

Abstract
Nbk/Bik (natural born killer/Bcl-2-interacting killer) is a tissue-specific BH3-only protein whose molecular function is still largely unknown. To investigate the mechanism of Nbk action, we established a single- vector adenoviral system based on the Tet-off conditional expression of Nbk. Upon Nbk expression, only Bax-positive, but not Bax-deficient cells were found to undergo apoptosis. Interestingly, Nbk failed to induce apoptosis in the absence of Bax, even despite expression of the related molecule Bak. Re-expression of Bax restored the sensitivity to Nbk. Similarly, Bax wild-type HCT116 cells were highly susceptible, whereas HCT116 Bax knock-out cells remained resistant to Nbk-induced apoptosis. In Bax-positive cells, Nbk induced a conformational switch in the Bax N-terminus coinciding with cytochrome c release, mitochondrial permeability transition and caspase-9 processing. Immunoprecipitation studies revealed that Nbk interacts with Bcl-xL and Bcl-2 but not with Bax. Since, in addition, Nbk did not localize to the mitochondria, our data suggest a model in which Nbk acts as an indirect killer to trigger Bax-dependent apoptosis, whereas Bak is not sufficient to confer sensitivity to Nbk.
Keywords: apoptosis/Bax/Bik/mitochondria/Nbk
Introduction
Bcl-2 proteins are essential mediators of death pathways initiated at the mitochondrial level. Structural analyses revealed that the apoptosis-promoting Bcl-2 members can be subdivided into two subfamilies: (i) the Bax homologs including Bax, Bak and Bok/Mtd, and (ii) the BH3-only subfamily (reviewed in Huang and Strasser, 2000; Puthalakath and Strasser, 2002; Borner, 2003; Daniel et al., 2003).
Unlike several other members of the BH3-only family, the function and regulation of Nbk/Bik (natural born killer/Bcl-2-interacting killer) (Boyd et al., 1995; Han et al., 1996) are still poorly defined. Nbk shows a rather tissue-specific expression pattern which is restricted to a subset of human epithelial tissues and activated lymphoid B cells (Daniel et al., 1999), suggesting that Nbk plays a role in tissue-specific regulation of apoptosis. Ectopic expression of Nbk restored sensitivity to anti-cancer drugs in resistant tumor cells and impaired tumorigenicity in a mouse xenotransplant model (Daniel et al., 1999; Radetzki et al., 2002).
A recent report showed that the Nbk BH3 domain is essential for apoptosis induction and its interaction with Bcl-xL (Tong et al., 2001). Similarly, the BH3-only protein Bad interacts via its BH3 domain with Bcl-xL (Kelekar et al., 1997). It has also been proposed that the activity of Nbk is regulated by phosphorylation. Unlike in the case of Bad, phosphorylation increases the pro-apoptotic potency of Nbk by a presently unknown mechanism that does not affect its affinity for anti-apoptotic Bcl-2 members (Verma et al., 2001). Altogether, the mechanisms by which Nbk is restrained in healthy cells and by which it induces apoptosis remain largely unclear.
In order to investigate the mechanism of Nbk action and to explore a potential use of Nbk in experimental cancer models, we established a single-vector conditional adenoviral expression system based on the Tet-off system (Gossen and Bujard, 1992). Employing this conditional expression system, we show that Nbk acts as an activator of the mitochondrial apoptotic pathway through a strictly Bax-dependent mechanism. Bax-negative carcinoma cells were completely refractory to Nbk-induced apoptosis, even though they expressed the Bax-related molecule Bak. Further analyses indicated that the proapoptotic effect of Nbk was mediated by an indirect effect on Bax: Nbk did not interact directly with Bax, but instead bound to Bcl-xL and Bcl-2. Thus, our data suggest a model in which Nbk acts as an indirect killer that triggers Bax-dependent, but Bak-independent apoptosis.
Results
Conditional expression of Nbk
We previously showed that Nbk enhances the sensitivity to different apoptosis stimuli such as CD95/Fas ligation or anti-cancer drugs (Daniel et al., 1999a; Radetzki et al., 2002). To investigate further the mechanism of this effect and to explore a potential use of Nbk gene transfer in cancer therapy, we aimed at establishing an adenoviral expression system. To circumvent the problem of induction of apoptosis in the HEK293 packaging cell line by Nbk, we constructed a conditional Tet-off expression system. To this end, the E3 region of Ad5 was replaced by the reverse tetracyclin-controlled transactivator (tTA) under the control of a cytomegalovirus (CMV) promoter and an SV40 poly(A) tail. To facilitate detection of the virally transduced Nbk, we introduced a myc tag 5′ of the Nbk cDNA and introduced the myc-Nbk cDNA into the E1 region under control of the Tet-off system to achieve conditional expression in the absence of doxycyclin (Figure 1A). The resulting Ad5-myc-Nbk-tTA DNA construct was transfected in HEK293 packaging cells to produce vector stocks. Transduction of DU145 prostate carcinoma cells with Ad5-myc-Nbk-tTA resulted in high expression of the 24 kDa myc-Nbk protein in the absence of doxycyclin, i.e. the Tet-on condition. Expression of Nbk was almost completely repressed at doxycyclin concentrations of ≥10 ng/ml (Tet-off condition; Figure 1B).

Fig. 1. Inducible Nbk expression mediated by Ad5-myc-Nbk-tTA. (A) Genomic structure of recombinant adenovirus Ad5-myc-Nbk-tTA. Ad5 sequences are indicated by black dashes. E1 and E3 regions of Ad5 are replaced by the myc-NBK expression cassette (white boxes) and tTA expression cassette (shaded boxes), respectively. [PCMV, immediate-early promoter of cytomegalovirus; tTA, tetracyclin- controlled (Tet-off) transactivator; PTRE, tetracyclin-responsive element located 5′ of the minimal immediate-early CMV promoter.] (B) Western blot analysis of Nbk expression in DU145 cells. Cells were infected with Ad5-myc-NBK-tTA at an m.o.i. of 25 (except control) and treated with increasing concentrations of doxycyclin (Dox) for 24 h. Equal protein loading was confirmed by immunoblotting using an anti-actin antibody. Strong induction of Nbk protein can be detected after doxycyclin withdrawal (Tet-on condition) whereas, in the presence of doxycyclin (Tet-off condition), Nbk expression is repressed to a weak background signal.
Induction of apoptosis by Nbk gene transfer
To analyze the effect of adenoviral gene transfer of Nbk, we transduced a panel of carcinoma cells including the colon carcinoma lines LoVo, SW48 and SW480, as well as the prostate carcinoma line DU145. Adenoviral transduction in the presence of doxycyclin did not induce apoptosis and resulted in no or only marginal expression of the myc-Nbk transgene (Figures 1B and 2A). In the absence of doxycyclin, high levels of myc-Nbk (24 kDa) were expressed in all cell lines (Figure 2A). As previously shown (Daniel et al., 1999), SW480 cells expressed low levels of endogenous (22 kDa) Nbk which was not affected by the adenoviral gene transfer of Nbk.
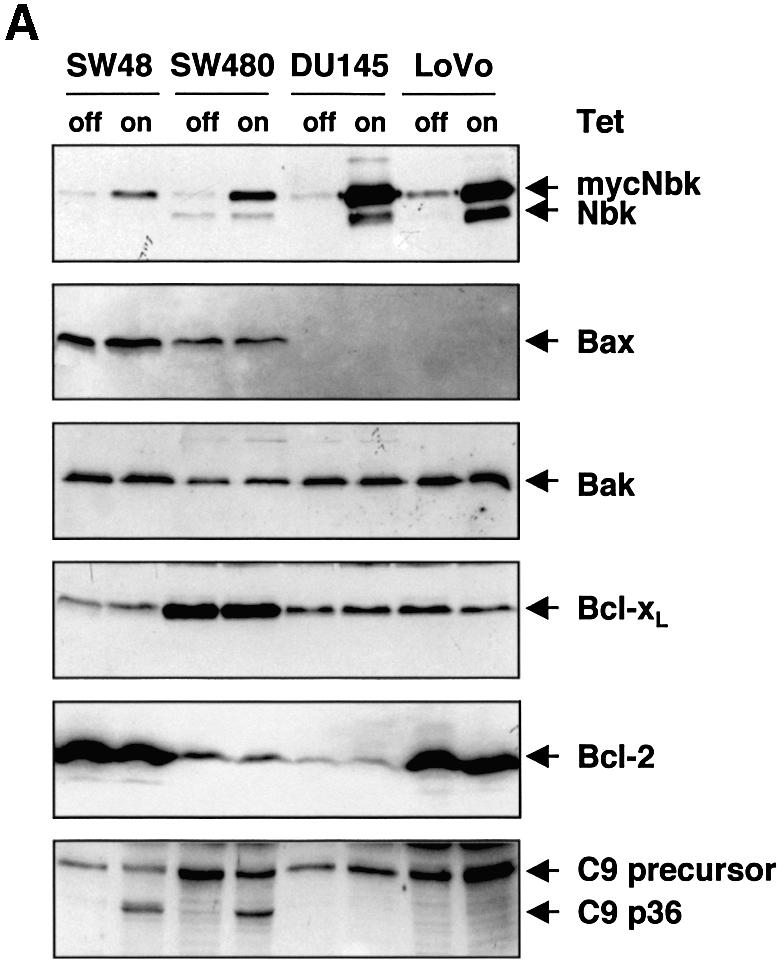
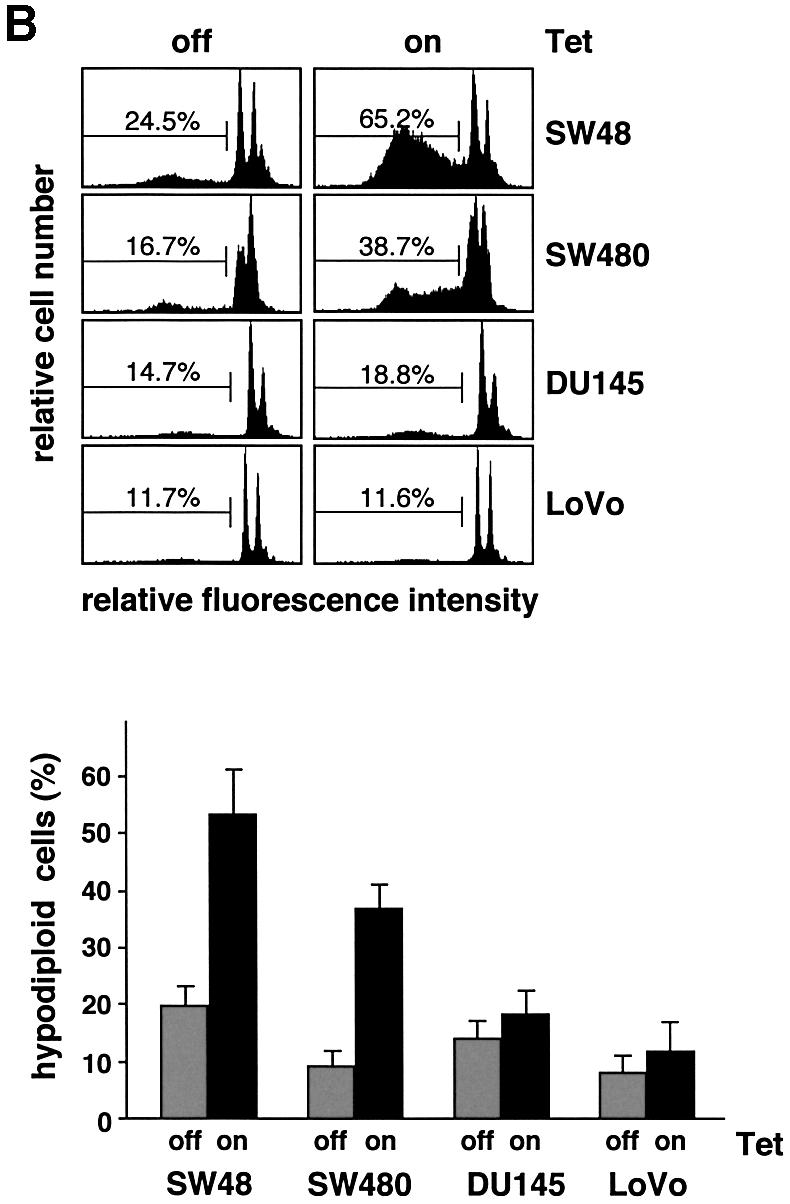
Fig. 2. Nbk expression induces apoptosis in Bax-positive but not in Bax-negative cells. Lovo, DU145, SW480 and SW48 cells were infected with Ad5-myc-NBK-tTA and cultured for 24 h in the presence (Tet-off condition) or absence of doxycyclin (Tet-on condition). (A) Western blot analysis for Nbk, Bax, Bak, Bcl-xL and Bcl-2 expression and processing of procaspase-9. (B) Flow cytometric detection of apoptotic cells based on measurement of the cellular DNA content. Upper panel: representative experiment. The percentage of hypodiploid, apoptotic cells is indicated between markers. Lower panel: means ± SD of the percentage of hypodiploid cells from three independent experiments.
In DU145 and LoVo cells, myc-Nbk gene transfer and expression of the 24 kDa myc-Nbk also increased the intensity of a 22 kDa band. Immunoprecipitation studies (see Figures 8 and 9) showed that only the upper band, i.e. myc-Nbk, could be precipitated by an anti-myc antibody. The lower band co-precipitated with Bcl-xL and was recognized by an anti-Nbk antibody. Neither the pan-caspase inhibitor zVAD-fmk nor calpain or cathepsin inhibitors prevented the appearance of the low molecular weight Nbk in transduced cells, suggesting that it was not generated by proteolytic cleavage (data not shown). It is known that eukaryotic ribosomes can ignore the first AUG and initiate translation at the next start codon, a phenomenon known as ‘leaky scanning’ (Saito and Tomita, 1999). As the start codon of endogenous Nbk is retained downstream and in-frame in the myc-tagged construct, the smaller form of Nbk was presumably generated from translation at the second AUG.

Fig. 8. Co-immunoprecipitation analyses show Nbk binding to Bcl-xL but not to Bax. SW480 cells were transduced with Ad5-myc-Nbk-tTA in the presence (Tet-off condition) or absence of doxycyclin (Tet-on condition) for 24 h. Lysates of cells solubilized in Triton X-100 buffer were immunoprecipitated with an anti-pan-Bax, anti-Bcl-xL or anti-c-Myc antibody. Immune complexes were resolved by SDS–PAGE and the presence of the respective protein was detected by immunoblotting, as indicated by arrows. Migration of the immunoglobulin light chain is indicated by an asterisk. S, supernatant; P, immunoprecipitate.

Fig. 9. Conformation-specific immunoprecipitation in CHAPS buffer. SW480 cells were infected with Ad5-myc-Nbk-tTA in the presence (Tet-off condition) or absence of doxycyclin (Tet-on condition) for 24 h, followed by cell lysis in CHAPS buffer. Immunoprecipitation was performed with antibodies against pan-Bax, activated Bax (conformation-specific antibody against the N-terminal epitope Bax-NT), Bcl-xL or c-myc. Immune complexes were resolved by SDS–PAGE, and the presence of each protein was detected by immunoblotting as indicated by arrows. Migration of the immunoglobulin light chain is indicated by an asterisk. S, supernatant; P, immunoprecipitate.
To measure induction of apoptosis upon Nbk expression, we performed flow cytometric analyses and assessed fragmentation of genomic DNA. Apoptotic cells were identified as cells with a hypodiploid, i.e. a sub-G1, DNA content. After 24 h of transduction with Ad5-myc-Nbk-tTA, a mean of 53.2% of the SW48 and 36.7% of the SW480 cells became apoptotic when doxycyclin was absent from the culture medium (Figure 2B). In contrast, a much smaller number of cells underwent cell death in the presence of doxycyclin, which inhibits Nbk transgene expression. To our surprise, transduction of Nbk did not significantly induce apoptosis in LoVo and DU145 cells, in both the absence and presence of doxycyclin. This was not due to defective Nbk expression, as both cell lines expressed large amounts of Nbk (Figure 2A). The observation that Nbk was even expressed at higher levels in apoptosis-resistant LoVo and DU145 cells as compared with the sensitive cell lines SW48 and SW480 suggests that there is a selection pressure against the expression of Nbk.
Since DU145 cells and LoVo cells carry frameshift mutations in the Bax gene, they do not express Bax protein (Figure 2A). In contrast, SW48 and SW480 cells do express Bax, and this clear difference indicated that Bax might be required for apoptosis induction by Nbk. In addition, the higher expression level of Bcl-xL (Figure 2A) could also contribute to the weaker induction of apoptosis in SW480 cells as compared with the SW48 cells (Figure 2B). However, there was no correlation between the expression levels of Bcl-2 and sensitivity to apoptosis induced by Nbk expression. Among the two Nbk-sensitive and Bax-positive lines, SW48 expressed high levels of Bcl-2, whereas SW480 showed low Bcl-2 expression. From the two Nbk-resistant and Bax-negative lines, DU145 cells marginally expressed Bcl-2, whereas LoVo cells showed high Bcl-2 expression (Figure 2A). Interestingly, both LoVo and DU145 cells expressed detectable levels of the Bax-related protein Bak (Figure 2A). Thus, the pro-apoptotic Bak alone was not sufficient to mediate Nbk-induced apoptosis. In turn, this indirectly indicated that Nbk might signal specifically via Bax and not through a Bak-dependent pathway.
Expression of Bax sensitizes for apoptosis induction by Nbk
To support the hypothesis that apoptosis induction by Nbk is mediated by a Bax-dependent mechanism, we overexpressed wild-type Bax in the Bax-mutated DU145 cells. To this end, we employed a retroviral vector, HyTK-Bax (Hemmati et al., 2002), containing the Bax-α cDNA under the control of a CMV promoter. Cells were retrovirally transduced, selected with hygromycin and screened for transgene expression by western blot analysis. Three clones were selected, DU145-Bax 2, 3 and 4, and compared with vector-transduced mock controls. The clones DU145 Bax 2 and 4 constitutively expressed high levels of Bax, whereas clone 3 showed a much lower expression level (Figure 3A).

Fig. 3. Apoptotic alterations induced by Nbk in Bax-expressing DU145 clones. DU145 clones expressing exogenous Bax were generated by retroviral transfer of the Bax cDNA under control of the CMV promoter. The DU145-mock clone was generated by transduction with control HyTK retrovirus. Stable clones were transduced with Ad5-myc-Nbk-tTA and cultured for 24 h in the presence (Tet-off condition) or absence of doxycyclin (Tet-on condition). Control cells were mock treated and grown in the absence of doxycyclin. (A) Western blot analysis of Bax and Nbk expression, cytochrome c release and procaspase-9 processing in DU145-Bax clones 24 h after infection with Ad5-myc-NBK-tTA. (B) Disruption of mitochondrial membrane potential (Δψm) by mycNbk in DU145-Bax cells. Cells were incubated with JC-1, a cationic dye that exhibits potential-dependent accumulation in mitochondria, and fluorescence intensity was measured by flow cytometry. Upper panel: representative experiment. The percentage of cells with Δψm loss is indicated between markers. Lower panel: means ± SD of the percentages of cells with Δψm loss from three independent experiments.
Consistent with a requirement for Bax for Nbk-induced apoptosis, adenoviral Nbk expression triggered the release of cytochrome c in the absence of doxycyclin (Tet-on condition, Figure 3A). Transduction with Ad5-myc-Nbk-tTA induced the release of cytochrome c to a similar extent in all three Bax clones, but not in the mock transfectants. This indicated that the relatively low Bax expression in clone 3 was sufficient to restore Nbk-induced mitochondrial activation. Likewise, all Bax clones exhibited processing of procaspase-9 upon expression of Nbk (Figure 3A).
To analyze mitochondrial permeability transition and loss of mitochondrial membrane potential (ΔΨm) during apoptosis, cells were incubated with JC-1 (5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethyl-benzimidazolylcarbocyanin iodide), a cationic dye that exhibits membrane potential-dependent accumulation in mitochondria. JC-1 fluorescence intensity was measured by flow cytometry on a single-cell level. The analysis of loss in ΔΨm showed that all DU145 Bax transfectants underwent disruption of the mitochondrial membrane potential upon myc-Nbk expression to a similar extent. This loss of ΔΨm occurred in a mean of 44.7–52.8% of the individual Bax transfectants as compared with 13.6% of the mock transfectants after Ad5-myc-Nbk-tTA transduction in the absence of doxycyclin (Figure 3B).
A sensitization for Nbk in the Bax transfectants was also apparent when apoptosis induction was analyzed by measurements of either DNA fragmentation or phosphatidylserine exposure. Adenoviral Nbk expression induced formation of hypodiploid DNA in the Bax transfectants: a mean of 31.3% (Bax clone 2), 36.2% (Bax clone 3) or 17.3% (Bax clone 4) of the cells underwent apoptosis in the absence of doxycyclin, while only 7.2% of the mock transfectants died as determined by flow cytometric analysis of apoptotic sub-G1 cells (Figure 4A). Likewise, after transduction with Ad5-myc-Nbk-tTA and 18 h of incubation in the absence of doxycyclin, all DU145 transfectants showed an increased percentage of propidium iodide (PI)-negative cells exhibiting exposure of phosphatidylserine to the outer cell membrane (Figure 4B).
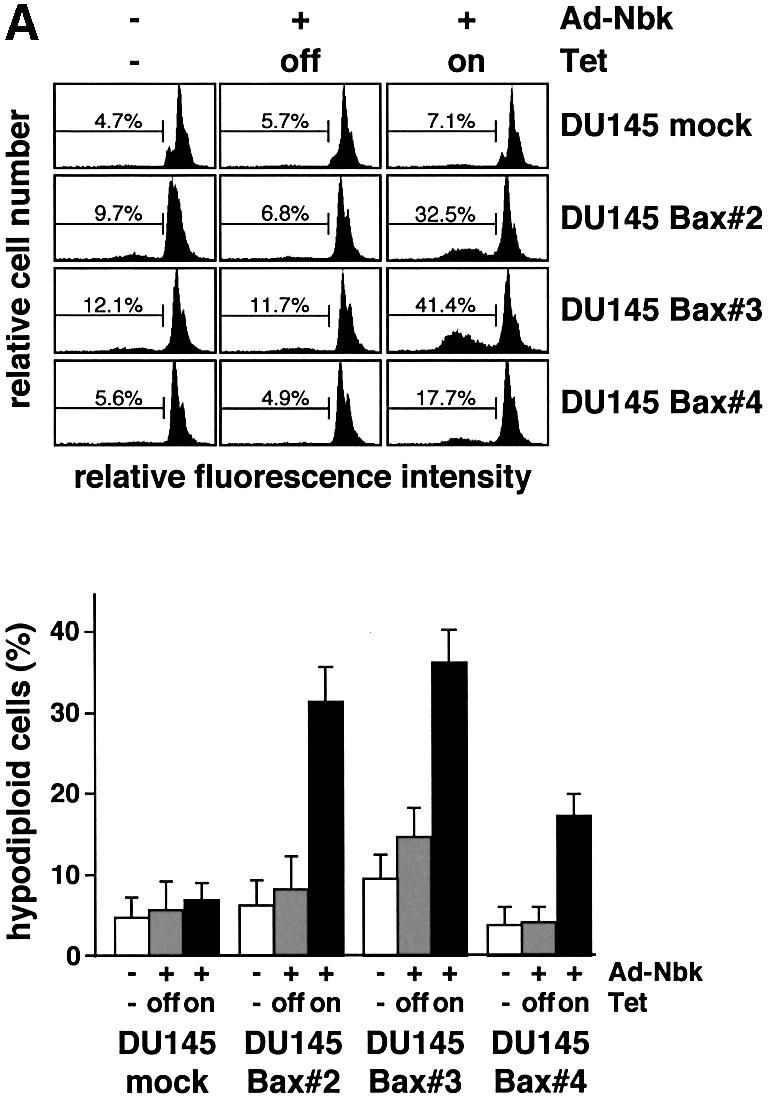

Fig. 4. Bax is required for Nbk-induced apoptosis. Stable Bax-expressing clones were transduced with Ad5-myc-Nbk-tTA as described in Figure 3 and cultured in the presence (Tet-off condition) or absence of doxycyclin (Tet-on condition). Control cells were mock treated and grown in the absence of doxycyclin. (A) Flow cytometric measurement of hypodiploid DNA. Upper panel: representative experiment. The percentage of apoptotic cells in a representative experiment was measured 24 h after adenoviral tranduction and is indicated between markers. Lower panel: means ± SD from three independent experiments. (B) Detection of phosphatidlyserine exposure: flow cytometric detection of apoptotic cells was performed by staining with annexin V–FITC to detect exposure of phosphatidylserine onto the cell surface 18 h after transduction. Cells were counterstained with propidium iodide (PI) to detect membrane damage. PI-positive cells were considered as necrotic and excluded from the analysis. The percentage of apoptotic cells is indicated between markers. Upper panel: representative experiment. Lower panel: means ± SD from three independent experiments.
The rates of apoptosis as measured by annexin V–fluorescein isothiocyanate (FITC) staining (Figure 4B) correlated with the extent of DNA fragmentation induced by Nbk in the DU145 clones (Figure 4A). As in the DNA fragmentation assays, clone 4 showed a lower induction of apoptosis as compared with clones 2 and 3 even despite high Bax expression. Nevertheless, Bax clone 4 revealed a loss of ΔΨm and release of cytochrome c similar to the other Bax clones. This observation is in line with an assumable selection disadvantage for cells with high Bax expression and an intact downstream signaling cascade. Clone 4 also showed impaired procaspase-9 cleavage (Figure 3A), indicating that it had presumably acquired a defect downstream of cytochrome c release resulting in decreased caspase activation and cell death.
To exclude potential clonal selection artifacts, we determined the role of Bax in an independent cell system. HCT116 cells express the wild-type Bax protein, whereas HCT116 Bax knock-out cells are devoid of Bax (Zhang et al., 2000). Similar to DU145 cells, we observed that conditional Nbk expression induced apoptosis in HCT116 colon carcinoma cells carrying the wild-type Bax gene, whereas Bax knock-out cells were resistant against induction of apoptosis (Figure 5A and D). Likewise, Ad5-myc-Nbk-tTA induced mitochondrial permeability transition, caspase-9 processing and poly(ADP-ribose)polymerase (PARP) cleavage only in the Bax wild-type cells, but not in the congeneic HCT116 Bax-deficient cells (Figure 5B and C). Notably, HCT116 cells expressed significant levels of Bak that were similar irrespective of the Bax status. These experiments therefore indicate that a sensitization to Nbk-induced apoptosis is observed not only after Bax overexpression, but also in the presence of endogenous Bax levels. Furthermore, the results are consistent with the assumption that Nbk-induced apoptosis proceeds in an entirely Bax-dependent manner which cannot be substituted by the presence of Bak.

Fig. 5. Induction of apoptosis by Nbk is entirely Bax dependent in HCT116 cells. HCT116 Bax wild-type (WT) and HCT116 Bax knock-out (k.o.) cells were transduced with Ad5-myc-Nbk-tTA and cultured for 24 h in the presence (Tet-off condition) or absence of doxycyclin (Tet-on condition). (A) Morphology of HCT116 cells. (B) Western blot analyses of Nbk, Bax, Bak expression, PARP and procaspase-9 cleavage. (C) Breakdown of the mitochondrial membrane potential. Loss of Δψm was measured on the single-cell level by the use of the cationic dye JC-1. Means ± SD were calculated from three independent experiments. (D) Apoptosis induction by Nbk. Genomic DNA fragmentation was measured on a single-cell level by determination of cells with a hypodiploid DNA content. Means ± SD were calculated from three independent experiments.
Nbk induces exposure of an N-terminal Bax epitope
The experiments described above clearly revealed that Nbk is not a direct activator of the mitochondrial pathway, but strongly depends on the presence of Bax. Recent studies reported that during apoptosis, Bax undergoes a conformational change leading to the exposure of an N-terminal epitope and insertion of cytosolic Bax into the outer mitochondrial membrane (Desagher et al., 1999; Eskes et al., 2000). Therefore, we performed immunofluorescent stainings with a conformation-specific antibody directed against the Bax N-terminus (Desagher et al., 1999). Figure 6 shows that this antibody stained permeabilized SW480 cells (mean 13.6%) after Ad5-myc-Nbk-tTA transduction in the absence of doxycyclin. In contrast, no staining was seen in Bax-negative DU145 cells or in Bax-positive SW480 cells in the presence of doxycyclin.

Fig. 6. Bax undergoes a conformational change in response to Nbk expression. SW480 cells, Bax-negative DU145 mock transfectants and DU145-Bax clones 2, 3 and 4 were transduced with Ad5-myc-NBK-tTA and cultured for 24 h in the presence (Tet-off condition) or absence of vector and doxycyclin (Tet-on condition). Control cells were grown in the absence of doxycyclin. (A) Cells were stained with a conformation-specific antibody against the Bax N-terminus and analyzed by flow cytometry. The percentage of immunostained cells is indicated between markers. (B) Bar chart showing mean ± SD of cells expressing activated Bax. Data were obtained from three independent experiments.
We also analyzed this conformational change in Bax-expressing DU145 cells after Ad5-myc-Nbk-tTA transduction. Staining of DU145 Bax clones showed an average of 28.2% positive cells for Bax clone 2, 18.4% for Bax clone 3 and 32.0% for Bax clone 4 in the absence of doxycyclin, i.e. the Tet-on condition (Figure 6). In contrast, only 1.8–3.8% of the cells displayed exposure of the N-terminal conformational Bax epitope in the presence of doxycyclin, i.e. in the absence of Nbk expression. Upon Nbk expression, the number of cells displaying a Bax conformational change was lower than the number of apoptotic cells. This can be explained by the fact that the exposure of the Bax N-terminus is a dynamic event. In addition, Bax can be cleaved by calpains at its N-terminus, generating a fragment that is not recognized by the antibody, but which is an even more potent inducer of apoptotic cell death than wild-type Bax (Toyota et al., 2003). Thus, the number of cells displaying the N-terminal switch of Bax may be lower than the extent of apoptosis detected by DNA fragmentation.
Interaction of Nbk with other Bcl-2 proteins
The conformational switch of the Bax N-terminus mediates its insertion into the outer mitochondrial membrane. We therefore determined the subcellular localization of Nbk in relation to staining of mitochondria with MitoTracker Green (Figure 7A and D). Confocal microscopy showed an almost complete suppression of Nbk expression in the presence of doxycyclin (Tet-off condition, Figure 7B) and strong expression of Nbk in the absence of doxycyclin (Tet-on condition, Figure 7E). Prior to the onset of apoptosis (e.g. 16 h after viral transduction) and also at later time points (data not shown), Nbk expression was confined to the cytoplasm. An overlay of the Nbk and the MitoTracker Green stainings demonstrated a complete segregation of the two signals (Figure 7F).

Fig. 7. Subcellular localization pattern of Nbk. Bax-expressing DU145 cells (clone 3) were transduced with Ad5-myc-Nbk-tTA. Cells were stained for Nbk protein expression after 16 h culture in the presence (Tet-off condition, A–C) or absence of doxycyclin (Tet-on condition, D–F). Nbk was visualized by goat-anti-Nbk followed by Alexa Fluor 594-conjugated chicken anti-goat IgG (red fluorescence; B and E). Cells were counterstained with MitoTracker Green (A and D). (C and F) The overlay of the Nbk and MitoTracker Green signals.
The immunocytochemical stainings indicated that Nbk was obviously not translocated to mitochondria. In contrast, Bid mediates exposure of the N-terminal Bax epitope upon its cleavage by caspase-8 and co-localizes with Bax to the mitochondria (Desagher et al., 1999). Therefore, Nbk mediates the conformational switch in the Bax N-terminus presumably through an indirect mechanism, e.g. by the interaction of Nbk with other Bcl-2 members. We therefore next performed co-immunoprecipitation experiments for Bcl-xL, Bax and Nbk. Immuno precipitation of Nbk was performed with an anti-myc antibody that precipitated the transduced myc-Nbk but not the endogenous Nbk (Figure 8A, lower panel). Bcl-xL readily co-immunoprecipitated with myc-Nbk (Figure 8A, middle panel). In contrast, immunoprecipitation of myc-Nbk showed no interaction with Bax (Figure 8A, upper panel). Bcl-xL, however, co-immunoprecipitated with Bax, which interestingly became less evident when Nbk was expressed (Figure 8B, middle panel). Also, in the reverse experiment, Bax showed co-immunoprecipitation with Bcl-xL and, again, this interaction was clearly weaker upon Nbk expression (Figure 8C, upper panel). Myc-Nbk also showed a strong co-precipitation with Bcl-xL, even when Triton X-100 was used as a detergent (Figure 8C, lower panel). Under these stringent conditions, Nbk did not co-immunoprecipitate with Bax (Figure 8B, lower panel). Notably, the co-immunoprecipitations between Bcl-xL and Bax or between Bcl-xL and Nbk were not complete, and part of the proteins remained in the supernatant. This might be explained by the possibilities that the proteins are not expressed in equimolar quantities or that additional interactions exist which prevented a quantitative immunoprecipitation.
Previous reports showed that detergents such as Triton X-100 trigger the conformational switch of the Bax N-terminus in vitro, facilitating Bax oligomerization (Hsu and Youle, 1998; Antonsson et al., 2000). Such Bax alterations do not occur when CHAPS is used as detergent, which should therefore allow a conformation-specific immunoprecipitation of Bax. Under CHAPS conditions, activated Bax was precipitated efficiently by a conformation-specific Bax antibody recognizing the N-terminal epitope when Nbk was expressed, but only weakly when Nbk expression was turned off by doxycyclin (Figure 9D, upper panel). Pan-Bax was precipitated efficiently by a pan-Bax antibody under both conditions (Figure 9B, upper panel). Similarly to the case in Triton X-100, in CHAPS buffer Bax did not co-precipitate with myc-Nbk, and vice versa (Figure 9A, upper panel; B, lower panel). Furthermore, the precipitation of activated Bax by the conformation-specific anti-Bax antibody did not result in immunoprecipitation of Nbk (Figure 9D, lower panel). Immunoprecipitation of Bcl-xL showed co-precipitation of Nbk, and vice versa (Figure 9A, middle panel; C, lower panel). In contrast to the experiments with Triton X-100 but consistent with previous reports (Hsu and Youle, 1998; Antonsson et al., 2000), we did not observe a co-immunoprecipitation of Bcl-xL with Bax and vice versa under CHAPS conditions (Figure 9B, middle panel; C, upper panel). As CHAPS can disrupt protein interactions, this does not exclude the possibility that Bax and Bcl-xL could interact in vivo. In this regard, Bcl-xL interaction with Bax was initially described in yeast two-hybrid studies (Sato et al., 1994; Yang et al., 1995) and confirmed by fluorescence resonance energy transfer (FRET) analyses in mammalian cells (Mahajan et al., 1998).
To address a potential interaction between Nbk and Bcl-2 in addition to Bcl-xL, we performed co-immunoprecipitation experiments in SW48 cells that demonstrate considerable expression of Bcl-2 (Figure 2). These results showed that Nbk interacts with both Bcl-xL and Bcl-2 under CHAPS or Triton X-100 conditions (Figure 10). No co-immunoprecipitation was observed between Nbk and Bcl-2 in SW480 cells that express low levels of Bcl-2 (data not shown). In summary, these results demonstrate that Nbk does not physically interact with Bax but induces the activation of Bax through an indirect mechanism. Moreover, Nbk interacts with Bcl-xL and Bcl-2 under both Triton X-100 and CHAPS buffer conditions, an event which presumably interferes with interaction of anti-apoptotic Bcl-2 proteins and Bax. Whether the interaction of Nbk with anti-apoptotic Bcl-2 homologs is strictly mandatory for the activation of Bax and the mitochondrial death cascade remains to be elucidated.

Fig. 10. Interaction between Nbk and Bcl-2. SW48 cells expressing high levels of endogenous Bcl-2 were transduced with Ad5-myc-Nbk-tTA in the presence (Tet-off condition) or absence of doxycyclin (Tet-on condition) for 24 h, followed by cell lysis in (A) Triton X-100 or (B) CHAPS buffer. Immunoprecipitation was performed with antibodies against pan-Bax, Bcl-xL, Bcl-2 or c-myc. Immune complexes were resolved by SDS–PAGE, and the presence of each protein was detected by immunoblotting as indicated by arrows. Migration of the immunoglobulin light chain is indicated by an asterisk. S, supernatant; P, immunoprecipitate.
Discussion
Members of the Bcl-2 family are key regulators of apoptosis. Tremendous progress has been made in elucidating the molecular basis of apoptosis regulation through both pro- and anti-apoptotic Bcl-2 family members. Nevertheless, the mechanism of action of the pro-apoptotic Bcl-2 homologs is still not completely understood. Bax has been shown to be a direct activator of mitochondria which triggers release of cytochrome c (Jürgensmeier et al., 1998) and other mitochondrial events such as permeability shift transition and the release of a variety of pro-apoptotic factors including AIF, Smac/Diablo and others. The Bax homologs Bak and Bok/Mtd that carry, like Bax, a BH1, BH2 and BH3 domain are believed to exert similar functions (Martinou and Green, 2001).
The mechanisms of apoptosis induction by Nbk, however, remain enigmatic. Nbk contains only one of the signature domains of the Bcl-2-family, the BH3 domain, and therefore might display a mode of action similar to other members of the BH3-only subfamily. Recently, an indirect mode of apoptosis induction was established for the BH3-only proteins Bad, Bid, Bim and Noxa that occurs via a Bax- or Bak-dependent pathway (Cheng et al., 2001; Zong et al., 2001). In the case of Bid, a truncated caspase cleavage product of Bid, tBid, triggers a conformational switch in the N-terminus of Bax (Desagher et al., 1999) or Bak (Wei et al., 2000), leading to activation of mitochondrial apoptosis signaling. Experiments in mouse embryonal fibroblasts showed that the activity of these BH3-only proteins in inducing apoptosis depends on the presence of Bax or Bak to trigger cytochrome c release and the mitochondrial apoptosis cascade (Cheng et al., 2001; Zong et al., 2001). This led to the hypothesis that all BH3-only proteins might share a similar mode of action, i.e. depend on Bax and its homologs to trigger apoptosis.
In the present study, we addressed the mechanism of action of Nbk in relation to Bax and the activation of the mitochondrial apoptosis cascade. To this end, we constructed a conditional adenoviral expression system based on the Tet-off system. The prominent finding of the present work is the fact that Nbk did not induce apoptosis in the Bax-negative carcinoma cells including Bax-mutated LoVo and DU145 cells, as well as HCT116 Bax knock-out cells.
The re-expression of Bax in DU145 cells restored the sensitivity for Ad5-myc-Nbk-tTA-induced apoptosis in these cells. Similarly, Bax wild-type HCT116 cells displayed high sensitivity to Nbk-induced apoptosis. Upon Nbk expression, the Bax-positive but not the Bax-negative cells showed release of cytochrome c, breakdown of the mitochondrial membrane potential and processing of procaspase-9. This formally demonstrates that Nbk acts via a Bax-dependent mechanism to activate the mitochondrial apoptosis pathway. Western blot analyses showed that the Bax-negative cell lines DU145, LoVo and HCT116 all express Bak. Thus, Bak could not compensate for the loss of Bax in these cells, indicating that Nbk signals preferentially via Bax and not Bak. In the case of other BH3-only proteins, such as Bad, Bid, Bim and Noxa, only the combined inactivation of both Bax and Bak significantly impaired apoptosis induction (Cheng et al., 2001; Zong et al., 2001). Altogether, Nbk appears to be an indirect inducer of mitochondrial apoptosis that entirely depends on Bax to exert its effect.
The BH3-only protein Bid induces cytochrome c release through a Bax-dependent mechanism (Desagher et al., 1999). This occurs by relieving inhibition of the Bax transmembrane signal anchor by the N-terminal domain, resulting in integration of Bax into the outer mitochondrial membrane (Eskes et al., 2000; Ruffolo et al., 2000). A recent report pointed out that Bax-independent mechanisms might also be relevant for Bid-induced mitochondrial activation. In this line, Bid was shown to activate mitochondria and cytochrome c release independently of Bax (Kim et al., 2000). Recently, it was even suggested that Bid by itself may possess channel-forming capabilities and may thus mediate cytochrome c release (Zhai et al., 2000) but does not interact with the voltage-dependent anion channel (Sugiyama et al., 2002).
Nbk differs from Bid in that it does not physically interact with Bax. In addition, Nbk does not localize to the mitochondria, whereas Bax does (Eskes et al., 2000; Ruffolo et al., 2000). Furthermore, so far we have found no evidence that Nbk is cleaved during apoptosis. This is in line with data from other BH3-only Bcl-2 homologs such as Bim and Bad. Both proteins have been shown to impair the anti-apoptotic effect of Bcl-2 or Bcl-xL and both also depend on Bax or Bak to exert their apoptosis-promoting effect (Cheng et al., 2001; Zong et al., 2001). Like in the case of Bad, Nbk might act via sequestration of Bcl-xL and Bcl-2, as suggested by the co-immunoprecipitation of Nbk and Bcl-2 or Bcl-xL, respectively, that was observed under both Triton X-100 and CHAPS buffer conditions. An interaction of Bax and Bcl-xL was observed, however, only in co-immunoprecipitation studies based on Triton X-100 as detergent, but not when CHAPS was employed.
It is conceivable that the binding of Nbk to Bcl-xL or Bcl-2 alone is sufficient to promote the conformational change of Bax, or that additional factors are required which would then, upon sequestration of Bcl-xL by Nbk, bind to Bax to trigger the pro-apoptotic conformation. Thus, the exact functional role of the interaction between Nbk and Bcl-xL or Bcl-2 remains to be determined. Evidence for a more complex mode of action comes from the observation that mutation of the phosphorylation sites at residues 33 (threonine) and 35 (serine) reduced the apoptotic activity of Nbk without significantly affecting its ability to heterodimerize with Bcl-2 (Verma et al., 2001). Thus, unlike Bad where serine phosphorylation results in inactivation and sequestration via 14-3-3 proteins, Nbk phosphorylation appears to result in a gain of function. A recent report showed that Nbk localizes to the endoplasmic reticulum (ER) membrane upon ectopic overexpression. Furthermore, ER-targeted Nbk could induce secondary activation of mitochondria and cytochrome c release in a cell-free system (Germain et al., 2002). This observation suggests that Nbk might be involved in a cross-talk of the ER with the mitochondrial apoptosis pathway. Moreover, the activation of mitochondria via artificially ER-targeted Nbk was independent from Bax as assessed in Bax–/– murine hepatocytes. This is in contrast to our data in several human Bax-negative carcinoma lines and the respective congeneic Bax-positive cells.
In conclusion, we provide evidence that Nbk induces apoptosis in Bax-proficient carcinoma cells tested, while Bax-negative cells were refractory. This corroborates a model of BH3-only proteins as indirect activators of the mitochondrial apoptosis signaling cascade that mediate their effects through Bax and its homologs. Interestingly, the Bax-negative cells investigated here express significant levels of Bak. Thus, Bak by itself appears to be insufficient to confer sensitivity for Nbk-induced apoptosis. We demonstrate that the BH3-only protein Nbk/Bik entirely depends on Bax but apparently not on Bak. Therefore, functional interactions between BH3-only proteins and Bax and its homologs might be more complex, as initially believed, and could occur in a subfamily-specific manner.
Recently published data support our notion that Bax and Bak differentially regulate apoptosis (Panaretakis et al., 2002). Interestingly, overexpression of Bcl-2 blocked the conformational activation of Bak upon treatment with the anti-cancer drug doxorubicin, whereas the Bax N-terminal switch was still partially activated. As doxorubicin also led to a strong increase in the expression of Nbk/Bik, this may indicate a differential regulation of Bax and Bak by Bcl-2 and Nbk.
Pro-apoptotic stimuli such as antigen receptor-mediated activation of B-lymphoid cells or induction of p53 by anti-cancer drugs may trigger induction of Nbk expression. The exact requirements for induction and transcriptional regulation of this killer protein remain, however, to be established. Evidence is accumulating that BH3 proteins link different cellular compartments or signal transduction machineries to the mitochondrial apoptosome. Our interest is therefore to elucidate the upstream signals regulating Nbk gene expression and activity, especially in view of its restricted tissue expression pattern.
Materials and methods
Cell culture
HEK293, SW48, SW480, DU145, LoVo and HCT116 cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) high-glucose supplemented with 10% fetal calf serum (FCS), 100 U/ml penicillin and 0.1 µg/ml streptomycin (all from Gibco, Karlsruhe, Germany). Cells were transduced with Ad5-myc-Nbk by incubation for 2 h at an m.o.i. of 25 in growth medium without FCS. Expression of myc-Nbk was suppressed by addition of 1 µg/ml doxycyclin to the culture medium (Tet-off condition).
Antibodies
Monoclonal mouse anti-Bax antibody (clone YTH-2D2, raised against a peptide corresponding to amino acids 3–16) was purchased from Trevigen (Gaithersburg, MD) and the conformation-specific polyclonal rabbit anti-Bax antibody (raised against a peptide corresponding to amino acids 1–21) was from Upstate Biotechnology (Lake Placid, NY). Polyclonal rabbit anti-myc antibody (A-14; raised against the 9E10 myc epitope) and goat anti-Nbk antibody (N-19; raised against an epitope mapping to the 19 amino acid N-terminus of human Nbk) were from Santa Cruz Biotechnology (Santa Cruz, CA), and polyclonal rabbit anti-Bcl-xS/L antibody (raised against amino acids 18–233 of rat Bcl-xL), monoclonal mouse anti-human cytochrome c (clone 7H8.2C12) and monoclonal mouse anti-human PARP antibody (clone C2-10) were from BD Biosciences Pharmingen (San Diego, CA). Monoclonal mouse anti-Bcl-2 antibody (NCL-bcl-2, raised against amino acids 41–55 of human Bcl-2) was purchased from Novocastra Laboratories (Newcastle, UK). Goat anti-caspase-9 antibody (raised against human caspase-9 amino acids 139–330) was from R&D Systems (Minneapolis, MN). The polyclonal rabbit anti-Bak antibody (raised against a peptide corresponding to amino acids 14–36) was from DAKO Corporation (Carpinteria, CA). Secondary anti-mouse, anti-rabbit and anti-goat IgG coupled to horseradish peroxidase were from Promega Corporation (Madison, WI) or Santa Cruz Biotechnology.
Construction of recombinant adenovirus
To insert the tTA expression unit into the adenovirus genome, the tTA expression cassette from pTet-Off (BD Biosciences Clontech, Palo Alto, CA) was first cloned as an XhoI–PvuII fragment into pHVAd3, an adenoviral shuttle vector for the E3 region. The virus genome containing the tTA expression unit was generated in the Escherichia coli strain BJ5183 RecBC-sbcB by homologous recombination of the shuttle plasmid with pHVAd1 containing the complete adenovirus genome, resulting in the plasmid pAd-tTA. To create an inducible myc-Nbk expression cassette, the XhoI–EcoRI fragment from pTRE containing the tetracyclin-responsive element (TRE) upstream of the CMV minimal promoter was inserted into the adenoviral shuttle vector pHVAd2. A myc-Nbk construct containing the full-length Nbk cDNA fused to an N-terminal myc tag was first cloned into pSL1180 (Amersham Pharmacia Biotech, Freiburg, Germany) and then inserted as a HindIII–SalI fragment into the TRE-containing pHVAd2 shuttle vector. The resulting TRE-myc-Nbk expression unit was inserted into the Ad5 virus genome by homologous recombination of the shuttle plasmid with pAd-tTA, thereby replacing the E1 region and creating pAd5-myc-Nbk-tTA (Figure 1A). The viral DNA was transfected into HEK293 cells and adenoviral plaques were propagated as described (Hemmati et al., 2002).
Stable expression of Bax by retroviral infection of DU145 cells
For expression of Bax in the Bax-negative DU145 cells, we employed the retroviral vector HyTK-Bax containing the human Bax-α cDNA under the control of a CMV promoter as described (Hemmati et al., 2002).
Measurement of apoptotic cell death by flow cytometry
Apoptosis was determined on a single-cell level by measuring the DNA content of individual cells with a FACScan (BD Biosciences) as described (Wieder et al., 2001). Alternatively, apoptotic cell death was determined by measuring binding of annexin V–FITC upon exposure of phosphatidylserine to the cell surface. PI-positive cells that had lost membrane integrity were considered as late apoptotic or necrotic cells and therefore excluded from the analysis. Data are given in percentage of cells with increased fluorescence reflecting the number of annexin V–FITC-stained, PI-negative cells.
Measurement of cytochrome c release
Cytosolic extracts were prepared according to a method described previously (von Haefen et al., 2003). After induction of apoptosis, cells were harvested in phosphate-buffered saline (PBS), equilibrated in hypotonic buffer (20 mM HEPES pH 7.4, 10 mM KCl, 2 mM MgCl2, 1 mM EDTA) supplemented with 0.1 mM phenylmethylsulfonyl fluoride (PMSF) and 0.75 mg/ml digitonin (Sigma-Aldrich) and incubated on ice for 3 min. Debris was pelleted by centrifugation at 10 000 g at 4°C for 5 min and the supernatant was subjected to western blot analysis.
Measurement of mitochondrial permeability transition
After infection with recombinant adenoviruses Ad5-myc-Nbk-tTA at an m.o.i. of 25, cells were collected by centrifugation at 300 g at 4°C for 5 min. The mitochondrial permeability transition was determined by staining the cells with JC-1 (Molecular Probes, Leiden, The Netherlands) as described (Wieder et al., 2001). Mitochondrial permeability transition was quantified by flow cytometric determination of cells with decreased red fluorescence, i.e. with mitochondria displaying a lower membrane potential (ΔΨm).
Measurement of conformational change of Bax by flow cytometry
A total of 2.5 × 105 cells (25 cm2 flask) were infected with Ad5-mycNbk-tTA in the presence or absence of doxycyclin and harvested 24 h after infection by trypsination. Quantification of cells showing exposure of the N-terminal epitope detected by the conformation-specific Bax antibody was performed by flow cytometry. Data are given in percentage of cells containing Bax with a conformational change in the N-terminal region.
Immunoblotting
After trypsination, cells were washed twice with ice-cold PBS and lysed in buffer L (10 mM Tris–HCl pH 7.5, 137 mM NaCl, 1% Triton X-100, 2 mM EDTA, 1 µM pepstatin, 1 µM leupeptin and 0.1 mM PMSF). Protein concentration was determined using the bicinchoninic acid assay. Equal amounts of protein (20 µg per lane) were separated by SDS–PAGE, electroblotted and visualized as described (Wieder et al., 2001).
Immunocytochemistry
SW480 cells were seeded on coverslips in 6-well plates and infected with Ad5-myc-Nbk-tTA in the presence or absence of doxycyclin. At 24 h post-infection, cells were washed three times with PBS and fixed for 30 min with ice-cold 1% paraformaldehyde. After two washing steps in PBS, the cells were permeabilized with ice-cold 100% methanol for 1 min. Cells were washed again twice and non-specific binding of antibodies was blocked by incubation with 8% bovine serum albumin (BSA) for 30 min at room temperature. The primary antibodies were diluted in 1% BSA in PBS and added to the cells overnight at 4°C. Incubation with secondary antibodies was performed for 1 h at room temperature. Then, cells were washed three times in PBS. The labeling of Nbk was performed by the use of goat anti-Nbk (1:50) followed by incubation with Alexa Fluor 594-conjugated chicken anti-goat IgG. Mitochondria were stained by the use of MitoTracker Green (Molecular Probes, Inc.) After staining, the cells were mounted in Aquamount and inspected in a Leica TCS SP2 confocal microscope.
Immunoprecipitation
A total of 1.5 × 106 cells per 75 cm2 flask were infected with Ad5-myc-Nbk-tTA and cultured for 24 h with or without doxycyclin. Cells were harvested, washed in ice-cold PBS and resuspended in 800 µl of either Triton X-100- or CHAPS-containing lysis buffer (20 mM Tris–HCl pH 7.5, 137 mM NaCl, 1% Triton X-100, 2 mM EDTA or 10 mM HEPES pH 7.4, 140 mM NaCl, 1% CHAPS) in the presence of protease inhibitors (1 µM pepstatin, 1 µM leupeptin and 0.1 mM PMSF). Lysates were pre-cleared with 40 µg of protein A– and protein G–Sepharose each (Sigma-Aldrich). A 150 µl aliquot of the pre-cleared cellular extract was shaken in the presence of 3 µg of the primary antibody and 0.5 mg of protein A/G–Sepharose at 4°C for 4 h. The protein A/G immune complex was sedimented by centrifugation at 15 000 g at 4°C for 20 s and washed three times in lysis buffer. Proteins bound to the protein A/G–Sepharose were eluted in 50 µl of sample buffer (62.5 mM Tris–HCl pH 6.8, 2% SDS, 2% β-mercaptoethanol, 10% glycerol and 1% bromophenol blue) and analyzed by western blot analysis.
Acknowledgments
Acknowledgements
We would like to thank Verena Lehmann and Karin Schmelz for expert technical assistance, the late Michael Strauss, Max Delbrück Center, Berlin for providing part of the Ad vector components, Dr Karsten Brand, Max Delbrück Center, Berlin for helpful discussions, Christopher Stroh, University of Düsseldorf for help with confocal microscopy, and Bert Vogelstein (Johns Hopkins University School of Medicine, Baltimore) for kindly providing HCT116 cells. This work was supported by the Deutsche Forschungsgemeinschaft grants SFB506 and Da 238/4.
References
- Antonsson B., Montessuit,S., Lauper,S., Eskes,R. and Martinou,J.C. (2000) Bax oligomerization is required for channel-forming activity in liposomes and to trigger cytochrome c release from mitochondria. Biochem. J., 345, 271–278. [PMC free article] [PubMed] [Google Scholar]
- Borner C. (2003) The Bcl-2 protein family: sensors and checkpoints for life-or-death decisions. Mol. Immunol., 39, 615–647. [DOI] [PubMed] [Google Scholar]
- Boyd J.M. et al. (1995) Bik, a novel death-inducing protein shares a distinct sequence motif with Bcl-2 family proteins and interacts with viral and cellular survival-promoting proteins. Oncogene, 11, 1921–1928. [PubMed] [Google Scholar]
- Cheng E.H., Wei,M.C., Weiler,S., Flavell,R.A., Mak,T.W., Lindsten,T. and Korsmeyer,S.J. (2001) Bcl-2, Bcl-xL sequester BH3 domain-only molecules preventing Bax- and Bak-mediated mitochondrial apoptosis. Mol. Cell, 8, 705–711. [DOI] [PubMed] [Google Scholar]
- Daniel P.T., Pun,K.T., Ritschel,S., Sturm,I., Holler,J., Dörken,B. and Brown,R. (1999) Expression of the death gene Bik/Nbk promotes sensitivity to drug-induced apoptosis in corticosteroid-resistant T-cell lymphoma and prevents tumor growth in severe combined immunodeficient mice. Blood, 94, 1100–1107. [PubMed] [Google Scholar]
- Daniel P.T., Schulze-Osthoff,K., Belka,C. and Güner,D. (2003) Guardians of cell death: the Bcl-2 family proteins. Essays Biochem., 39, in press. [DOI] [PubMed] [Google Scholar]
- Desagher S., Osen-Sand,A., Nichols,A., Eskes,R., Montessuit,S., Lauper,S., Maundrell,K., Antonsson,B. and Martinou,J. (1999) Bid-induced conformational change of Bax is responsible for mitochondrial cytochrome c release during apoptosis. J. Cell Biol., 144, 891–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eskes R., Desagher,S., Antonsson,B. and Martinou,J.C. (2000) Bid induces the oligomerization and insertion of Bax into the outer mitochondrial membrane. Mol. Cell. Biol., 20, 929–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Germain M., Mathai,J.P. and Shore,G.C. (2002) BH-3 only BIK functions at the endoplasmic reticulum to stimulate cytochrome c release from mitochondria. J. Biol. Chem., 277, 18053–18060. [DOI] [PubMed] [Google Scholar]
- Gossen M. and Bujard,H. (1992) Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc. Natl Acad. Sci. USA, 89, 5547–5551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J., Sabbatini,P. and White,E. (1996) Induction of apoptosis by human Nbk/Bik, a BH3-containing protein that interacts with E1B 19K. Mol. Cell. Biol., 16, 5857–5864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemmati P.G., Gillissen,B., von Haefen,C., Wendt,J., Stärck,L., Güner,D., Dörken,B. and Daniel,P.T. (2002) Adenovirus-mediated overexpression of p14ARF induces p53 and Bax-independent apoptosis. Oncogene, 21, 3149–3161. [DOI] [PubMed] [Google Scholar]
- Hsu Y.T. and Youle,R.J. (1998) Bax in murine thymus is a soluble monomeric protein that displays differential detergent-induced conformations. J. Biol. Chem., 273, 10777–10783. [DOI] [PubMed] [Google Scholar]
- Huang D.C. and Strasser,A. (2000) BH3-only proteins—essential initiators of apoptotic cell death. Cell, 103, 839–842. [DOI] [PubMed] [Google Scholar]
- Jürgensmeier J.M., Xie,Z.H., Deveraux,Q., Ellerby,L., Bredesen,D. and Reed,J.C. (1998) Bax directly induces release of cytochrome c from isolated mitochondria. Proc. Natl Acad. Sci. USA, 95, 4997–5002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelekar A., Chang,B.S., Harlan,J.E., Fesik,S.W. and Thompson,C.B. (1997) Bad is a BH3 domain-containing protein that forms an inactivating dimer with Bcl-xL. Mol. Cell. Biol., 17, 7040–7046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim T.H., Zhao,Y., Barber,M.J., Kuharsky,D.K. and Yin,X.M. (2000) Bid-induced cytochrome c release is mediated by a pathway independent of mitochondrial permeability transition pore and Bax. J. Biol. Chem., 275, 39474–39481. [DOI] [PubMed] [Google Scholar]
- Mahajan N.P., Linder,K., Berry,G., Gordon,G.W., Heim,R. and Herman,B. (1998) Bcl-2 and Bax interactions in mitochondria probed with green fluorescent protein and fluorescence resonance energy transfer. Nat. Biotechnol., 16, 547–552. [DOI] [PubMed] [Google Scholar]
- Martinou J.C. and Green,D.R. (2001) Breaking the mitochondrial barrier. Nat. Rev. Mol. Cell Biol., 2, 63–67. [DOI] [PubMed] [Google Scholar]
- Panaretakis T., Pokrovskaja,K., Shoshan,M.C. and Grander,D. (2002) Activation of Bak, Bax and BH3-only proteins in the apoptotic response to doxorubicin. J. Biol. Chem., 277, 44317–44326. [DOI] [PubMed] [Google Scholar]
- Puthalakath H. and Strasser,A. (2002) Keeping killers on a tight leash: transcriptional and post-translational control of the pro-apoptotic activity of BH3-only proteins. Cell Death Differ., 9, 505–512. [DOI] [PubMed] [Google Scholar]
- Radetzki S., Köhne,C.H., von Haefen,C., Gillissen,B., Sturm,I., Dörken,B. and Daniel,P.T. (2002) The apoptosis promoting Bcl-2 homologues Bak and Nbk/Bik overcome drug resistance in Mdr-1-negative and Mdr-1 overexpressing breast cancer cell lines. Oncogene, 21, 227–238. [DOI] [PubMed] [Google Scholar]
- Ruffolo S.C., Breckenridge,D.G., Nguyen,M., Goping,I.S., Gross,A., Korsmeyer,S.J., Li,H., Yuan,J. and Shore,G.C. (2000) Bid-dependent and Bid-independent pathways for Bax insertion into mitochondria. Cell Death Differ., 7, 1101–1108. [DOI] [PubMed] [Google Scholar]
- Saito R. and Tomita,M. (1999) On negative selection against ATG triplets near start codons in eukaryotic and prokaryotic genomes. J. Mol. Evol., 48, 213–217. [DOI] [PubMed] [Google Scholar]
- Sato T. et al. (1994) Interactions among members of the Bcl-2 protein family analyzed with a yeast two-hybrid system. Proc. Natl Acad. Sci. USA, 91, 9238–9242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiyama T., Shimizu,S., Matsuoka,Y., Yoneda,Y. and Tsujimoto,Y. (2002) Activation of mitochondrial voltage-dependent anion channel by a pro-apoptotic BH3-only protein Bim. Oncogene, 21, 4944–4956. [DOI] [PubMed] [Google Scholar]
- Tong Y., Yang,Q., Vater,C., Venkateah,L.K., Custeau,D., Chittenden,T., Chinnadurai,G. and Gourdeau,H. (2001) The pro-apoptotic protein Bik exhibits potent antitumor activity that is dependent on its BH3 domain. Mol. Cancer Ther., 1, 95–102. [PubMed] [Google Scholar]
- Toyota H., Yanase,N., Yoshimoto,T., Moriyama,M., Sudo,T. and Mizuguchi,J. (2003) Calpain-induced Bax-cleavage product is a more potent inducer of apoptotic cell death than wild-type Bax. Cancer Lett., 189, 221–230. [DOI] [PubMed] [Google Scholar]
- Verma S., Zhao,L. and Chinnadurai,G. (2001) Phosphorylation of the pro-apoptotic protein Bik: mapping of phosphorylation sites and effect on apoptosis. J. Biol. Chem., 276, 4671–4676. [DOI] [PubMed] [Google Scholar]
- von Haefen C., Wieder,T., Essmann,F., Schulze-Osthoff,K., Dörken,B. and Daniel,P.T. (2003) Paclitaxel-induced apoptosis in BJAB cells proceeds via a death receptor-independent, caspase-3/caspase-8-driven mitochondrial amplification loop. Oncogene, 22, 2236–2247. [DOI] [PubMed] [Google Scholar]
- Wei M.C., Lindsten,T., Mootha,V.K., Weiler,S., Gross,A., Ashiya,M., Thompson,C.B. and Korsmeyer,S.J. (2000) tBID, a membrane-targeted death ligand, oligomerizes BAK to release cytochrome c. Genes Dev., 14, 2060–2071. [PMC free article] [PubMed] [Google Scholar]
- Wieder T., Essmann,F., Prokop,A., Schmelz,K., Schulze-Osthoff,K., Beyaert,R., Dörken,B. and Daniel,P.T. (2001) Activation of caspase-8 in drug-induced apoptosis of B-lymphoid cells is independent of CD95/Fas receptor ligand interaction and occurs downstream of caspase-3. Blood, 97, 1378–1387. [DOI] [PubMed] [Google Scholar]
- Yang E., Zha,J., Jockel,J., Boise,L.H., Thompson,C.B. and Korsmeyer,S.J. (1995) Bad, a heterodimeric partner for Bcl-xL and Bcl-2, displaces Bax and promotes cell death. Cell, 80, 285–291. [DOI] [PubMed] [Google Scholar]
- Zhai D., Huang,X., Han,X. and Yang,F. (2000) Characterization of tBid-induced cytochrome c release from mitochondria and liposomes. FEBS Lett., 472, 293–296. [DOI] [PubMed] [Google Scholar]
- Zhang L., Yu,J., Park,B.H., Kinzler,K.W. and Vogelstein,B. (2000) Role of Bax in the apoptotic response to anticancer agents. Science, 290, 989–992. [DOI] [PubMed] [Google Scholar]
- Zong W.X., Lindsten,T., Ross,A.J., MacGregor,G.R. and Thompson,C.B. (2001) BH3-only proteins that bind pro-survival Bcl-2 family members fail to induce apoptosis in the absence of Bax and Bak. Genes Dev., 15, 1481–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]