].
Enzyme stability
DHDPS was incubated at selected temperatures in storage buffer (20 mM Tris/HCl, pH 8.0) in a solid heat block. Aliquots were taken at varying time intervals, and stored on ice or added directly to initiate the coupled assay. DHDPS was also incubated at selected urea concentrations at 25 °C. Aliquots were taken at varying time intervals and used, without delay, to initiate the coupled assay.
CD spectroscopy
CD spectroscopy data were generated using an Aviv 62DS CD spectrophotometer. Wavelength scans were collected using a 1 mm path length cuvette, 1.0 nm bandwidth, 0.5 nm step size and 2 s averaging time. Temperature scans were monitored at 222 nm and data were collected at 0.5 °C intervals between 20 and 80 °C with a 5 s averaging time. Cuvettes were stoppered during temperature scans to avoid evaporation. DHDPS spectra were collected at a concentration of 0.3–0.4 mg·ml−1 in a buffer containing 20 mM Tris/HCl and 150 mM NaCl (pH 8.0).
RESULTS AND DISCUSSION
X-ray crystal structure
Structural co-ordinates for T. maritima DHDPS have been deposited in PDB (accession number 1O5K). As with DHDPS from E. coli and N. sylvestris, DHDPS from T. maritima appears to be a homotetramer in the crystal, with each monomer consisting of a (β/α)8-barrel, and the active site located at the centre of each β-barrel. The quaternary structure reveals a dimer of dimers (Figure 1A), with the enzyme from T. maritima resembling the E. coli enzyme in the packing of the dimer subunits, rather than the N. sylvestris enzyme (Figure 1). The residues and contact areas within the tight dimer are similar for E. coli and N. sylvestris DHDPS [17] and T. maritima DHDPS. However, there is little conservation of sequence or contact area at the dimer–dimer interface.
Recent work in our laboratory has identified several residues that are involved in the dimer–dimer interface of E. coli DHDPS [40], which differs greatly from that of the N. sylvestris enzyme [17]. Of these residues, only Arg230 is conserved between the E. coli and T. maritima forms of the DHDPS enzyme, suggesting that although the tetrameric structure is required, the particular network of residues comprising that network is unimportant for activity. Examination of the T. maritima DHDPS crystal structure using JavaProtein Dossier [50] suggests that there are many more residues involved in inter-subunit contacts at the dimer–dimer interface compared with the E. coli DHDPS enzyme, with 20 residues involved in many interactions (Figure 2). This is entirely consistent with the increased thermal stability of the enzyme [14,15].
Figure 2. Residue interactions at the dimer–dimer interface.
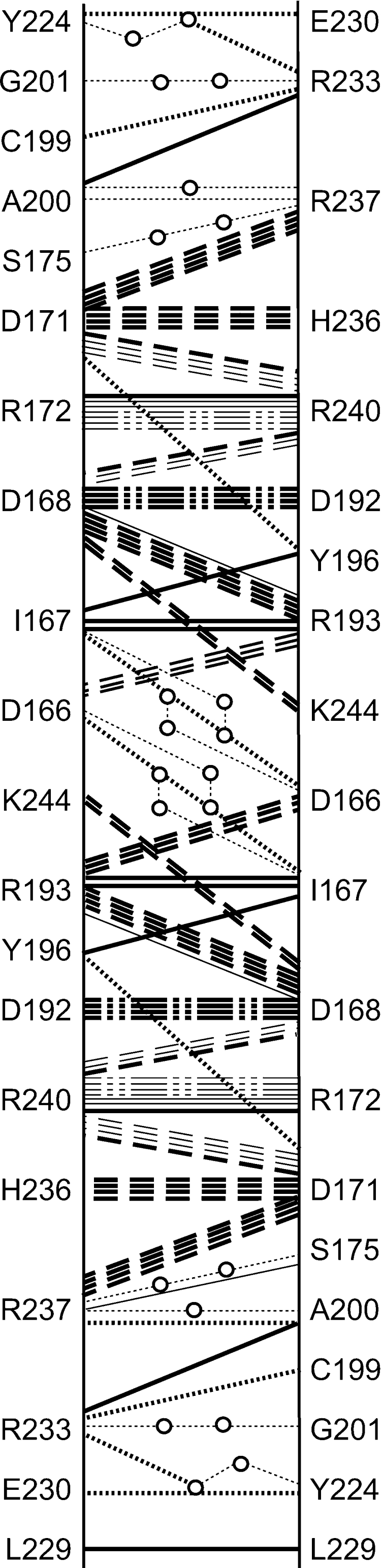
Twenty residues are involved in hydrogen bonds (dotted lines), water-mediated hydrogen bonds (dotted lines with circles), hydrophobic interactions (solid black lines), attractive charges (dashed lines) and repulsive charges (dash-dot-dot-dashed lines). Boldface lines indicate interactions that occur in both the A/D and B/C interface. Interactions were determined using JavaProtein Dossier [50].
At the dimer interface, Cys199 from the A subunit is located close to the complementary Cys199 residue from the D subunit (Figure 3). This allows for the potential formation of an inter-subunit disulfide bridge at each of the dimer interfaces. There is no density in the crystal structure to suggest the formation of this bridge, and experiments using blue native gel electrophoresis failed to identify a disulfide bridge (results not shown); however, the possibility of this linkage adding to the stability of the enzyme in vivo remains, if an appropriate protein disulfide oxidoreductase is present in T. maritima [13].
Figure 3. Dimer–dimer interface of T. maritima DHDPS.

The Cys199 residue from each of the A and D subunits is shown in grey and black. Electron density (2Fo−Fc) is contoured to 1 σ. This Figure was drawn using Pymol (http://www.pymol.org).
All of the residues that have been identified as being important in the active site of E. coli DHDPS [40] are also present in the T. maritima DHDPS active site. Overlaying the active site from the two different crystal structures reveals the orientation of residues to be highly conserved (Figure 4). However, the allosteric binding site for (S)-lysine is not conserved between the two forms of the enzyme (Figure 5). A comparison of the sequences reveals that few of the residues involved in binding (S)-lysine [40] are conserved. Among the differences are the lack of Ala49, His53, His56 and Glu84 residues, which have been shown to be involved in the binding of (S)-lysine to the E. coli DHDPS enzyme [40].
Figure 4. Overlay of the active site of E. coli (black) and T. maritima (grey) DHDPS structures.

Numbering is shown for the T. maritima enzyme. This Figure was drawn using Pymol (http://www.pymol.org).
Figure 5. Overlay of the (S)-lysine binding site of E. coli (black) and T. maritima (grey) DHDPS structures.

Numbering is shown for the T. maritima enzyme, except where the residues are not conserved, in which case the E. coli numbering and residue are indicated by an asterisk. This Figure was drawn using Pymol (http://www.pymol.org).
Quaternary structure in solution
To confirm the oligomeric state of T. maritima DHDPS in solution, sedimentation velocity experiments were initially conducted in the analytical ultracentrifuge. The sedimenting boundary of the T. maritima enzyme shows minimal spreading with time, which is consistent with a largely homogeneous sample (Figure 6A). The data were fitted to a continuous size-distribution model [46], which confirmed that the T. maritima enzyme was a single species in solution with a standardized sedimentation coefficient of 7.2 S and a molar mass of 128 kDa taken from the ordinate maximum of the single peak observed in the c(M) distribution (Figure 6B, Table 1). The axial ratio assuming a prolate ellipsoid shape is also estimated to be 2.6 (Table 1). This compares well with that of the E. coli DHDPS tetrameric enzyme [51]. The quality of the fit is indicated by the low rmsd (root mean square deviation) of 0.0066 and random distribution of residuals with a Runs test Z value of 14.4. The tetrameric nature of the enzyme was also confirmed by sedimentation equilibrium analysis (Table 1), native gel electrophoresis and size-exclusion HPLC experiments (results not shown).
Figure 6. Sedimentation velocity analysis of T. maritima DHDPS.

(A) Absorbance at 235 nm plotted as a function of radial position from the axis of rotation (cm) for T. maritima DHDPS at a concentration of 0.5 mg·ml−1. The raw data are presented as open symbols (○) plotted at time intervals of 8 min overlaid with the non-linear least squares best fit (solid line) to a continuous size distribution model [46] as described in (B). (B) The c(M) distribution is plotted as a function of molar mass (kDa) for T. maritima DHDPS. The fit was obtained using a resolution of 200 species between Mmin of 10 kDa and Mmax of 200 kDa with v=0.743, ρ=1.005 g·ml−1, η=1.021 cp and f/f0=1.21. The rmsd and run test averages for the fit were 0.00657 and 14.4 respectively. Top: the residuals for the c(M) distribution best fit described in (B) plotted as a function of radial position (cm) from the axis of rotation.
Table 1. Hydrodynamic properties of DHDPS from T. maritima.
*Standardized sedimentation coefficient taken from the ordinate maximum of the c(s) distribution (results not shown).
†Relative monomeric molar mass (Mr) determined from the amino acid sequence.
‡Apparent molar mass (M) taken from the ordinate maximum of the c(M) distribution (Figure 6B).
§Axial ratio (a/b) assuming a prolate ellipsoidal structure calculated using the v method [47].
∥Equivalent molar mass assuming a single species calculated from global analysis of sedimentation equilibrium data at 10000 and 16000 rev.·min−1 using eqn (1).
Kinetics
Data from the characterization of T. maritima DHDPS fitted the Ping Pong kinetic mechanism (Figure 7). The Vmax was 1.01 μmol·s−1·mg−1, while the Michaelis–Menten constants for pyruvate and (S)-ASA were 0.053 (±0.006) and 0.16 (±0.01) mM respectively. These compare with the E. coli DHDPS, which has a Vmax of 0.58 μmol·s−1·mg−1 and Michaelis–Menten constants for pyruvate and (S)-ASA of 0.25 and 0.11 mM respectively [44]. This is consistent with the similar values observed for the kinetic constants for the different forms of the DHDPS enzyme [39,52] and the highly conserved active site structure. It was not possible to obtain kinetic parameters for the Thermotoga enzyme at thermophilic temperatures, due to the inherent instability of the substrate (S)-ASA at these temperatures [53].
Figure 7. Kinetics of enzyme activity.

Initial velocity was measured at varying pyruvate and (S)-ASA concentrations. Each data point was measured at least in duplicate and data were fitted to the Ping Pong model [30] and the R2 for the fit was 0.960.
Varying the (S)-lysine concentration did not have any effect on the activity of T. maritima DHDPS, while the enzyme from E. coli showed inhibition that was consistent with that previously observed [30] (Figure 8). This was expected from the X-ray crystal structure, since the allosteric binding site for (S)-lysine is not present, and suggests that feedback regulation by lysine evolved after the divergence of E. coli DHDPS enzymes from the T. maritima lineage.
Figure 8. Inhibition of DHDPS by (S)-lysine.

T. maritima (○) and E. coli (△) DHDPS enzymes were assayed for activity in the presence of varying concentrations of (S)-lysine and saturating concentrations of pyruvate and (S)-ASA using the coupled assay. The apparent rate (vapp) was then compared with the rate of non-incubated enzyme (vcon). Each data point was measured in duplicate or triplicate and error bars show the standard deviation.
Enzyme stability
When DHDPS was incubated at 90 °C, the enzyme from E. coli lost nearly all activity within 30 s, while the enzyme from T. maritima still retained over 60% of the original activity after 7 h (Figure 9A). Similarly, when DHDPS was incubated with 8 M urea, the E. coli enzyme showed a rapid decrease in activity, with little activity remaining after 1 min, whereas T. maritima DHDPS proved more of a lummox, with 40% of the original activity remaining after 90 min (Figure 9B).
Figure 9. Stability of DHDPS enzymes.

T. maritima (○) and E. coli (△) DHDPS enzymes were incubated for varying times at 90 °C (A) or with 8 M urea (B) and then assayed for activity at 30 °C using the coupled assay. The rate (v) was then compared with the rate of non-incubated enzyme (vcon). Each data point was carried out in duplicate or triplicate and error bars show the standard deviation. Data were fitted to an equation for exponential decay.
The unfolding of T. maritima DHDPS enzyme was monitored at 222 nm using CD spectroscopy, and showed little change from 20 to 80 °C, unlike the E. coli enzyme, which underwent a dramatic increase in mean residue ellipticity at 60–70 °C (Figure 10). This is consistent with the decrease in activity shown by the E. coli DHDPS enzyme being associated with a general decrease in ordered protein structure, while few changes occurred in the protein structure and activity of T. maritima DHDPS. Since no disulfide bond was present in the enzyme in vitro, the additional heat and chemical stability of the enzyme would seem to be explained by the additional inter-subunit contacts at the dimer–dimer interface, when compared with the E. coli form of the enzyme.
Figure 10. CD spectroscopy of DHDPS thermal stability.

The mean residue ellipticity at 222 nm of T. maritima (○) and E. coli (■) DHDPS was measured using an AVID 62DS CD spectrophotometer. Data were collected at 0.5 °C intervals between 20 and 80 °C in a buffer containing 20 mM Tris/HCl and 150 mM NaCl (pH 8.0). °, degrees·.
Conclusions
It is thought that T. maritima is one of the most slowly evolving lineages in the bacteria [2] and may provide the closest modern relative of ancestral DHDPS enzymes. Modern DHDPS enzymes can be classified by their regulatory properties with respect to (S)-lysine, with plant enzymes showing strong inhibition, Gram-negative bacteria showing only weak inhibition and Gram-positive bacteria showing little or no feedback inhibition. T. maritima DHDPS does not show any inhibition by (S)-lysine, suggesting that this feedback regulation must have evolved later in bacteria. Given the lack of feedback inhibition of DHDPS, it is likely that (S)-lysine biosynthesis in T. maritima is regulated by aspartate kinase or at the level of gene regulation [54], but further work would be required to investigate this possibility.
Thermostability of enzymes is consistent with the increase in electrostatic interactions in the enzyme. This is demonstrated in the T. maritima DHDPS enzyme, which has many more residues involved in inter-subunit contacts at the dimer–dimer interface when compared with the E. coli DHDPS enzyme. The T. maritima DHDPS enzyme also has the potential for an inter-subunit disulfide bridge, which has been shown to improve the thermal stability of several other enzymes [10–12].
An enigma in the evolution of DHDPS is the difference in dimer arrangement between the plant and bacterial forms of the enzyme. T. maritima DHDPS is unequivocally a tetramer in the crystal structure and in solution, raising the possibility that the plant and bacterial enzymes evolved from a dimeric form of the enzyme that predated the T. maritima enzyme. How the novel architecture of the plant form of the enzyme evolved from the bacterial protein remains a mystery.
Acknowledgments
This work was funded, in part, by the Royal Society of New Zealand Marsden Fund and, in part, by the Foundation of Research, Science and Technology via a fellowship to F.G.P. We thank Dr Scott Lesley and Heath Klock for supplying the plasmid and for useful discussions and comments, Dr Ren Dobson and Sean Devenish for comments, and Jackie Healy for supersonic technical support. J.A.G. thanks the University of Canterbury for funding and the Bio21 Institute, University of Melbourne, for hosting a sabbatical leave.
References
- 1.Huber R., Langworthy T. A., Konig H., Thomm M., Woese C. R., Sleytr U. B., Stetter K. O. Thermotoga maritima sp represents a new genus of unique extremely thermophilic eubacteria growing up to 90 °C. Arch. Microbiol. 1986;144:324–333. [Google Scholar]
- 2.Achenbach-Richter L., Gupta R., Stetter K. O., Woese C. R. Were the original Eubacteria thermophiles? Syst. Appl. Microbiol. 1989;9:34–39. doi: 10.1016/s0723-2020(87)80053-x. [DOI] [PubMed] [Google Scholar]
- 3.Lesley S. A., Kuhn P., Godzik A., Deacon A. M., Mathews I., Kreusch A., Spraggon G., Klock H. E., McMullan D., Shin T., et al. Structural genomics of the Thermotoga maritima proteome implemented in a high-throughput structure determination pipeline. Proc. Natl. Acad. Sci. U.S.A. 2002;99:11664–11669. doi: 10.1073/pnas.142413399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Daniel R. M. The upper limits of enzyme thermal stability. Enzyme Microb. Tech. 1996;19:74–79. [Google Scholar]
- 5.Wrba A., Schweiger A., Schultes V., Jaenicke R., Zavodszky P. Extremely thermostable D-glyceraldehyde-3-phosphate dehydrogenase from the eubacterium Thermotoga maritima. Biochemistry. 1990;29:7584–7592. doi: 10.1021/bi00485a007. [DOI] [PubMed] [Google Scholar]
- 6.Varley P. G., Pain R. H. Relation between stability, dynamics and enzyme-activity in 3-phosphoglycerate kinases from yeast and Thermus thermophilus. J. Mol. Biol. 1991;220:531–538. doi: 10.1016/0022-2836(91)90028-5. [DOI] [PubMed] [Google Scholar]
- 7.Daniel R. M., Dines M., Petach H. H. The denaturation and degradation of stable enzymes at high temperatures. Biochem. J. 1996;317:1–11. doi: 10.1042/bj3170001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maes D., Zeelen J. P., Thanki N., Beaucamp N., Alvarez M., Thi M. H. D., Backmann J., Martial J. A., Wyns L., Jaenicke R., Wierenga R. K. The crystal structure of triosephosphate isomerase (TIM) from Thermotoga maritima: a comparative thermostability structural analysis of ten different TIM structures. Proteins. 1999;37:441–453. [PubMed] [Google Scholar]
- 9.Walden H., Bell G. S., Russell R. J. M., Siebers B., Hensel R., Taylor G. L. Tiny TIM: a small, tetrameric, hyperthermostable triosephosphate isomerase. J. Mol. Biol. 2001;306:745–757. doi: 10.1006/jmbi.2000.4433. [DOI] [PubMed] [Google Scholar]
- 10.Mallick P., Boutz D. R., Eisenberg D., Yeates T. O. Genomic evidence that the intracellular proteins of archaeal microbes contain disulfide bonds. Proc. Natl. Acad. Sci. U.S.A. 2002;99:9679–9684. doi: 10.1073/pnas.142310499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cacciapuoti G., Moretti M. A., Forte S., Brio A., Camardella L., Zappia V., Porcelli M. Methylthioadenosine phosphorylase from the archaeon Pyrococcus furiosus. Mechanism of the reaction and assignment of disulfide bonds. Eur. J. Biochem. 2004;271:4834–4844. doi: 10.1111/j.1432-1033.2004.04449.x. [DOI] [PubMed] [Google Scholar]
- 12.Mansfeld J., Vriend G., Dijkstra B. W., Veltman O. R., Van den Burg B., Venema G., Ulbrich-Hofmann R., Eijsink V. G. H. Extreme stabilization of a thermolysin-like protease by an engineered disulfide bond. J. Biol. Chem. 1997;272:11152–11156. doi: 10.1074/jbc.272.17.11152. [DOI] [PubMed] [Google Scholar]
- 13.Beeby M., Connor B. D., Ryttersgaard C., Boutz D. R., Perry L. J., Yeates T. O. The genomics of disulfide bonding and protein stabilization in thermophiles. PLoS Biol. 2005;3:e309. doi: 10.1371/journal.pbio.0030309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Robinson-Rechavi M., Godzik A. Structural genomics of Thermotoga maritima proteins shows that contact order is a major determinant of protein thermostability. Structure. 2005;13:857–860. doi: 10.1016/j.str.2005.03.011. [DOI] [PubMed] [Google Scholar]
- 15.Robinson-Rechavi M., Alibes A., Godzik A. Contribution of electrostatic interactions, compactness and quaternary structure to protein thermostability: lessons from structural genomics of Thermotoga maritima. J. Mol. Biol. 2006;356:547–557. doi: 10.1016/j.jmb.2005.11.065. [DOI] [PubMed] [Google Scholar]
- 16.Cox R., Sutherland A., Vederas J. Bacterial diaminopimelate metabolism as a target for antibiotic design. Bioorg. Med. Chem. 2000;8:843–871. doi: 10.1016/s0968-0896(00)00044-4. [DOI] [PubMed] [Google Scholar]
- 17.Blickling S., Renner C., Laber B., Pohlenz H., Holak T., Huber R. Reaction mechanism of Escherichia coli dihydrodipicolinate synthase investigated by X-ray crystallography and NMR spectroscopy. Biochemistry. 1997;36:24–33. doi: 10.1021/bi962272d. [DOI] [PubMed] [Google Scholar]
- 18.Galili G. Regulation of lysine and threonine synthesis. Plant Cell. 1995;7:899–906. doi: 10.1105/tpc.7.7.899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Galili G. New insights into the regulation and functional significance of lysine metabolism in plants. Annu. Rev. Plant Biol. 2002;53:27–43. doi: 10.1146/annurev.arplant.53.091401.110929. [DOI] [PubMed] [Google Scholar]
- 20.Frisch D. A., Gengenbach B. G., Tommey A. M., Sellner J. M., Somers D. A., Myers D. E. Isolation and characterization of dihydrodipicolinate synthase from maize. Plant Physiol. 1991;96:444–452. doi: 10.1104/pp.96.2.444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kumpaisal R., Hashimoto T., Yamada Y. Inactivation of wheat dihydrodipicolinate synthase by 3-bromopyruvate. Agric. Biol. Chem. 1989;53:355–359. [Google Scholar]
- 22.Mathews B., Widholm J. Regulation of lysin and threonine synthesis in carrot cell suspension cultures and whole carrot roots. Planta. 1978;141:315–321. doi: 10.1007/BF00388350. [DOI] [PubMed] [Google Scholar]
- 23.Wallsgrove R. M., Mazelis M. Spinach leaf dihydrodipicolinate synthase: partial purification and characterization. Biochemistry. 1981;20:2651–2655. [Google Scholar]
- 24.Ghislain M., Frankard V., Jacobs M. Dihydrodipicolinate synthase of Nicotiana sylvestris, a chloroplast-localized enzyme of the lysine pathway. Planta. 1990;180:480–486. doi: 10.1007/BF02411444. [DOI] [PubMed] [Google Scholar]
- 25.Dereppe C., Bold G., Ghisalba O., Ebert E., Schar H.-P. Purification and characterization of dihydrodipicolinate synthase from pea. Plant Physiol. 1992;98:813–821. doi: 10.1104/pp.98.3.813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Karchi H., Miron D., Ben-Yaacov S., Galili G. The lysine-dependent stimulation of lysine catabolism in tobacco seed requires calcium and protein phosphorylation. Plant Cell. 1995;7:1963–1970. doi: 10.1105/tpc.7.11.1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bartlett A., White P. Regulation of the enzymes of lysine biosynthesis in Brevibacterium lactofermentum NCTC 9602 during vegetative growth. J. Gen. Microbiol. 1986;132:3169–3177. [Google Scholar]
- 28.Bakhiet N., Forney F., Stahly D., Daniels L. Lysine biosynthesis in Methanobacterium thermoautotropicum is by the diaminopimelic acid pathway. Curr. Microbiol. 1984;10:195–198. [Google Scholar]
- 29.Yugari Y., Gilvarg C. The condensation step in diaminopimelate synthesis. J. Biol. Chem. 1965;240:4710–4716. [PubMed] [Google Scholar]
- 30.Dobson R. C. J., Griffin M. D. W., Roberts S. J., Gerrard J. A. Dihydrodipicolinate synthase (DHDPS) from Escherichia coli displays partial mixed inhibition with respect to its first substrate, pyruvate. Biochimie. 2004;86:311–315. doi: 10.1016/j.biochi.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 31.Stahly D. Dihydrodipicolinate synthase of Bacillus lichenifornis. Biochem. Biophys. Acta. 1969;191:439–451. doi: 10.1016/0005-2744(69)90263-0. [DOI] [PubMed] [Google Scholar]
- 32.Webster F., Lechowich R. Partial purification and characterization of dihydrodipicolinate synthase from sporulating Bacillus megaterium. J. Bacteriol. 1970;101:118–126. doi: 10.1128/jb.101.1.118-126.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yamakura F., Ikeda Y., Kimura K., Sasakawa T. Partial purification and some properties of pyruvate-aspartic semialdehyde condensing enzyme from sporulating Bacillus subtilis. J. Biochem. 1974;76:611–621. doi: 10.1093/oxfordjournals.jbchem.a130605. [DOI] [PubMed] [Google Scholar]
- 34.Cremer J., Eggeling L., Sahm H. Cloning the dapA dapB cluster of the lysine-secreting bacterium Corynebacterium glutamicum. Mol. Gen. Genet. 1990;229:478–480. [Google Scholar]
- 35.Hoganson D., Stahly D. Regulation of dihydrodipicolinate synthase during growth and sporulation of Bacillus cereus. J. Bacteriol. 1975;124:1344–1350. doi: 10.1128/jb.124.3.1344-1350.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tosaka O., Takinami K. Pathway and regulation of lysine biosynthesis in Brevibacterium lactofermentum. Agric. Biol. Chem. 1978;42:95–100. [Google Scholar]
- 37.Coulter C. V., Gerrard J. A., Kraunsoe J. A. E., Pratt A. J. Escherichia coli dihydrodipicolinate synthase and dihydrodipicolinate reductase: kinetic and inhibition studies of two putative herbicide targets. Pesticide Sci. 1999;55:887–895. [Google Scholar]
- 38.Mirwaldt C., Korndorfer I., Huber R. The crystal structure of dihydrodipicolinate synthase from Escherichia coli at 2.5 Å resolution. J. Mol. Biol. 1995;246:227–239. doi: 10.1006/jmbi.1994.0078. [DOI] [PubMed] [Google Scholar]
- 39.Dobson R. C. J., Valegard K., Gerrard J. A. The crystal structure of three site-directed mutants of Escherichia coli dihydrodipicolinate synthase: further evidence for a catalytic triad. J. Mol. Biol. 2004;338:329–339. doi: 10.1016/j.jmb.2004.02.060. [DOI] [PubMed] [Google Scholar]
- 40.Dobson R. C. J., Griffin M. D. W., Jameson G. B., Gerrard J. A. The crystal structures of native and (S)-lysine-bound dihydrodipicolinate synthase from Escherichia coli with improved resolution show new features of biological significance. Acta Crystallogr. Sect. D Biol. Crystallogr. 2005;61:1116–1124. doi: 10.1107/S0907444905016318. [DOI] [PubMed] [Google Scholar]
- 41.Blickling S., Beisel H., Bozic D., Knablein J., Laber B., Huber R. Structure of dihydrodipicolinate synthase of Nicotiana sylvestris reveals novel quaternary structure. J. Mol. Biol. 1998;274:608–621. doi: 10.1006/jmbi.1997.1393. [DOI] [PubMed] [Google Scholar]
- 42.Bradford M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein–dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 43.Roberts S. J., Morris J. C., Dobson R. C. J., Gerrard J. A. The preparation of (S)-aspartate semi-aldehyde appropriate for use in biochemical studies. Bioorg. Med. Chem. Lett. 2003;13:265–267. doi: 10.1016/s0960-894x(02)00923-x. [DOI] [PubMed] [Google Scholar]
- 44.Dobson R. C. J., Gerrard J. A., Pearce F. G. Dihydrodipicolinate synthase is not inhibited by its substrate, (S)-aspartate beta-semialdehyde. Biochem. J. 2004;377:757–762. doi: 10.1042/BJ20031389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nelson K. E., Clayton R. A., Gill S. R., Gwinn M. L., Dodson R. J., Haft D. H., Hickey E. K., Peterson J. D., Nelson W. C., Ketchum K. A., et al. Evidence for lateral gene transfer between Archaea and Bacteria from genome sequence of Thermotoga maritima. Nature. 1999;399:323–329. doi: 10.1038/20601. [DOI] [PubMed] [Google Scholar]
- 46.Schuck P. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophys. J. 2000;78:1606–1619. doi: 10.1016/S0006-3495(00)76713-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Laue T. M., Shah D. B., Ridgeway T. M., Pelletier S. L. Computer-aided interpretation of analytical sedimentation data for proteins. In: Harding S. E., Rowe A. J., Horton J. C., editors. Analytical Ultracentrifugation in Biochemistry and Protein Science. Cambridge: The Royal Society of Chemistry; 1992. pp. 90–125. [Google Scholar]
- 48.Vistica J., Dam J., Balbo A., Yikilmaz E., Mariuzza R. A., Rouault T. A., Schuck P. Sedimentation equilibrium analysis of protein interactions with global implicit mass conservation constraints and systematic noise decomposition. Anal. Biochem. 2004;326:234–256. doi: 10.1016/j.ab.2003.12.014. [DOI] [PubMed] [Google Scholar]
- 49.Cornish-Bowden A. London: Portland Press Ltd; 1999. Fundamentals of Enzyme Kinetics. [Google Scholar]
- 50.Neshich G., Rocchia W., Mancini A. L., Yamagishi M. E. B., Kuser P. R., Fileto R., Baudet C., Pinto I. P., Montagner A. J., Palandrani J. F., et al. JavaProtein Dossier: a novel web-based data visualization tool for comprehensive analysis of protein structure. Nucleic Acids Res. 2004;32:W595–W601. doi: 10.1093/nar/gkh480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Perugini M. A., Griffin M. D. W., Smith B. J., Webb L. E., Davis A. J., Handman E., Gerrard J. A. Insight into the self-association of key enzymes from pathogenic species. Eur. Biophys. J. Biophy. 2005;34:469–476. doi: 10.1007/s00249-005-0491-y. [DOI] [PubMed] [Google Scholar]
- 52.Dobson R. C. J., Devenish S. R. A., Turner L. A., Clifford V. R., Pearce F. G., Jameson G. B., Gerrard J. A. Role of arginine 138 in the catalysis and regulation of Escherichia coli dihydrodipicolinate synthase. Biochemistry. 2005;44:13007–13013. doi: 10.1021/bi051281w. [DOI] [PubMed] [Google Scholar]
- 53.Coulter C. V., Gerrard J. A., Kraunsoe J. A. E., Pratt A. J. (S)-aspartate semi-aldehyde: synthetic and structural studies. Tetrahedron. 1996;52:7127–7136. [Google Scholar]
- 54.Rodionov D. A., Vitreschak A. G., Mironov A. A., Gelfand M. S. Regulation of lysine biosynthesis and transport genes in bacteria: yet another RNA riboswitch? Nucleic Acids Res. 2003;31:6748–6757. doi: 10.1093/nar/gkg900. [DOI] [PMC free article] [PubMed] [Google Scholar]