

Abstract
The transcriptional regulation of the Hoxc8 gene is controlled during early mouse embryogenesis by an enhanceosome-like control region, termed the early enhancer (EE), located 3 kb upstream from the Hoxc8 translation start site. The EE is involved in establishing the posterior expression pattern of Hoxc8 at embryonic day (E) 8.5–9.0. Genetic and biochemical data have shown that nuclear factors interact with this region in a sequence-specific manner. We have used a yeast one-hybrid screen in a search for transcription factors that bind to EE motifs and have isolated a novel murine DNA-binding protein, termed BEN (binding factor for early enhancer). The ORF of BEN encodes a protein of 1072 amino acids and contains six helix–loop–helix domains, a hydrophobic leucine zipper-like motif, and a serine-rich repeat. The murine BEN gene is structurally similar to the human gene TFII-I in that both genes encode unique 95-amino acid long helix–loop/span–helix domains. The BEN gene produces several major transcripts (3.6, 4.4, and 5.9 kb) present in most adult tissues and shows discrete spatial and temporal domains of expression in areas of epithelial-mesenchymal interaction during mouse embryogenesis from E9.5 to E12.5. Several BEN-encoded polypeptides of different sizes ranging from 165 to 40 kDa were identified by Western blot analysis using BEN-specific polyclonal Abs. We propose, on the bases of sequence homology, that BEN is the mouse ortholog of the recently described human gene, WBSCR11, known also as GTF2IRD1, GTF3, Cream1, and MusTRD1. This gene is deleted hemizygously in individuals with Williams Syndrome, an autosomal dominant genetic condition characterized by complex physical, cognitive, and behavioral traits resulting from a perturbed developmental process.
Keywords: Williams Syndrome, Hoxc8
Homeobox genes encode transcription factors involved in positional specification during embryonic development in diverse organisms (1). These proteins contain a highly conserved helix–turn–helix motif, termed the homeodomain, responsible for binding to specific DNA sequences. In amniotes, there are four clusters of homeobox genes, termed Hox genes, which share a high degree of sequence, structural, and functional similarity with the Drosophila HOM-C genes (2–4). Hox genes determine body plan features on the anterior/posterior axis. Each gene contributes to the positional identity of a particular region on the axis. The order of the genes within a cluster corresponds to the order of anatomical features on the anterior/posterior axis. Mutations in Hox genes result in homeotic transformations, deficiencies, and other morphological abnormalities.
Hox gene expression, itself, is critically important in the proper regulation of development and depends in large measure on the orderly expression of a variety of “upstream” transcription factors. We have reported previously on control regions that regulate Hoxc8 gene expression. The expression of Hoxc8 can be divided into an early, “establishment” phase and a late “maintenance” phase (5, 6). Hoxc8 early expression at embryonic day (E) 8.5 extends from the base of the allantois anteriorly within the segmental plate mesoderm, and similarly within the neurectoderm to more anterior position. The anterior boundary of Hoxc8 at E9.5 is located in the neural tube at the level of the 9th somite, in the paraxial mesoderm at the 14th somite, and in the lateral plate mesoderm at the 12th somite. Later in development, posterior expression of Hoxc8 decreases, whereas intense expression is maintained at previously determined anterior limits within the thorax (somites and lateral plate mesoderm) and in the brachial region (neural tube) (7).
Transgenic reporter analysis was used to identify cis-regulatory domains critical for the normal expression of Hoxc8. Two distinct genomic regions were identified that regulate the early and late phases of Hoxc8 expression (5, 6). The early phase is regulated by DNA elements located 3 kb upstream from the translation start site of the gene, whereas the late expression is regulated by elements present 11–20 kb downstream of the start site. The Hoxc8 early enhancer (EE) was limited by progressive deletions to a 200-bp fragment, necessary and sufficient to direct expression to the neural tube, somites, and lateral plate mesoderm (6). There are at least nine distinct cis-regulatory elements within the EE 200-bp region that are partially redundant and interdependent, based on the reporter gene analysis in transgenic mouse experiments. These elements, designated A to H, were mapped from the 5′ end to the 3′ end of the EE, respectively. Mutational analysis showed that different combinations of these elements govern both the anterior limits and the tissue-specific pattern of reporter gene expression along the anterior/posterior embryonic axis. A comparison of the DNA sequence of protein binding motifs within the EE with binding motifs for known transcription factors revealed several potential binding sites for cdx, HNF, Lef-1/Tcf, signal transducer and activator of transcription (STAT), and Hox transcription factors.
We have used a yeast one-hybrid screen to isolate candidate transcription factor genes that regulate the Hoxc8 gene through interactions with the EE. We report here on one such gene encoding a novel mouse nuclear protein that is structurally related to the human TFII-I gene. This gene, which we have named binding factor for early enhancer (BEN), is also an ortholog of the recently described human gene termed WBSCR11, also known as GTF2IRD1, GTF3, Cream1, and MusTRD1 (8–12). WBSCR11 is a candidate gene involved in the pathogenesis of Williams Syndrome (WS). We describe here the physical characteristics and possible functional role of BEN and compare it with its previously reported paralogs and orthologs. We also consider its possible interaction with the EE of Hoxc8.
Materials and Methods
Oligonucleotides.
T7OH40-1, cca agc ttc taa tac gac tca ctA TAG GCC TCA ACA TTG CT; OH40-2, TGT GGA CAG ACG CCT GCA GGG CCT; OH40-3, CCT CTG AGG GCA GAT GCC CAG GTA ACG AGT CA; OH40-4, CGC ACC GCG TCC GCC CTC CTT; EFG-2s, aat tCT TTC CTT TGA AAT CGG ATT ATA GGA ATG TTT TGT CTC TTT CCT TTG AAA TCG GAT TAT AGG AAT GTT TTG TCT CTT TCC TTT GAA ATC GGA TTA TAG GAA TGT TTT GTC T; EFG-2a, cta gAG ACA AAA CAT TCC TAT AAT CCG ATT TCA AAG GAA AGA GAC AAA ACA TTC CTA TAA TCC GAT TTC AAA GGA AAG AGA CAA AAC ATT CCT ATA ATC CGA TTT CAA AGG AAA G; EFG-1s, aat tCC TTT GAA ATC GGA TTA TAG TTC CTT TGA AAT CGG ATT ATA GTT CCT TTG AAA TCG GAT TAT AGT TCC TTT GAA ATC GGA TTA TAG; EFG-1a, tct aCT ATA ATC CGA TTT CAA AGG AAC TAT AAT CCG ATT TCA AAG GAA CTA TAA TCC GAT TTC AAA GGA ACT ATA ATC CGA TTT CAA AGG; 3xUCD1, aat tCT GGC TAG ACG TCT GGG CTT CTG GCT AGA CGT CTG GGC TTC TGG CTA GAC GTC TGG GCT T; 3xUCD2, cta gAA GCC CAG ACG TCT AGC CAG AAG CCC AGA CGT CTA GCC AGA AGC CCA GAC GTC TAG CCA G; pET1, CCG ACT CGA GCG CCA CCA TGG GCA GCA; BEN1, GCG GAA TTC GTG CTG TGG ACA GAC GCC T; BEN2, GTG CGG CCG CGG CCC GGA AGC TGC ACG T; B80, TTC CTT TGA AAT CGG ATT ATA GGA ATG T; B81, AGA CAA AAC ATT CCT ATA ATC CGA TTT C.
Yeast One-Hybrid Screening.
We used the Matchmaker yeast one-hybrid system kit (CLONTECH), and procedure was performed according to the manufacturer's protocol. The bait plasmids, pHisi-1-3EFG and pLacZi-3EFG, were constructed by ligation of double-stranded oligonucleotides, containing three tandem repeats of the EFG site of the EE, to sites within the multiple cloning region of the reporter plasmids. The oligonucleotides containing three copies of the EFG site were synthesized with EcoRI/XbaI sites at the 5′ and 3′ ends (sense, EFG-2s; antisense, EFG-2a), annealed, and cloned into the EcoRI/XbaI sites of the HIS3 reporter plasmid, pHisi-1, to produce pHisi-1-3EFG. Another set of oligonucleotides was synthesized with EcoRI/XhoI sites (sense, EFG-1s; antisense, EFG-1a) and cloned into the EcoRI/XhoI sites of the β-galactosidase reporter plasmid pLacZi to generate pLacZi-3EFG. To obtain yeast reporter strain YM-3EFG, the bait plasmids were linearized and integrated into the yeast genome of the YM4271 strain by homologous recombination. One-hybrid screening with the 11-day-old mouse embryo MATCHMAKER cDNA library (CLONTECH) was carried out on yeast-selective media SD/-His/-Ura/-Leu plates supplemented with 30 mM 3-amino-1,2,4-triazole (3-AT) to suppress leaky His expression. From the initial 4 × 106 transformants screened, 120 were histidine prototrophs, and 21 clones turned out to be positive for LacZ expression. Plasmids were isolated from these latter clones, and cDNA inserts were PCR amplified with 5′AD and 3′AD primers (CLONTECH). The PCR-amplified fragments were cloned into pGEM-T Easy vector (Promega) and partially sequenced. The clone OH40 was fully sequenced.
Isolation of the 5′ End Sequence.
The National Center for Biotechnology Information (NCBI) GenBank database was searched for expressed sequence tags (EST) bearing sequence similarity to the 5′-end portion of the OH40 cDNA clone. The mouse EST designated 555547 (GenBank accession no. AA111609) was identified as such a clone. The additional 5′ sequence of this EST was used to design an oligonucleotide primer OH40-4 for PCR amplification. The missing 5′-end of OH40 cDNA was obtained by PCR amplification using mouse E11 Marathon-Ready cDNA (CLONTECH) with OH40-4 and OH40-3 primers. The thermal cycle was 30 sec at 94°C, 30 sec at 60°C, and 2 min at 68°C for 30 cycles followed by 10 min extension at 72°C. A resulting 1.55-kb fragment was purified on 1% agarose gel with the Qiagen kit and subcloned into the pGEM-T Easy vector for further sequencing.
Production of His-Tag Fusion Proteins and Preparation of Protein Extracts from Different Mouse Tissues.
The truncated BEN fragment from 1098 to 3390 bp was amplified by using OH40 cDNA, derived from a one-hybrid screen, with BEN1 and BEN2 primers, which carry the EcoRI and NotI restriction sites, respectively. The PCR fragment was digested and cloned into the EcoRI and NotI sites of the pET-33b vector (Novagen) to generate a pET-BEN construct for the production of a His-tagged fusion protein. Affinity purification was done according to Novagen protocols. Protein extracts were isolated for Western blot analysis from different adult mouse tissues as described (13).
Abs and Western Blots.
Two polypeptides, derived from the BEN ORF, GNKFTKDPMKLEPASP (amino acids 434–449) and CNNAKVPAKDNIPKRK (amino acids 1000–1015), were used to produce a polyclonal antiserum. Rabbits (New Zeland White, SPF females) were immunized against these polypeptides, and affinity-purified sera were screened for quality by Western blots, using an affinity-purified hexahistidine-tagged BEN fusion protein, produced from the bacterial expression vector pET-BEN. Western blot analysis was performed essentially as described (14). Briefly, the proteins isolated from different adult mouse tissues (10–20 μg/lane) were resolved on a 7.5% SDS/PAGE gel, transferred to a nylon membrane, and hybridized in a final volume of 5 ml with anti-BEN Abs at 1:500 dilution and anti-goat alkaline phosphatase secondary Abs at 1:10,000 dilution. In the case of the affinity-purified His-tagged BEN proteins, hybridization was performed with T7-Tag Abs (Novagen) at 1:5,000 dilution. All further steps were processed according to Novagen protocols.
In Vitro Transcription/Translation.
The TnT coupled reticulocyte lysate system was used for in vitro transcription/translation according to the Promega protocol. The pCI-BEN, used as a template, was constructed by subcloning the DNA fragment, amplified from pET-BEN with pET-1 and BEN2 primers, into the NotI and XhoI sites of pCI vector (Promega), respectively.
Northern and RNA Blot Analysis.
A filter with mRNA from various mouse tissues was obtained from Origene Technologies (Rockville, MD). The mouse RNA dot blot was purchased from CLONTECH. Prehybridization and hybridization steps were done according to the manufacturer's recommendations. The 32P-labeled probe was derived from the original OH40 clone.
Whole-Mount in Situ Hybridizations.
Whole-mount in situ hybridizations were performed essentially as described (15). Embryos were fixed in 4% paraformaldehyde in PBS for 24 h at 4°C and dehydrated through a methanol series. Digoxigenin-labeled antisense BEN probe, spanning positions 1098 to 1666 bp of the BEN cDNA sequence, was generated from the PCR fragment, amplified with primers T7OH40-1 and OH40-2, according to the manufacturer's instructions (Boehringer Mannheim).
DNA-Binding Studies: Electrophoretic Mobility-Shift Assay (EMSA) and Southwestern Blot Analysis.
The double-stranded B8 oligonucleotide was made by annealing B81 and B82 oligonucleotides, and the UCD oligonucleotide was made by annealing 3xUCD1 and 3xUCD2. Double-stranded B8, UCD, and EFG-2 oligonucleotides were labeled with [α-32P]dATP (Amersham) by end-filling with Klenow enzyme according to New-England Biolabs protocols. In vitro translated protein (1–2 μl) and 2 × 104 cpm of labeled B8 oligonucleotide were used in binding reactions. The protein-DNA complexes were identified by running samples on 5% nondenaturing PAGE (29:1, acrylamide/bisacrylamide) in 0.5 × TBE [90 mM Tris/64.6 mM boric acid/2.5 mM EDTA (pH 8.3)] for 2 h at 200 V at 4°C. For Southwestern blot analysis, the affinity-purified BEN protein (500 ng per lane) was resolved on a 10% SDS/polyacrylamide gel. All further steps in the EMSA and Southwestern blots were carried out according to the previously reported procedures (16, 17).
Computer Homology Searches.
The homologous genes were identified by using the blast search at the National Center for Biotechnology Information. Potential protein motifs and phosphorylation sites were identified with prosite at ExPASy (University of Geneva). The sequence alignments were done by using clustal w.
Results
Isolation of cDNA Clones That Express Proteins That Bind Hoxc8 EE.
The EFG site is located at the 3′ end of the Hoxc8 EE and was shown (6) to be responsible for the somitic and lateral plate mesoderm expression of the gene. To identify transcription factors that interact with this site, we performed a yeast one-hybrid screen. The genetically modified yeast strain YM-3EFG, carrying three copies of the EFG site upstream of HIS3 and LacZ reporters, was transformed with an E11-day mouse embryo cDNA library fused to the GAL4 activation domain. One hundred twenty histidine prototrophs were isolated in selective medium from a total of 4 × 106 transformants. A subset of 21 colonies was confirmed as true positives on the basis of β-galactosidase expression (Table 1). These clones were analyzed by AluI digestion to classify by restriction pattern, and only two clones, OH40 and OH101, were shown to be identical and probably derived from the same gene. The genes were classified subsequently as strong to weak interactors on the basis of LacZ reaction intensity and then partially sequenced from both ends. Three clones turned out to represent novel genes, whereas other clones were either previously reported enzymes, ribosomal proteins, or were without obvious homology to existing entries in the database (Table 1). The one-hybrid clone OH40 was fully sequenced and is the subject of this report. The two remaining clones will be reported elsewhere.
Table 1.
One-hybrid clones that interact with the EFG site of Hoxc8
| Clone no. | β-Galactosidase activity | Homology | |
|---|---|---|---|
| 1. | 40 | + + + + + | MusTRD1, TFII-I |
| 2. | 101 | + + + + + | MusTRD1, TFII-I |
| 3. | 10 | + + + + + | Homeodomain |
| 4. | 112 | + + + + | Unknown |
| 5. | 109 | + + + + | RNP |
| 6. | 50 | + + + | Unknown |
| 7. | 90 | + + + | Unknown |
| 8. | 108 | + + + | Ribosomal protein L7 |
| 9. | 102 | + + + | Ribosomal protein L7 |
| 10. | 116 | + + + | β-Globin |
| 11. | 80 | + + | Unknown |
| 12. | 104 | + + | Y1 globin |
| 13. | 30 | + | ATP synthase alpha subunit |
| 14. | 107 | + | Unknown |
| 15. | 110 | + | Unknown |
| 16. | 111 | + | RNP |
| 17. | 113 | + | Unknown |
| 18. | 114 | + | Unknown |
| 19. | 115 | + | VMP-CMP kinase |
| 20. | 117 | + | Unknown |
| 21. | 119 | + | RNP |
Determination of Full-Length Gene Sequence.
The OH40 clone had an insert of 2.5 kb and was sequenced completely from both ends. Its nucleotide sequence revealed a TFII-I-type helix–loop–helix (HLH) domain, as assessed by blast analysis (18). The ORF of OH40 encoded a polypeptide without a putative translation initiation codon. To isolate the full-length gene, an EST database was searched with the OH40 cDNA sequence, and several homologous mouse EST sequences were found. One of the entries, 555547 (GenBank accession no. AA111609), appeared to have the missing 5′portion. This EST sequence has two stop codons in front of the first methionine, and we concluded that it was most probably a nearly complete upstream sequence. We then designed a specific primer that corresponded to the 5′ end of this EST. The missing portion of the gene was obtained by PCR using E11 mouse embryo cDNA as a template. This reaction allowed the isolation of the 5′-noncoding sequence and the portion of the BEN cDNA ORF that was not present in the original OH40 clone.
Structural Characterization of BEN.
The complete coding sequence was deduced from the combination of the two sequences derived from the PCR fragment and clone OH40. We named this gene BEN, signifying the binding factor for the early enhancer. The full-length mouse cDNA has 3572 bp and encodes a protein of 1072 amino acids, beginning at the first ATG codon at position 186 nt (Fig. 1A). The putative methionine codon is in an appropriate Kozak sequence (19). The ORF ended at a stop codon at position 3391 nt, suggesting that the cDNA clone contains a complete BEN coding sequence. This protein has a predicted molecular mass of 120.6 kDa and an isoelectric point of 6.2. It contains six unusually long HLH domains, found first in TFII-I, a transcription factor that binds to both Inr and E-box elements (18). A prosite search revealed multiple potential phosphorylation sites for protein kinase C and casein kinase 2 and single phosphorylation sites for cAMP/cGMP-dependent protein kinase and tyrosine kinases, respectively. Additional features include a hydrophobic leucine zipper dimerization motif at the N-terminal end at 276–341 nt, a nuclear localization signal at 3219–3239 nt, and a serine-rich repeat at the C-terminal end at 3243–3304 nt. One ATTTA motif, associated with rapid mRNA degradation (20) and two polyadenylation-like sequences (AATAAG and AATGAA) are located in the 3′ untranslated region (Fig. 1A).
Figure 1.

Nucleotide, deduced amino acid sequence, and structural organization of BEN. (A) The nucleotide and amino acid sequence of BEN. The six HLH repeat domains are highlighted in black. The putative nuclear localization signal is double underlined and the serine-rich region is single underlined. The amino acid residues in the hydrophobic leucine-zipper motif are indicated by circles. The mRNA destabilization motif is marked by asterisks. Two polyadenylation-like sequences are marked by dots. (B) Domain organization of BEN. LZ, potential leucine zipper motif; R1–R6, HLH repeat domains; NLS, putative nuclear localization signal; and SR, serine-rich region.
BEN-Related Proteins.
Amino acid analysis of BEN and mouse TFII-I (21) shows that homology between the two proteins is mostly restricted to the HLH domains (Fig. 2A). In addition, an N-terminal domain of BEN (amino acids 30–88) shows a 52.5% identity to the TFII-I sequence (amino acids 22–80) at the same location (Fig. 2B). A novel human gene, termed WBSCR11, was reported recently by Osborne et al. (8), while our project was underway. blast analysis between our murine clone and WBSCR11 revealed a high level of sequence similarity, greater than 76% identity at the nucleotide level. This homology is significantly higher than that of TFII-I at 30% identity. Murine BEN is longer than human WBSCR11 with 1072 aa vs. 944 aa, and it has six HLH domains, whereas WBSCR11 has only five (8). Identity at the amino acid level is 84% in the first 773 residues, whereas there is only 34% identity between BEN and mouse TFII-I throughout the entire protein. We view TFII-I as being an evolutionarily different gene from BEN, but related, whereas human WBSCR11 is a direct ortholog of murine BEN.
Figure 2.

Sequence alignments of BEN and TFII-I. (A) Amino acid sequence alignments of the HLH repeat domains of BEN and TFII-I (R1–R6). Conserved residues are highlighted with black, and consensus amino acid sequences are indicated below. (B) Alignment of the N-terminal 59-aa residues of BEN (amino acids 30–88) and TFII-I (amino acids 22–80).
BEN Embryonic Expression and Tissue Distribution.
The tissue distribution of BEN mRNA was determined by Northern blot and RNA dot blot analysis. The BEN transcript is expressed in all mouse tissues that have been examined. Northern blots probed at high stringency showed three predominant transcripts of about 3.6, 4.4, and 5.9 kb (Fig. 3A). The ratio and intensity of these transcripts varied among different tissues. BEN is expressed in E7 mouse embryos based on RNA dot blot analysis (data not shown).
Figure 3.

BEN expression in mouse tissues. (A) BEN mRNA was detected by Northern blot analysis of mRNA from adult mouse tissues (2 μg per lane) with BEN cDNA probe. The positions of RNA molecular weight markers are shown on the left. (B) Immunodetection of the BEN protein in mouse tissues. Western blot analysis was performed with BEN-specific polyclonal Abs. Total cellular extracts were run on 7.5% SDS/PAGE gel and processed as described in Materials and Methods. The protein standards (Novagene) are shown on the left.
The embryonic expression pattern of BEN in the mouse was examined by whole-mount in situ hybridizations using antisense RNA probe (summarized in Table 2).
Table 2.
Embryonic expression of BEN (whole-mount in situ hybridization with antisense RNA)
| Embryonic day | Forebrain | Midbrain | Hindbrain | Branchial arches (1st + 2nd) | Otic vesicle | Limb buds | Somites | Neural tube |
|---|---|---|---|---|---|---|---|---|
| 8.0–8.5 | + | + | − | − | − | − | + | + |
| 9.5 | + | + | + | + | − | + | + | + |
| 10.5 | + | + | + | + | + | + | − | + |
| 11.5 | + | + | − | + | − | + | − | − |
| 12.5 | − | + | + | + | − | + | − | − |
At an E8–8.5, BEN is already expressed in somites, neural tube, and brain. The BEN expression pattern is constant from E9.5 to E12.5, with the highest expression levels in the limb buds, branchial arches, craniofacial area, brain, and spinal cord.
Western blotting with BEN-specific Abs was performed to detect endogenous protein expression in adult tissues. Several BEN-specific polypeptides of 165, 145, 120, 100, 71, 62, and 40 kDa were detected in different tissues. These proteins were not detected by using preimmune serum (Fig. 3B). The predicted size of the intact BEN protein is 120.6 kDa, which corresponds to the observed 120-kDa band. Polypeptides of dissimilar size were also observed on Western blots. The quantity and molecular masses of these polypeptides are different in various tissues. Proteins of 120, 100, 71, and 62 kDa are expressed in most tissues. The 145-kDa isoform is expressed mostly in thymus, spleen, and liver. The 40-kDa band is present in spleen, heart, and lung, and there is an expression of a unique 165-kDa polypeptide in heart tissues. Several factors, such as differently spliced transcripts and posttranslational modifications, could explain these size differences.
BEN Binds to the EFG Site Within the Early Enhancer of Hoxc8.
TFII-I protein has been reported previously to bind to both E-box and Inr-elements in a sequence-specific manner (18). To examine the binding activity of the protein encoded by the clone OH40, we performed an in vitro transcription/translation reaction to generate BEN-specific polypeptides and used them in mobility shift experiments. A specific band was formed when the EFG oligonucleotides were incubated in the binding reaction in the presence of in vitro translated products. This band could be competed away by increasing amounts of unlabeled EFG oligonucleotide (Fig. 4A), but not with nonspecific oligonucleotides (data not shown).
Figure 4.
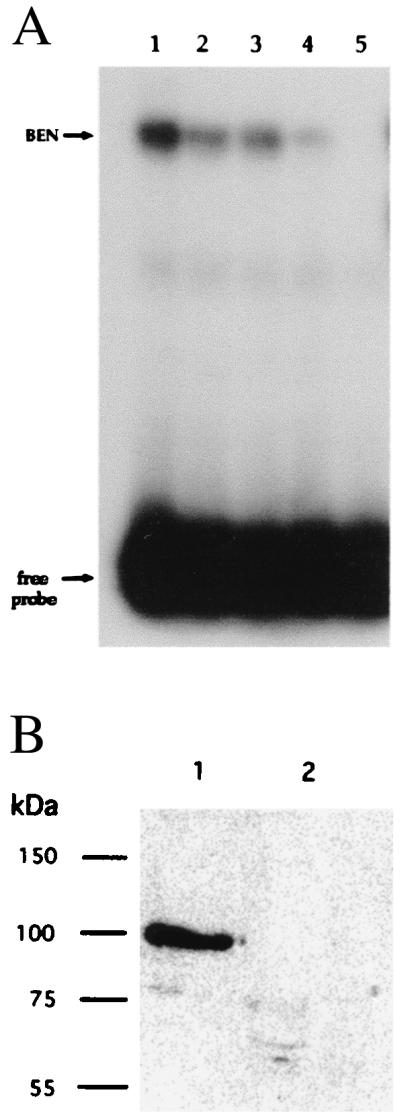
DNA-binding studies of BEN. (A) EMSA of the binding of in vitro translated BEN protein to the EFG probe. All lanes contain labeled EFG oligonucleotide and in vitro translated BEN protein. Lane 1, no competitor; lane 2 to 5 have 5-, 10-, 20-, and 40-fold excess of cold EFG oligonucleotide. (B) Southwestern blot analysis of BEN shows that affinity-purified His-tagged BEN binds the EFG sequence. The proteins purified on a nickel column were run on 10% SDS/PAGE and electroblotted to a nitrocellulose membrane. The filters were probed with labeled EFG (lane 1) or UCD (lane 2) oligonucleotides, respectively. The single band around 90 kDa is the truncated His-tagged BEN fusion protein.
We also used a His-tagged BEN fusion protein, purified on an affinity column, in Southwestern experiments. The results showed that the fusion protein bound preferentially to the EFG oligonucleotide, but not to a nonspecific DNA sequence (Fig. 4B). These DNA-binding studies show that BEN binds specifically and with high affinity to the EFG sequence derived from the EE of Hoxc8.
Discussion
The present study reports on the cloning and characterization of BEN, a murine sequence-specific DNA-binding protein. The partial cDNA encoding BEN was isolated employing a yeast one-hybrid screen of an E11 mouse embryo cDNA library using the EFG motif of the Hoxc8 EE as bait. Using this approach, we were able to identify several DNA-binding proteins, some of which represent different transcription factor families (Table 1). One of the clones, termed BEN, encodes a member of the TFII-I transcription factor family of HLH proteins. TFII-I, also known as SPIN and BAP-135, is distinctly different from other known basic HLH (bHLH) proteins in that it contains multiple HLH domains (18, 21–23). These domains, measuring 95-aa residues with 69-aa loops, are unusually large in size compared with known bHLH proteins. The significance of the long loops is not yet clear, but they may facilitate contacts outside of the core DNA binding motifs as suggested for certain bHLH transcription factors (24). The loop domains within the bHLH proteins Max, USF, PHO4, and Mlx were shown to contact the phosphate backbone in the DNA sequence surrounding the E-box (24).
BEN has a leucine zipper-like motif at its N-terminal end and six HLH domains. This configuration may allow it to bind to DNA as either a homo- or heterodimer. The multiple and long HLH domains could then possibly recognize a number of different nucleotide motifs, depending on several factors, such as other interacting proteins or conformational changes within BEN, possibly potentiated by specific posttranslational modifications. The loop in the HLH domain could also be involved in stabilizing protein–protein contacts. It was reported that TFII-I binds specifically to several different transcription factors, for example, SRF, Phox, UBF, NF-κB, STAT1, STAT3, and c-myc (22, 25–28), as well as signaling molecules such as Bruton's tyrosine kinase (23, 29, 30). The presence of multiple HLH repeat domains suggests that the DNA-binding activities of BEN and functional properties, like those of TFII-I, could be multiple and possibly more complex than other members of bHLH families.
TFII-I has been studied in depth and may provide insight into the functional roles of BEN. TFII-I was originally identified as a nuclear factor that interacts cooperatively with USF and c-myc at both Inr- and E-box sites and supports basal transcription from the adenovirus major late promoter (25, 28). TFII-I also cooperates with TBP on an Inr-containing TATA-less promoter that requires TFIID (28). Another transcription factor, NF-κB, was shown to recruit TFII-I to the Inr element and to complete preinitiation complex formation on HIV Inr (26). TFII-I promotes the formation of stable higher-order complexes of SRF and Phox and is responsible for serum-inducible transcription of downstream target genes (22). It also forms in vivo protein–protein complexes with c-fos upstream activators SRF, STAT1, and STAT3 (27). The ras pathway is involved in this TFII-I regulation of the c-fos gene (27). It was shown that Ras and RhoA synergize with TFII-I in supporting c-fos promoter activation (31). The TFII-I function is also dependent on an active mitogen-activated protein (MAP) kinase pathway (31). Another interesting interaction of TFII-I is in a signaling cascade originating at the B cell Ag receptor, in which TFII-I is a downstream target for Bruton's tyrosine kinase, a member of the non-receptor protein-tyrosine kinases (23, 29). Many studies implicate TFII-I in the formation of multiprotein complexes (enhanceosomes) that interact with cis-regulatory motifs. BEN may play a similar role with respect to the EFG region of the Hoxc8 EE. It is interesting in this respect that STAT binding sequences are present in the EFG region, and as described above, TFII-I has been shown to interact with STAT proteins in the activation of c-fos.
The DNA-binding mechanism of TFII-I is different from known bHLH proteins in that it can interact both with E-box and Inr elements (18). The molecular basis for these interactions is not yet well elucidated. The homology and structural similarity between BEN and TFII-I support the conclusion that they are members of a unique family of HLH transcription factors (8).
WBSCR11 is the human counterpart of murine BEN, and information on WBSCR11 can be expected to provide further insight into the functional role of BEN. There are five recent reports on WBSCR11, four of which use pseudonyms such as GTF2IRD, GTF3, Cream1, and MusTRD1. Presumably, the authors were unaware of each other's work, or believed that slight differences in gene structure could be interpreted as a different gene at another genetic locus. Our comparisons of DNA sequence, derived amino acid sequence, and gene map position suggest to us that all five names describe the same human gene, which is counterpart of murine BEN. MusTRD1 was described as being expressed predominately in muscle (Mus = muscle) although we believe the authors overlooked its more universal expression. MusTRD1 is reported to bind to the human troponin I slow upstream enhancer B1 element. This element is essential for high level troponin I expression in slow-twitch muscles (12). The major difference from WBSCR11 is that MusTRD1 contains only two HLH repeat domains instead of the five found in WBSCR11 (8). A possible explanation for this discrepancy is sequencing error, resulting in a truncated product, as explained recently by Franke et al. (9).
The N-terminal domain and HLH domains most probably have critical functional importance related to protein–protein/protein–DNA interactions. It was shown that the N-terminal part of Cream1 (WBSCR11) is involved in protein interactions with transcriptional machinery proteins (11). The Cream1 protein has also been reported to bind to the retinoblastoma protein through its C-terminal end and may play a role in cell cycle regulation (11). These recent studies reinforce those on TFII-I and WBSCR11, suggesting that BEN may potentiate the formation of multiprotein complexes that interact with DNA control motifs and that these interactions may take place in a broad array of transcriptional regulatory systems.
The possible role of BEN as a developmental factor has been brought into focus by the recent reports linking WBSCR11 as a putative causal factor in the human developmental abnormality described as WS (8–10). WS is an autosomal dominant genetic condition characterized by an ensemble of physical, cognitive, and behavioral traits (32). Typical pathologies include facial dysmorphology, vascular stenoses, growth deficiencies, dental anomalies, and neurologic and musculoskeletal abnormalities (33, 34). Most affected individuals demonstrate an uneven cognitive profile, having mild to moderate mental retardation, with relative strengths in verbal processing and selected language skills, but dramatic weaknesses in visual-spatial skills (34). Persons with WS also display a unique combination of behavioral traits, often showing a friendly engaging demeanor that coexists with an anxiety disorder, and a shortened attention span (35). The syndrome has been mapped to 7q11.23, where genetic causation is attributed to a microdeletion ranging up to 1.5 Mb in length, producing a haploinsufficiency condition for genes that map to this region (36, 37). Interestingly, WBSCR11, the human ortholog of murine BEN, maps to the critical deleted area. Two other genes have been shown to map to this region. Surprisingly, one of these is TFII-I, a homolog of BEN. The other is CLIP-115, reported to link specific cellular organelles to the cytoskeleton via microtubules (38).
Transcription factors are often dosage sensitive, and haploinsufficiency syndromes have previously been shown to express as dominant developmental disorders. For example, several mutations within the Pitx2 homeodomain region are specifically responsible for the development of Rieger syndrome, characterized by ocular and dental malformations (39, 40). Mutations in PAX2 result in Renal-Coloboma syndrome, an autosomal dominant disorder characterized by colobomatous eye defects, vesicoureteral reflux, and abnormal kidneys (41). Another example is the Saethre-Chotzen syndrome, which is characterized by craniosynostosis and limb abnormalities and is associated with mutations in TWIST (42). Additional haploinsufficiency conditions associated with transcription factors are Greig cephalopolysyndactyly, Pallister-Hall, Waardenburg syndrome type 2, Boston-type craniosynostosis, and Townes-Brocks syndrome (43–46).
Although BEN expression is widespread in adult mouse tissues, in developing embryos (E9.5–13) the highest levels are found in domains of epithelial-mesenchymal interactions such as limb buds, branchial arches, and craniofacial areas. This expression pattern raises the possibility that BEN may participate in regulating mesoderm induction or differentiation at these sites. We have also seen expression in different regions of the developing brain at E8.5–12.0. The expression pattern of BEN in mice is certainly consistent with its possible role in craniofacial development. The expression pattern of BEN is also consistent with a possible interaction with the Hoxc8 EE, because the expression of BEN and Hoxc8 overlap to a considerable degree.
In conclusion, BEN emerges as an interesting new transcription factor that may mediate transcription complex formation and interaction at multiple cis regulatory sites. It will be of interest to determine its specific functional properties in mice by transgenic and knockout methodologies and by these means to provide insight into the functional role of the human WBSCR11 gene in normal and abnormal development in man.
Acknowledgments
We thank Barbara Pober for helpful discussions, and Trevor Williams, Steve Irvine, and Lewis Lukens for critical reading of the manuscript. This work was supported by National Institutes of Health Grant GM0966-37.
Abbreviations
- EST
expressed sequence tag
- HLH
helix–loop–helix
- bHLH
basic HLH
- EE
early enhancer
- BEN
binding factor for EE
- STAT
signal transducer and activator of transcription
- WS
Williams Syndrome
- En
embryonic day n
Footnotes
Data deposition: The sequence reported in this paper has been deposited in the GenBank database (accession no. AF 260133)
References
- 1.Krumlauf R. Cell. 1994;78:191–201. doi: 10.1016/0092-8674(94)90290-9. [DOI] [PubMed] [Google Scholar]
- 2.Scott M, Weiner A. Proc Natl Acad Sci USA. 1984;81:4115–4119. doi: 10.1073/pnas.81.13.4115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McGinnis W R, Garber J W, Kuroiwa A, Gehring W. Cell. 1984;37:403–408. doi: 10.1016/0092-8674(84)90370-2. [DOI] [PubMed] [Google Scholar]
- 4.Ruddle F H, Bartels J L, Bentley K L, Kappen C, Murtha M T, Pendleton J W. Annu Rev Genet. 1994;28:432–442. doi: 10.1146/annurev.ge.28.120194.002231. [DOI] [PubMed] [Google Scholar]
- 5.Shashikant C S, Bieberich C J, Belting H G, Wang C H, Borbely M A, Ruddle F H. Development. 1995;121:4339–4347. doi: 10.1242/dev.121.12.4339. [DOI] [PubMed] [Google Scholar]
- 6.Shashikant C S, Ruddle F H. Proc Natl Acad Sci USA. 1996;93:12364–12369. doi: 10.1073/pnas.93.22.12364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bradshaw M S, Shashikant C S, Belting H G, Bollekens J A, Ruddle F H. Proc Natl Acad Sci USA. 1996;93:2426–2430. doi: 10.1073/pnas.93.6.2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Osborne L R, Campbell T, Daradich A, Scherer S W, Tsui L-C. Genomics. 1999;57:279–284. doi: 10.1006/geno.1999.5784. [DOI] [PubMed] [Google Scholar]
- 9.Franke Y, Peoples R J, Francke U. Cytogenet Cell Genet. 1999;86:296–304. doi: 10.1159/000015322. [DOI] [PubMed] [Google Scholar]
- 10.Tassabejhi M, Carette M, Wilmot C, Donnai D, Read A P, Metcalfe K. Eur J Hum Genet. 1999;7:737–747. doi: 10.1038/sj.ejhg.5200396. [DOI] [PubMed] [Google Scholar]
- 11.Yan X, Zhao X, Quan M, Guo N, Gong X, Zhu X. Biochem J. 2000;345:749–757. [PMC free article] [PubMed] [Google Scholar]
- 12.O'Mahoney J V, Guven K L, Lin J, Joya J E, Robinson C S, Wade R P, Hardeman E C. Mol Cell Biol. 1998;18:6641–6652. doi: 10.1128/mcb.18.11.6641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Andrews N C, Faller D V. Nucleic Acids Res. 1991;19:2499. doi: 10.1093/nar/19.9.2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sambrook J, Fritsch E F, Maniatis T. Molecular Cloning: A Laboratory Manual. 2nd Ed. Plainview, NY: Cold Spring Harbor Lab. Press; 1989. [Google Scholar]
- 15.Wilkinson D G. In: In Situ Hybridization. Wilkinson D, editor. Oxford: IRL; 1992. pp. 75–83. [Google Scholar]
- 16.Miskimins W K, Roberts M P, McClelland A, Ruddle F H. Proc Natl Acad Sci USA. 1985;82:6741–6744. doi: 10.1073/pnas.82.20.6741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bayarsaihan D, Enkhmandakh B, Lukens L. Biochem J. 1996;319:203–207. doi: 10.1042/bj3190203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roy A L, Du H, Gregor P D, Novina C D, Martinez E, Roeder R G. EMBO J. 1997;16:7091–7104. doi: 10.1093/emboj/16.23.7091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kozak M. Mamm Genome. 1996;6:563–574. doi: 10.1007/s003359900171. [DOI] [PubMed] [Google Scholar]
- 20.Reeves R, Nissen M S. J Biol Chem. 1990;265:8573–8582. [PubMed] [Google Scholar]
- 21.Wang Y-K, Perez Juardo L A, Francke U. Genomics. 1998;48:163–170. doi: 10.1006/geno.1997.5182. [DOI] [PubMed] [Google Scholar]
- 22.Grueneberg D A, Henry R W, Brauer A, Novina C D, Cheriyath V, Roy A L, Gilman M. Genes Dev. 1997;11:2482–2493. doi: 10.1101/gad.11.19.2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang W, Desiderio S. Proc Natl Acad Sci USA. 1997;94:604–609. doi: 10.1073/pnas.94.2.604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Billin A N, Eilers A L, Queva C, Ayer D E. J Biol Chem. 1999;274:36344–36350. doi: 10.1074/jbc.274.51.36344. [DOI] [PubMed] [Google Scholar]
- 25.Roy A L, Meisterernst M, Pognonec P, Roeder R G. Nature (London) 1991;354:245–248. doi: 10.1038/354245a0. [DOI] [PubMed] [Google Scholar]
- 26.Montano M A, Kripke K, Novina C D, Achacoso P, Herzenberg L A, Roy A L, Nolan G P. Proc Natl Acad Sci USA. 1996;93:12376–12381. doi: 10.1073/pnas.93.22.12376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim D W, Cheriyath V, Roy A L, Cochran B H. Mol Cell Biol. 1998;18:3310–3320. doi: 10.1128/mcb.18.6.3310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roy A L, Carruthers C, Gutjahr T, Roeder R G. Nature (London) 1993;365:359–361. doi: 10.1038/365359a0. [DOI] [PubMed] [Google Scholar]
- 29.Mano H. Cytokine Growth Factor Rev. 1999;10:267–280. doi: 10.1016/s1359-6101(99)00019-2. [DOI] [PubMed] [Google Scholar]
- 30.Novina C D, Kumar S, Bajpai U, Cheriyath V, Zhang K, Pillai S, Wortis H H, Roy A L. Mol Cell Biol. 1999;19:5014–5024. doi: 10.1128/mcb.19.7.5014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim D W, Cochran B H. Mol Cell Biol. 2000;20:1140–1148. doi: 10.1128/mcb.20.4.1140-1148.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morris C A, Demsey S A, Leonard C D, Dilts C, Blackburn B L. J Pediatr. 1988;113:318–326. doi: 10.1016/s0022-3476(88)80272-5. [DOI] [PubMed] [Google Scholar]
- 33.Lashkari A, Smith A K, Graham J M. Clin Pediatr. 1999;38:189–208. doi: 10.1177/000992289903800401. [DOI] [PubMed] [Google Scholar]
- 34.Bellugi U, Lichtenberger L, Mills D, Garaburda A, Korenberg J R. Trends Neurosci. 1999;22:197–207. doi: 10.1016/s0166-2236(99)01397-1. [DOI] [PubMed] [Google Scholar]
- 35.Pober B R, Dykens E M. Child Adolesc Psychiatr Clin North Am. 1996;5:929–943. [Google Scholar]
- 36.Francke U. Hum Mol Genet. 1999;8:1947–1954. doi: 10.1093/hmg/8.10.1947. [DOI] [PubMed] [Google Scholar]
- 37.Osborne L. Mol Genet Metabol. 1999;67:1–10. doi: 10.1006/mgme.1999.2844. [DOI] [PubMed] [Google Scholar]
- 38.De Zeeuw C I, Hoogenraad C C, Goedknegt E, Hertzberg E, Neubauer A, Grosveld F, Galjart N. Neuron. 1997;19:1187–1199. doi: 10.1016/s0896-6273(00)80411-0. [DOI] [PubMed] [Google Scholar]
- 39.Semina E V, Reiter R, Leysens N J, Alward W L, Small K W, Datson N A, Siegel-Bartelt J, Bierke-Nelson D, Bitoun P, Zabel B U, Carey J C, Murray J C. Nat Genet. 1996;14:392–399. doi: 10.1038/ng1296-392. [DOI] [PubMed] [Google Scholar]
- 40.Gage P J, Suh H, Camper S A. Development. 1999;126:4643–4651. doi: 10.1242/dev.126.20.4643. [DOI] [PubMed] [Google Scholar]
- 41.Eccles M R, Schimmenti L A. Clin Genet. 1999;56:1–9. doi: 10.1034/j.1399-0004.1999.560101.x. [DOI] [PubMed] [Google Scholar]
- 42.Howard T D, Paznekas W A, Green E D, Chiang L C, Ma N, Ortiz de Luna R I, Garcia Delgado C, Gonzalez-Ramos M, Kline A D, Jabs E W. Nat Genet. 1997;15:36–41. doi: 10.1038/ng0197-36. [DOI] [PubMed] [Google Scholar]
- 43.Kalff-Suske M, Wild A, Topp J, Wessling M, Jacobsen E M, Bornholdt D, Engel H, Heuer H, Aalfs C M, Ausems M G, et al. Hum Mol Genet. 1999;8:1769–1777. doi: 10.1093/hmg/8.9.1769. [DOI] [PubMed] [Google Scholar]
- 44.Shibahara S, Yasumoto K, Amae S, Fuse N, Udono T, Takahashi K. J Invest Dermatol Symp Proc. 1999;4:101–104. [PubMed] [Google Scholar]
- 45.Hehr U, Muenke M. Mol Genet Metabol. 1999;68:139–151. doi: 10.1006/mgme.1999.2915. [DOI] [PubMed] [Google Scholar]
- 46.Doray B, Langer B, Stoll C. Genet Couns. 1999;10:359–367. [PubMed] [Google Scholar]