

Abstract
During polyadenylation of mRNA precursors in metazoan cells, poly(A) polymerase is stimulated by the nuclear poly(A) binding protein PABPN1. We report that stimulation depends on binding of PABPN1 to the substrate RNA directly adjacent to poly(A) polymerase and results in an ∼80-fold increase in the apparent affinity of poly(A) polymerase for RNA without significant effect on catalytic efficiency. PABPN1 associates directly with poly(A) polymerase either upon allosteric activation by oligo(A) or, in the absence of RNA, upon deletion of its N-terminal domain. The N-terminal domain of PABPN1 may function to inhibit undesirable interactions of the protein; the inhibition is relieved upon RNA binding. Tethering of poly(A) polymerase is mediated largely by the C-terminal domain of PABPN1 and is necessary but not sufficient for stimulation of the enzyme; an additional interaction dependent on a coiled-coil structure located within the N-terminal domain of PABPN1 is required for a productive interaction.
Keywords: poly(A) binding protein/poly(A) polymerase/3′ processing/processivity
Introduction
In 3′-end processing of eukaryotic mRNAs, the transcript is first cleaved by an endonuclease, then poly(A) polymerase adds a poly(A) tail to the upstream cleavage product. In mammalian cells, the cleavage reaction takes place between a highly conserved sequence, AAUAAA, upstream and a degenerate GU-rich sequence downstream of the cleavage site. The AAUAAA signal also directs the subsequent polyadenylation. The complex protein machinery carrying out the two reactions has been reviewed (Minvielle-Sebastia and Keller, 1999; Wahle and Rüegsegger, 1999; Zhao et al., 1999; Edmonds, 2002).
In vitro, polyadenylation can be assayed independently of cleavage through the use of so-called pre-cleaved precursor RNAs, which end at or close to the cleavage site (Manley, 1983; Moore et al., 1986; Zarkower et al., 1986). Under physiological reaction conditions, addition of a poly(A) tail depends on the AAUAAA sequence in the substrate RNA (‘specific polyadenylation reaction’). This sequence is recognized by cleavage and polyadenylation specificity factor (CPSF), a complex of at least four polypeptides (Bienroth et al., 1991). CPSF and a further protein, the nuclear poly(A) binding protein (PABPN1; previously called PABII or PABP2) (Wahle, 1991a; Bienroth et al., 1993) greatly stimulate the activity of poly(A) polymerase, which is nearly inactive on its own. Poly(A) polymerase depends on CPSF and PABPN1 because the enzyme has a very low inherent affinity for its RNA substrate under specific polyadenylation conditions, i.e. in the presence of Mg2+ as opposed to Mn2+. Poly(A) polymerase also lacks sequence specificity (Wahle, 1991b). CPSF, binding specifically to AAUAAA-containing RNAs (Keller et al., 1991), recruits the polymerase and thus endows the enzyme with specificity for such RNAs. Similarly, PABPN1 binds the growing poly(A) tail and permits the polyadenylation of RNAs carrying a tail of at least 10 adenylate residues (Wahle, 1991a). Significantly, CPSF and PABPN1 cooperate in the stimulation of poly(A) polymerase, as seen most clearly in the processivity of the reaction: whereas poly(A) polymerase by itself is completely distributive, the presence of either CPSF or PABPN1 leads to a small degree of processivity. Only in the presence of both stimulatory factors is the processivity high: a poly(A) tail of ∼250 nucleotides, corresponding to the length of a newly synthesized poly(A) tail in vivo, is synthesized without dissociation of the polyadenylation complex (Bienroth et al., 1993). Thus, CPSF and PABPN1 can be considered both processivity factors and, by virtue of their ability to bind specific RNA sequences, specificity factors for poly(A) polymerase. After ∼250 adenylate residues have been added, the reaction becomes distributive, resulting effectively in a termination of polyadenylation (Wahle, 1995).
The increased processivity of polyadenylation is probably caused by an increased stability of the RNA–protein complexes: a complex of CPSF, poly(A) polymerase and substrate RNA is more stable than a similar complex lacking poly(A) polymerase, suggesting that the two proteins mutually stabilize their interactions with the RNA. A quaternary complex containing PABPN1 in addition is even more stable (Bienroth et al., 1993). The simplest basis for this cooperative type of RNA–protein complex formation would be a direct interaction between the participating proteins, leading to a ‘tethering’ of poly(A) polymerase. Indeed, poly(A) polymerase interacts with the 160 kDa subunit of CPSF (Murthy and Manley, 1995). In contrast, no physical interaction has so far been reported for poly(A) polymerase and PABPN1. In fact, not even a ternary complex of poly(A) polymerase, PABPN1 and RNA has been demonstrated; such an interaction can only be inferred from the strong stimulation of the polymerase by PABPN1.
PABPN1 (32.3 kDa) contains an RNP-type RNA binding domain, a very acidic N-terminus and a basic C-terminus rich in dimethylated arginine residues (Nemeth et al., 1995; Smith et al., 1999). Whereas the RNP domain and the C-terminal domain are required for binding to poly(A), the N-terminus is essential for the stimulation of polyadenylation (Kühn et al., 2003). Here we have characterized the mechanism by which PABPN1 stimulates poly(A) polymerase. We demonstrate that the two proteins associate directly, but only in the presence of RNA. The interaction strongly increases the RNA affinity of the polymerase. The analysis of mutant proteins suggests that tethering of the polymerase to the RNA primer through PABPN1 is necessary but not sufficient for the stimulation of polyadenylation.
Results
PABPN1 increases the affinity of poly(A) polymerase for RNA
The binding of PABPN1 to the RNA primer is an essential feature of the ‘tethering model’ for the stimulation of poly(A) polymerase by PABPN1. The model predicts that mutants of PABPN1 that are compromised in RNA binding should have a defect in the stimulation of poly(A) polymerase. Two such mutants carrying single amino acid substitutions in the RNP domain (Kühn et al., 2003) were tested for their ability to stimulate the extension of radiolabeled primer RNAs. Under standard reaction conditions (3 nM primer concentration), ∼4-fold higher concentrations of the F215A mutant were required to effect the same extent of elongation of A80 as the wild-type protein (Figure 1A). The mutant Y175A, which has a slightly smaller defect in RNA binding than F215A, was functional in the stimulation of poly(A) polymerase on A80, but deficient on an A25 primer (Figure 1B). The properties of the two mutants thus confirm the prediction of the tethering model. The difference between the two primers in the case of the Y175A mutant is probably due to the pronounced cooperativity of RNA binding of such mutants (Kühn et al., 2003); cooperative binding requires long poly(A).

Fig. 1. Stimulation of poly(A) polymerase by PABPN1 mutants with reduced affinity for RNA. (A) Eighty fmol of 5′-labeled poly(A) (average chain length 80 nucleotides) were elongated by 50 fmol of poly(A) polymerase for 15 min in the presence of wild-type PABPN1 or the F215A mutant as indicated and analyzed on a denaturing polyacrylamide gel. Lane 1, incubation without poly(A) polymerase. Lane 2, reaction in the absence of PABPN1, lanes 3–7 and 8–12, reactions containing 100, 200, 400, 800 and 1600 fmol of PABPN1. Sizes of DNA markers (lane M) are indicated on the left. PAP, poly(A) polymerase. (B) 5′-labeled oligo(A) (∼25 nucleotides) was elongated in the presence of 200, 400, 800 and 1600 fmol of wild-type PABPN1 or Y174A mutant. Incubation was for 30 min.
The dependence of poly(A) polymerase activity on the concentration of a poly(A) primer in the absence of any stimulatory factor was determined with an assay that measures the polymerization of labeled AMP (from ATP). A hyperbolic dependence on poly(A) concentration was observed. Double reciprocal plots (Figure 2A) revealed a Km of 4.6 ± 2.9 µM 3′-ends. Vmax was 3.6 ± 1.2 µmol/min/mg, corresponding to a kcat of 3.6 s–1. In a similar titration of poly(A) completely covered by PABPN1, the Km was decreased ∼80-fold to 0.058 ± 0.017 µM 3′-ends. The kcat was 2.6 s–1, similar to that measured in the absence of PABPN1 (Figure 2B). Thus, the stimulatory effect of PABPN1 is due to an increase in the apparent affinity of poly(A) polymerase for RNA, as predicted by the tethering model. PABPN1 does not increase the catalytic efficiency of poly(A) polymerase.

Fig. 2. Kinetic constants of poly(A) polymerase. Reaction rates of poly(A) polymerase were determined by measuring the incorporation of [α-32P]ATP into RNA as described in Materials and methods. (A) Double-reciprocal plot of reaction rates in the presence of increasing amounts of unfractionated poly(A). (B) Double-reciprocal plot of reaction rates in the presence of increasing amounts of unfractionated poly(A) saturated with PABPN1.
A putative α-helical region of PABPN1 is essential for the stimulation of poly(A) polymerase
A deletion of the first 160 amino acids of PABPN1 (ΔN160) does not affect poly(A) binding but destroys the ability to stimulate poly(A) polymerase (Kühn et al., 2003). Within this region, a sequence of ∼30 amino acids (L119–Q147) is predicted to form an amphipathic α-helix or a coiled-coil domain: when the sequence is written as a heptad repeat, hydrophobic side chains are found in positions a and d, whereas mostly charged residues are found in the other positions (Figure 3A). In contrast to ΔN160, a deletion mutant (ΔN113) retaining this putative helix stimulated poly(A) polymerase as strongly as the wild type (Figure 3B).

Fig. 3. The helical region in the N-terminus of PABPN1 is necessary for poly(A) polymerase stimulation. (A) Heptad repeat representation of the helical region (amino acids 119–147). Hydrophobic amino acids are shaded gray, amino acids in positions a and d are boxed. (B) Polyadenylation with N-terminal truncation mutants of PABPN1: radioactively labeled A80 was incubated for 15 min with poly(A) polymerase (15 fmol) in the presence of PABPN1 as indicated and analyzed on a denaturing polyacrylamide gel. Lane 1, incubation without poly(A) polymerase (PAP); lanes 2–6, incubations with 0, 100, 200, 400 and 800 fmol of wild-type PABPN1; lanes 7–9, incubations with 200, 400 and 800 fmol of ΔN160; lanes 10–15, incubations with 50, 100, 200, 400, 800 and 1600 fmol of ΔN113. Sizes of DNA markers (lane M) are indicated on the left. A reduction of the extent of stimulation at high concentrations of PABPN1 (lanes 5, 6 and 13–15) is usually seen in such assays (for example, Wahle, 1995) and may reflect covering of the RNA to the extent that poly(A) polymerase cannot get access. (C) Polyadenylation of radioactively labeled A80 by 9 fmol of poly(A) polymerase for 15 min. PABPN1 wt, and the substitution mutants A133S, V143A and L136S were added as indicated. Lane 1, poly(A) substrate; lane 2, incubation with poly(A) polymerase; lanes 3–6, 8–12, 13–17, 18–21, incubations with 100, 200, 400, 800 and 1600 fmol of the respective PABPN1 variant. Products were analyzed on a denaturing polyacrylamide gel. Sizes of DNA markers (lane M) are indicated on the left.
Point mutations were generated in the putative α-helix, and purified mutant proteins were tested for the stimulation of poly(A) polymerase. Examples of these assays are shown in Figure 3C and the results are summarized in Table I. Mutations at five positions had no effect. Mutations at six positions, however, led to a partial defect in the stimulation of polyadenylation, and either a serine or an alanine substitution of L136 abolished the activity almost completely. Almost all mutations resulting in reduced activity affected hydrophobic amino acid side chains, whereas none of the mutations of charged amino acids had a strong effect. Four of the inactivating mutations caused a change in amino acids that occupy every seventh position in the sequence and are thus located along a continuous hydrophobic surface of the α-helix (I122, M129, L136 and V143; compare Figure 3A). The mutant L136S, which had the most severe defect in the stimulation of poly(A) polymerase, was also deficient in processive polyadenylation of an oligoadenylated, AAUAAA-containing RNA (L3preA15) in the presence of CPSF: whereas the addition of wild-type PABPN1 led to a further stimulation of polyadenylation beyond that caused by CPSF alone, only the CPSF effect was seen in the presence of the L136S mutant (Figure 4). The behavior of the six other mutations tested in processive polyadenyl ation also reflected their activity in the simple poly(A) extension assay (data not shown).
Table I. Effect of point mutations in the coiled-coil domain of PABPN1 on the stimulation of poly(A) polymerase.
| No defect | E120A, K123A, V126S, A133S, K135A |
| Slight defect | A124S, M129A, E131A, V143A |
| Strong defect | L119A, I122Q |
| Inactive | L136S, L136A |
The activity of poly(A) polymerase was measured by the extension of a radiolabeled A80 primer as described in the legend to Figure 3C.
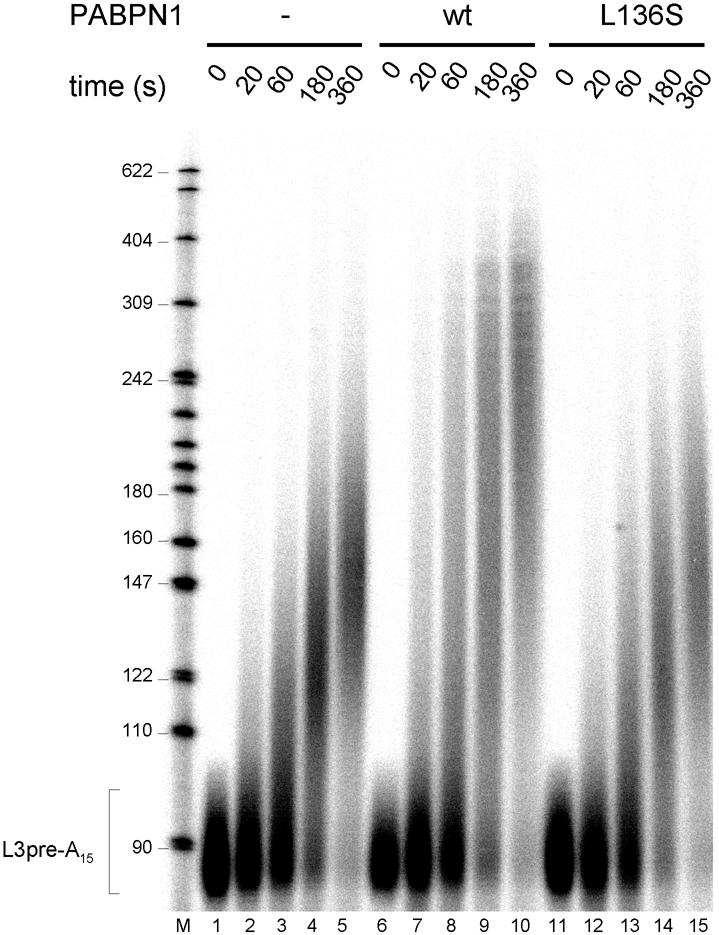
Fig. 4. PABPN1 L136A is inactive in processive polyadenylation. Reaction mixtures contained 80 fmol of L3pre-A15, 12 fmol of poly(A) polymerase and 120 fmol of CPSF. Wild-type PABPN1 or L136S mutant (300 fmol) was added to the reactions shown in lanes 6–10 and 11–15. After preincubation for 2 min at 37°C, polyadenylation was initiated by the addition of ATP. Reactions were stopped at the time points indicated. RNAs were recovered and analyzed by denaturing gel electrophoresis.
All point mutants had an affinity for poly(A) similar to that of the wild-type protein, with the possible exception of A124S (data not shown). Thus, a defect in RNA binding or a gross folding defect does not account for the inability of the mutant proteins to stimulate the polymerase. Addition of the L136S mutant to a processive polyadenylation reaction in slight excess (∼3-fold) over wild-type PABPN1 caused a strong inhibition of processive polyadenylation, whereas additional wild-type protein marginally decreased the efficiency of the reaction (data not shown). Thus, the mutant protein is incorporated into the polyadenylation complex, again excluding gross folding defects.
In quantitative assays, the L136S mutant reduced the activity of poly(A) polymerase ∼10-fold in comparison with wild-type PABPN1 (see below; Figure 5). As PABPN1 stimulates poly(A) polymerase by increasing its apparent affinity for the primer RNA, a simple prediction is that the mutation L136S should reverse this effect, i.e. the Km of poly(A) polymerase for poly(A) complexed with the mutant protein should be higher than that for poly(A) complexed with wild-type PABPN1. Surprisingly, this was not the case: in preliminary experiments, the mutant protein reduced the Km of the polymerase for the primer RNA as strongly as wild-type PABPN1, but led to a strong reduction in Vmax instead (data not shown).

Fig. 5. Activity of poly(A) polymerase at different ratios of wild-type PABPN1 to L136S mutant. A95 (1.7 pmol, 3′-ends) were incubated with 0.2 pmol of poly(A) polymerase and 8.5 pmol of wild-type PABPN1 and L136S in different ratios as indicated. Reactions were pre-warmed, started by addition of [α-32P]ATP and incubated for 30 min. The reactions were stopped by TCA precipitation, and the incorporated radioactivity was determined. The data are averaged from two independent experiments. Incorporation in the presence of only wild-type PABPN1 was set to 100%. Statistical model: poly(A) polymerase activity was calculated using the Bernoulli distribution for three different models in which random replacement of one (black line), two (dotted line) or three (dashed line) PABPN1 molecules by L136S reduces the activity of poly(A) polymerase to 8.9% of that in the presence of only wild-type PABPN1.
PABPN1 has to bind RNA adjacent to poly(A) polymerase
When mixtures of wild-type protein and L136S were tested for the stimulation of poly(A) polymerase, a progressive inhibition by increasing proportions of mutant protein was seen (Figure 5). PABPN1 was used at a molar ratio of 5:1 compared with the A80 primer, corresponding to ∼80% occupancy (Meyer et al., 2002). Five different theoretical inhibition curves were modeled, based on the assumption that random replacement of either one, two, three, four or five molecules of wild-type PABPN1 by mutant protein blocked the stimulation of polyadenylation. All theoretical curves were non-linear and deviated strongly from the experimental data (Figure 5; data not shown). Thus, the strictly linear relationship of inhibition to the percentage of mutant protein cannot be explained by the assumption that the replacement of one or more random PABPN1 molecules by mutant protein disrupts the stimulation; rather, it must be the substitution of one particular PABPN1 molecule that blocks stimulation. Evidently, the ratio of mutant to wild-type protein at any particular position is equal to the ratio in the total population. This means that poly(A) polymerase is stimulated by interaction with one particular molecule of PABPN1 among those arrayed on the A80 primer. The most likely candidate for this particular PABPN1 molecule is the one closest to the 3′-end of the RNA, the enzyme’s direct neighbor.
The following experiment strongly supports this conclusion: derivatives of the pre-cleaved polyadenylation substrate L3pre were generated carrying either poly(A) sequences at the 3′-end or internal poly(A) sequences followed by an additional heteropolymeric sequence of 63 nucleotides (see Materials and methods). Electrophoretic mobility shift assays (EMSAs) confirmed that the internal poly(A) sequences bound PABPN1 in proportion to the poly(A) length (Figure 6A). Binding of two molecules of PABPN1, rather than the expected single molecule, to the RNA carrying an A15 sequence may be explained by binding to a purine-rich internal RNA sequence. The RNA molecules were then tested for their ability to support the stimulation of polyadenylation by PABPN1. Surprisingly, internal poly(A) sequences did not permit the slightest stimulation of poly(A) polymerase (Figure 6B, compare lanes 8 and 9, 11 and 12, 14 and 15). Thus, the direct neighborhood of PABPN1 and poly(A) polymerase is essential for the stimulation. As a positive control, a 3′-terminal A15 sequence resulted in the expected stimulation (Figure 6B, compare lanes 5 and 6). As further controls, CPSF was able to stimulate the extension of RNAs containing internal oligo(A) sequences, and the same RNAs supported processive polyadenylation in the presence of both CPSF and PABPN1 provided that an additional 3′ oligo(A) tract was added (data not shown).

Fig. 6. PABPN1 cannot stimulate poly(A) polymerase from internal binding sites. (A) Binding of PABPN1 to different RNA substrates (80 fmol each) in the presence of 1.25 µg of tRNA was measured by native gel shift assays. RNA was briefly heated to 95°C and chilled on ice before it was added to the binding reaction. Circumstantial evidence indicates that the double band observed in lanes 2–5 is due to conformational heterogeneity of the RNA. It is almost certainly not due to the binding of different numbers of proteins. (B) Specific polyadenylation assays using the same RNA substrates as in (A). Reactions contained 80 fmol of RNA, 20 fmol of poly(A) polymerase and 350 fmol of PABPN1, as indicated. RNA was heated as in (A). Incubation time was 30 min. Products were analyzed on a denaturing polyacrylamide gel.
Circular dichroism analyses confirm the predicted α-helical structure
A PABPN1 variant (α-RNP) comprising a major part of the predicted α-helical region and the RNP domain, amino acids 126–263, was overproduced and purified without any tag. Val126 was chosen as the N-terminus of this protein fragment based on the analysis of stable proteolysis products upon limited tryptic digestion (T.Scheuermann, A.Blume, B.Schulz, E.Wahle, R.Rudolph and E.Schwarz, submitted for publication). For comparison, the isolated RNP domain (amino acids 161–263) was purified and circular dichroism (CD) spectra of both fragments were recorded (Figure 7A). Deconvolution of the spectra predicted 21.8% α-helix and 26.3% β-sheet contribution for α-RNP, whereas 13.4% α-helix and 32.4% β-sheet were predicted for the RNP domain alone. Accordingly, the difference spectrum of the two fragments confirmed that amino acids 126–160 contribute α-helical elements to the overall secondary structure (Figure 7B).

Fig. 7. CD spectra of PABPN1 fragments. (A) The CD spectra of the RNP domain (rectangles) and the α-RNP variant (circles) are shown. (B) Difference spectrum calculated from (A).
Coiled-coil domains often serve the purpose of homodimer formation. However, in analytical ultracentrifugation, α-RNP behaved as a monomer in sedimentation equilibrium and sedimentation velocity experiments at concentrations of up to 67 µM (data not shown). Moreover, analytical ultracentrifugation of wild-type PABPN1 indicated that the protein is predominantly monomeric at concentrations up to 2 µM (H.Lilie and S.Meyer, unpublished data; see also below), and other experiments have shown that the RNA binding unit is a monomer (Nemeth et al., 1995; Meyer et al., 2002; Kühn et al., 2003). Thus, the requirement for the coiled-coil domain in the stimulation of poly(A) polymerase is not due to an involvement in PABPN1 dimerization. At concentrations >2 µM, far higher than in polyadenylation assays, PABPN1 can assume various states of oligomerization (Nemeth et al., 1995; Keller et al., 2000), and a homotypic interaction appears to be responsible for the slight cooperativity of RNA binding (Kühn et al., 2003). However, this interaction is mediated by the C-terminal domain of PABPN1, not the coiled-coil domain (Fan et al., 2001; Kühn et al., 2003).
The α-helical domain is not sufficient for the stimulation of poly(A) polymerase
In order to test whether the α-helical domain is sufficient for the stimulation of poly(A) polymerase or whether other domains also contribute, chimeric proteins were generated that contained the α-helical domain of PABPN1 and had the RNP domain and the C-terminal domain, singly or in combination, replaced by RNP domains from the cytoplasmic poly(A) binding protein (PABPC) (see Materials and methods). PABPC is unable to stimulate polyadenyl ation (Wahle et al., 1993). All fusion proteins bound tightly to poly(A), but were inactive in the stimulation of poly(A) polymerase (data not shown). Thus, the α-helical domain is not an independent module that can be used to transfer the ability to stimulate poly(A) polymerase to other proteins; both the C-terminal domain of PABPN1 and the RNP domain directly or indirectly contribute to the stimulation of polyadenylation.
A set of progressive C-terminal deletions of PABPN1 (Kühn et al., 2003) were assayed for their ability to stimulate poly(A) polymerase. Even though these mutations showed only a gradual loss of poly(A) binding activity (Kühn et al., 2003), all except ΔC8 were deficient in the stimulation of poly(A) polymerase, even at the highest concentration tested (Figure 8). The affinity of ΔC20 and ΔC27 for poly(A) was not weaker than that of the point mutant F215A, which is able to stimulate polyadenylation at an elevated concentration (Figure 1A). Thus, the defect of the deletion mutants in polyadenylation can probably not be explained by their reduced affinity for poly(A) alone and supports a direct role of the C-terminal domain in the stimulation of poly(A) polymerase.

Fig. 8. The C-terminus of PABPN1 is necessary for stimulation of poly(A) polymerase. Polyadenylation reactions containing radioactively labeled A70 (40 fmol of 3′-ends) and poly(A) polymerase (30 fmol) were incubated with increasing amounts of wild-type or C-terminal truncation variants of PABPN1 as indicated. Lane 1, untreated A70; lane 2, incubation with a 10-fold higher amount of poly(A) polymerase; lanes 3–11, 13–20, 21–28, 29–35, reactions with 0, 100, 200, 500, 1000, 2000, 5000 and 10000 fmol of PABPN1 variants, as indicated. The reaction in the presence of 100 fmol of PABPN1-ΔC27 was left out. Products were analyzed on a denaturing polyacrylamide gel.
PABPN1 interacts directly with poly(A) polymerase; the α-helical domain is not essential
The data presented so far suggest that tethering is involved in the stimulation of poly(A) polymerase, but provide no evidence for a direct contact between the enzyme and PABPN1. Analytical ultracentrifugation experiments were carried out in order to detect protein associations. A C-terminal truncation mutant of poly(A) polymerase, PAP513 (Martin and Keller, 1996), was used, which has full catalytic activity and can be stimulated by PABPN1. At concentrations of 2 µM, the individual proteins, PABPN1 and poly(A) polymerase, behaved as monomeric species, characterized by apparent s-values of 1.06 and 2.23S, respectively (Table II; data not shown). Sedimentation equilibrium runs revealed molecular masses of 32 190 and 57 380 Da, in good agreement with the prediction from the amino acid sequence (33 790 and 59 970 Da, respectively, for the His-tagged proteins). Upon combination, the proteins did not associate; the resulting apparent sedimentation coefficient (Table II) was lower than that of poly(A) polymerase and corresponded to a mixture of the two monomeric proteins. Sedimentation was also measured in the presence of stoichiometric amounts of oligo(A) (A8 or A10). Simultaneous measurement at 230 and 260 nm allowed both RNA and protein to be detected. Poly(A) polymerase did not interact with oligo(A): sedimentation velocity was not affected (Table II), and the absorbance at 260 nm did not show co-sedimentation of RNA (see Supplementary data available at The EMBO Journal Online). In contrast, binding of oligo(A) to PABPN1 was clearly detectable by the increased sedimentation constant (Table II), and the absorbance at 260 nm showed quantitative association of oligo(A) with the protein (see Supplementary data). Combination of all three components, oligo(A), PABPN1 and poly(A) polymerase, resulted in the appearence of a faster sedimenting complex characterized by an apparent s-value of 2.73S (Table II) and complete co-sedimentation of RNA (Supplementary data). Although no titration experiments were performed, the sedimentation velocity of the complex is consistent with a 1:1 stoichiometry of the two polypeptides. Taken together, the sedimentation data show that poly(A) polymerase indeed associates with PABPN1, but does so only in the presence of oligo(A).
Table II. Analyses of the sedimentation behavior of poly(A) polymerase and PABPN1 dependent on oligo(A).
| Protein/RNA | Apparent sedimentation constant |
|---|---|
| PABPN1 | 1.06S |
| PAP | 2.23S |
| PABPN1/PAP | 2.10S |
| PABPN1/A10 | 1.33S |
| PAP/A10 | 2.20S |
| PAP/PABPN1/A10 | 2.73S |
All components were used at a concentration of 2 µM each (see Materials and methods). Under the conditions used, sedimentation of A10 was not sufficient for the determination of a sedimentation constant. Very similar data were obtained with A8. PAP, poly(A) polymerase.
Since poly(A) polymerase does not bind oligo(A), the RNA permits formation of the PABPN1–polymerase complex not by independent and simultaneous binding of both proteins but most likely through an allosteric effect on PABPN1. This interpretation of the data is supported by the observation that elongation by poly(A) polymerase of the oligo(A) used in the sedimentation experiments could not be stimulated by PABPN1; a primer length of at least 14 nucleotides is necessary for PABPN1 stimulation of poly(A) polymerase (data not shown). Thus, A8 or A10 seems to be too short to allow simultaneous binding of both PABPN1 and poly(A) polymerase.
The interaction between poly(A) polymerase and PABPN1 was also investigated by glutathione S-transferase (GST) pull-down experiments: soluble radioactively labeled PABPN1 or mutant versions thereof were tested for binding to a GST–poly(A) polymerase fusion protein (GST–PAP) immobilized on glutathione beads. Both binding partners were treated with nucleases to eliminate the possibility of soluble protein binding to RNA retained by the immobilized polymerase. Binding of wild-type PABPN1 to GST–PAP was barely detectable (data not shown). Even in the presence of oligo(A) (after inactivation of the nucleases), no association was observed, presumably due to a high off-rate under these non-equilibrium conditions. However, an RNA-independent interaction with poly(A) polymerase was found with an N-terminal deletion variant of PABPN1 (ΔN113) (Figure 9). The interaction seemed to be of limited specificity: in repeated experiments, binding to GST–PAP was between 2- and 5-fold stronger than to the GST control. Xenopus PABPC, used as an internal specificity control, did not bind GST–PAP (Figure 9). The association of PABPN1 and GST–PAP was dependent on the C-terminal domain of PABPN1: while the ΔC8 mutation reduced binding but weakly, ΔC20 and more extended deletions led to a complete loss of binding (Figure 9). The agreement between the loss of binding in the C-terminal deletion mutants and the loss of stimulation (Figure 8) argues that the protein–protein interaction is required for stimulation. Even though the α-helical region between amino acids 119 and 147 is absolutely required for the stimulation of polyadenylation, its deletion had no effect on the interaction with poly(A) polymerase (data not shown). This is in agreement with the fact that the point mutation in the α-helical region, L136S, does not interfere with the ability of PABPN1 to increase the affinity of poly(A) polymerase for poly(A).
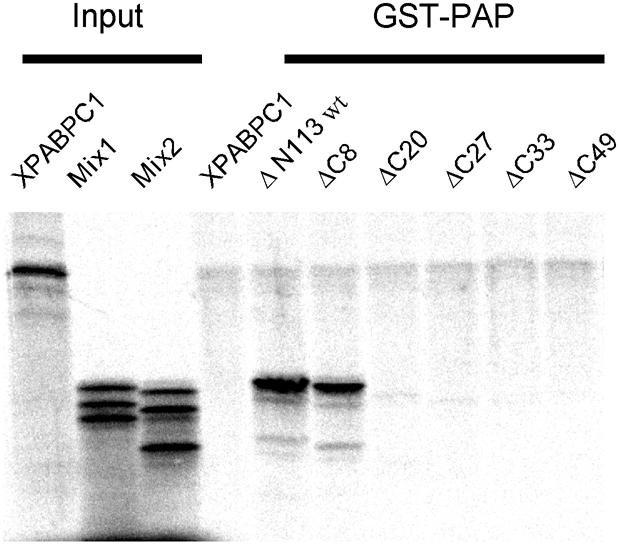
Fig. 9. The C-terminus of PABPN1 is necessary for poly(A) polymerase binding. GST–PAP was immobilized on GST–Sepharose beads and incubated with 35S-labeled PABPN1-ΔN113 and variants in which the ΔN113 mutation was combined with progressive C-terminal truncations. Labeled cytoplasmic poly(A) binding protein from Xenopus laevis was included as a negative control. Bound proteins as well as 10% of the protein input (mix1: ΔN113, ΔN113-ΔC20, ΔN113-ΔC33 and mix2: ΔN113-ΔC8, ΔN113-ΔC27, ΔN113-ΔC49) were analyzed on a 13% SDS–polyacrylamide gel.
Discussion
The data presented in this paper support the following conclusions: PABPN1 stimulates poly(A) polymerase exclusively by recruiting the enzyme to its substrate RNA; there is no enhancement of the catalytic activity. Recruitment involves a direct interaction between the two proteins bound adjacent to each other on the RNA. RNA allosterically activates PABPN1 to interact with poly(A) polymerase. Simple tethering, mediated directly or indirectly through the C-terminal domain of PABPN1, is not sufficient for stimulation, though; additional interactions, depending on a coiled-coil structure within the N-terminal domain, are needed for a productive orientation or conformation of the polymerase on its substrate.
Under physiological reaction conditions, the activity of poly(A) polymerase, in the absence of stimulatory factors, is limited by a very low affinity for the 3′-end of the RNA (Wahle, 1991b). The tethering model posits that PABPN1 increases this affinity by binding tightly to the growing poly(A) tail and providing a second contact for the polymerase. Within the context of the complete polyadenylation reaction, a further contact for the polymerase is thought to be provided by CPSF. The enzyme is thus held in place by cooperative interactions. Two types of earlier experiments suggested that RNA binding by PABPN1 is required for the stimulation of poly(A) polymerase, in agreement with the tethering model. First, the optimum concentration of PABPN1 for stimulation of the poly(A) polymerase was proportional to the concentration of the poly(A) primer and consistent with its complete coating (Wahle, 1995). Secondly, the elongation of non-poly(A) RNA depended on the presence of an oligo(A) tail serving as a binding site for PABPN1 (Wahle, 1991a). As further support of this model, we now find that PABPN1 mutants with a reduced affinity for poly(A) stimulate the polymerase only at elevated concentration. In the case of the F215A mutant, the increase in concentration required to bring about stimulation (∼4-fold) is not as high as expected based on the mutant’s reduced affinity for RNA (∼100-fold) (Kühn et al., 2003). However, in the polyadenylation reaction, poly(A) polymerase provides an additional contact for PABPN1, presumably stabilizing the binding of the protein to the substrate RNA, and buffer conditions in the polyadenyl ation reaction differ from those in the binding assays.
The kinetic measurements reported here revealed that PABPN1 increases the apparent affinity of poly(A) polymerase for its RNA substrate ∼80-fold with no increase in the catalytic efficiency. Thus, an increase in RNA affinity is the only mechanism through which PABPN1 activates the polymerase.
Both RNA binding domains of PABPN1 and, in addition, a coiled-coil domain are required for the stimulation of poly(A) polymerase. As coiled-coil domains are typically involved in protein–protein interactions and a single point mutation in this region strongly interferes with the stimulation of poly(A) polymerase, it is tempting to speculate that the helix directly contacts the polymerase. However, association through a 30 amino acid coiled-coil would be expected to result in a much tighter complex than is seen here, and the crystal structure of poly(A) polymerase (Martin et al., 2000) does not reveal a helix of an equally pronounced amphipathic nature. Thus, the effect of the coiled-coil domain on the activity of poly(A) polymerase may be indirect. Even though the α-helical region is essential for the stimulation of poly(A) polymerase, it is dispensable for the binding interaction. In agreement with this, the L136S point mutation does not affect the ability of PABPN1 to increase the affinity of poly(A) polymerase for the RNA; instead, the mutation causes a reduction in Vmax. Both observations suggest that the C-terminal domain is the dominant binding partner for poly(A) polymerase, but binding through the C-terminal domain alone results in a complex that is catalytically inefficient, e.g. due to a structural distortion or an unfavorable orientation on the RNA. The α-helix appears to be required to position the polymerase such that it is catalytically efficient. This is reminiscent of the interaction between the yeast polyadenylation factor Fip1p and the cognate poly(A) polymerase: binding of Fip1p alone to poly(A) polymerase in a tight 1:1 complex (Preker et al., 1995) inhibits (Zhelkovsky et al., 1998), while the Fip1p-containing complex, CPF, activates the enzyme (Preker et al., 1997). Different regions of Fip1p are involved in binding and inhibition of the polymerase as opposed to its activation in the complete polyadenylation complex (Helmling et al., 2001). Likewise, the 160 kDa subunit of mammalian CPSF binds poly(A) polymerase and inhibits it. Presumably, additional interactions in the complete CPSF complex are responsible for the interaction to result in stimulation of the enzyme (Murthy and Manley, 1995).
Experiments in which the L136S mutant was mixed with the wild-type protein indicated that one particular PABPN1 molecule is responsible for stimulation of poly(A) polymerase, most likely the enzyme’s immediate neighbor on the RNA. Even so, the extension of longer poly(A) is more efficient when it is nearly completely coated by PABPN1 (Wahle, 1995). Presumably, additional copies of PABPN1 prevent the one molecule directly adjacent to the polymerase from sliding away. The inability of PABPN1 to stimulate the enzyme from internal RNA binding sites supports the idea that direct neighborhood on the RNA is essential for a productive interaction. This is surprising in view of the flexibility of single-stranded RNA. For example, CPSF can stimulate the polyadenylation of one RNA molecule even when its binding site, AAUAAA, is on a separate RNA molecule base-paired to the one receiving the poly(A) tail (Bienroth et al., 1993). Both the dependence of stimulation on direct neighborhood of the proteins and the ability of mutant PABPN1 versions, like L136S or ΔN160, to bind but not stimulate poly(A) polymerase suggest that tethering, while necessary, is not sufficient for the stimulation of polyadenylation.
Analytical ultracentrifugation revealed an association between PABPN1 and poly(A) polymerase. However, no complex formation could be seen in the absence of RNA when the two proteins were used at 2 µM. Any RNA-independent interaction must therefore be extremely weak. Complex formation was observed in the presence of A8 or A10. Two results suggest that these RNA molecules cannot bind PABPN1 and poly(A) polymerase independently and simultaneously. First, no association of poly(A) polymerase with oligo(A) could be seen at the same concentrations at which oligo(A) promoted the formation of the PABPN1–poly(A) polymerase complex. Secondly, PABPN1 was unable to stimulate the extension of these short oligo(A) molecules by poly(A) polymerase. Thus, we propose that oligo(A) permits a direct protein–protein interaction through an allosteric effect on PABPN1. Alternatively, it is conceivable that the polymerase recognizes the combined surface of PABPN1 and oligo(A) exposed on the β-sheet of the RNP domain. A possible mechanism for an allosteric effect is suggested by the RNA-independent interaction of poly(A) polymerase with an N-terminal truncation variant of PABPN1 (ΔN113). Similarly, the self-interaction of the protein, which is mediated by the C-terminus and likely to be responsible for cooperative RNA binding, was detectable only with an N-terminal truncation (Kühn et al., 2003), and methylation of arginine residues in the C-terminal domain by recombinant protein arginine methyltransferases was more efficient when the N-terminus was deleted (Smith et al., 1999). Possibly, access to the basic C-terminus is blocked by an intramolecular interaction with the acidic N-terminus. Binding of poly(A) to the C-terminal domain may displace the N-terminus and permit the C-terminus to interact with other partners. The interactions permitted by an N-terminal deletion may be relatively promiscuous due to the ‘sticky’ nature of the C-terminal domain. This limited specificity may be tolerable if, in the cell, the C-terminal domain of PABPN1 is made available for protein–protein interactions by its association with RNA, as this probably restricts the choice of potential interaction partners to proteins acting on the same RNA molecule. One might speculate that the N-terminal domain acts as an intra molecular chaperone that prevents illegitimate C-terminal interactions unless the protein is bound to poly(A).
The C-terminal domain of PABPN1 has been reported to contribute to three interactions: with poly(A) (Kühn et al., 2003), with other PABPN1 molecules (Kühn et al., 2003) and with poly(A) polymerase (this paper). The interaction between the C-terminal domain of PABPN1 and poly(A) polymerase is likely to be functionally important, as C-terminal deletions lead not only to a loss of binding but also to a defect in the stimulation of poly(A) polymerase. Recently, we have found that the interaction between PABPN1 and the 30 kDa subunit of CPSF (Chen et al., 1999) is mediated by the C-terminal domain as well (C.Böhm, U.Kühn and E.Wahle, unpublished data). While the functional importance of this latter interaction is still uncertain, the various interactions of the C-terminus can be reconciled in a model of the polyadenylation complex in which the growing poly(A) tail is saturated with PABPN1 molecules touching each other. Whereas all internal molecules have an identical partner on either side, the first and the last molecule in this row have only one PABPN1 neighbor. The first molecule binds the 30 kDa subunit of CPSF in addition to its single PABPN1 neighbor, and the last PABPN1 molecule binds poly(A) polymerase. This model is also supported by the data discussed above, suggesting that the polymerase may interact only with its immediate neighbor in a row of PABPN1 molecules lined up on poly(A). Such an array of protein–protein interactions from CPSF through PABPN1 to poly(A) polymerase would result in a contiguous coverage of the growing poly(A) tail by PABPN1, which is probably important for measurement of the tail length.
Materials and methods
DNA
Point mutations were introduced into a partially synthetic bovine PABPN1 coding sequence (Kühn et al., 2003) either with the Gene Editor™ site-directed mutagenesis system (Promega) or a PCR-based protocol provided by Stratagene using Pwo (AGS) as a proofreading polymerase and DpnI to destroy parental DNA. Mutants were subcloned into the pUK vector (Kühn et al., 2003) using NdeI and BamHI restrictions sites. PABPN1-ΔN160 and the C-terminal deletion mutants have been described (Kühn et al., 2003). PABPN1-ΔN113 was obtained by PCR with an upstream primer containing the new start codon as part of an NdeI site and a second primer covering the single MscI site of the PABPN1 coding sequence. The PCR product was restricted with NdeI–MscI and ligated into a NdeI–MscI-opened pGM10(His6)-PABPN1-plasmid (Smith et al., 1999).
The following PABPN1/PABPC domain swaps were generated. H12 contains the α-helical domain of PABPN1 (amino acids 114–160) and the first two RNP domains (RBD 1 and 2) of Xenopus PABPC (amino acids 1–190; DDBJ/EMBL/GenBank accession No. X57483). H4C contains the α-helical domain of PABPN1, RBD 4 of Xenopus PABPC (amino acids 295–395) and the C-terminal domain of PABPN1 (amino acids 258–306). HR4 contains the α-helical domain and the RNP domain of PABPN1 (amino acids 114–257) and RBD 4 of Xenopus PABPC. All chimeric proteins carried N-terminal His6 tags. DNA sequences and details of the construction are available upon request.
The DNA fragments encoding the untagged RNP domain (amino acids 161–263) and the RNP domain plus α-helix (α-RNP; amino acids 126–263) were amplified by PCR. The upstream primer introduced an NdeI site at the 5′-end and, in the case of α-RNP, an additional methionine; the downstream primer generated a BamHI site at the 3′-end. After digestion, the DNA fragments were ligated into the NdeI–BamHI opened vector pET11a (Novagen).
A His6-GST–poly(A) polymerase fusion was generated by cloning of the bovine poly(A) polymerase cDNA into pUK, followed by insertion of the GST coding sequence as described previously for a GST–PABPN1 fusion (Kühn et al., 2003).
The plasmid pSP64-L3preA15 was generated as follows: a DNA oligonucleotide (A15CCGTCTTCCCGGGAATTCC, recognition sequences for BbsI and EcoRI underlined) and a complementary strand were synthesized, phosphorylated, annealed and digested with EcoRI. The plasmid pSP64-L3pre (Christofori and Keller, 1989) was opened by a limited RsaI digestion at the polyadenylation site (position 65 after the transcription start site) and completely digested with EcoRI. After gel purification of the vector fragment, the EcoRI-digested oligonucleotide was inserted. Positive clones were identified by restriction enzyme digestions. A45 and A110 sequences were integrated at the same site of pSP64-L3preA15 by annealing of phosphorylated dA34 and dT34 in the presence of BbsI-cut, dephosphorylated pSP64-L3pre and ligation. Run-off transcription of these plasmids after digestion with BbsI results in the synthesis of the polyadenylation substrate L3pre containing an A15, A45 or A110 tail without any additional nucleotide at its 3′-end. Transcription after digestion with MbiI leads to RNAs containing internal A15, A45 or A110 sequences followed by 63 nucleotides of heterogeneous sequence.
All DNA constructs were verified by sequencing.
Protein purification and analysis
His-tagged PABPN1 variants were expressed and purified on Ni2+-NTA columns as described (Kühn et al., 2003). H12, ΔN113 and HR4 were further purified on Mono Q columns (Pharmacia) and the PABPN1 point mutants and H4C on Mono S (Pharmacia). The ΔN160 mutant and C-terminal deletion mutants were purified as described (Kühn et al., 2003). Protein concentrations were determined either by the Bio-Rad protein assay (Bradford, 1976) or by comparison of the band in a Coomassie Blue-stained SDS gel to a BSA standard. For some protein preparations, UV spectroscopy was used to confirm the absence of contaminating nucleic acids. The untagged RNP domain was purified by successive chromatography on Q-Sepharose (Pharmacia), Phenyl Fractogel (Merck), high performance SP Sepharose (Pharmacia) and Superdex 75 prep grade (Pharmacia). α-RNP was purified in the same manner except that a second Q-Sepharose column replaced the SP Sepharose column. Concentrations of both proteins were determined from the UV spectrum. CPSF was purified from calf thymus (Wahle, 1995). Full-length bovine poly(A) polymerase was kindly provided by Georges Martin (Basel). The C-terminal truncated variant PAP513 was expressed in Escherichia coli and purified as described (Martin and Keller, 1996).
RNA
Poly(A) (Roche; average chain length ∼170 nucleotides) was dissolved in water, extracted with phenol/chloroform, ethanol precipitated and dissolved in water. Concentration was determined by UV spectroscopy with ε = 10 300 cm–1 M–1 (AMP). Gel-purified A80 was 5′-end labeled with T4 polynucleotide kinase (NEB) and [γ-32P]ATP (Amersham). Derivatives of the polyadenylation substrate L3pre were generated with SP6 polymerase (Roche) after appropriate digestion of the template DNA as described (Wahle, 1995). Product lengths were slightly heterogeneous, presumably due to polymerase slippage on the poly(dT) sections of the templates. Synthetic oligo(A) was obtained from IBA, Göttingen.
CD spectroscopy
CD spectra were recorded at 20°C on an Aviv model 62A DS spectropolarimeter in 5 mM MOPS pH 6.5, in 0.1 mm quartz cuvettes. The spectra of α-RNP were measured at a protein concentration of 1.74 mg/ml (112 µM), those of the RNP domain at 1 mg/ml (86.9 µM). The spectra shown are the average of 10 scans. They are plotted as residual ellipticities. The difference spectrum was obtained using the molar ellipticities of the RNP domains and calculated back to residual ellipticity. Spectra were deconvoluted by means of the CDNN program (Böhm et al., 1992).
Analytical ultracentrifugation
An Optima XL-A ultracentrifuge (Beckman Instruments) equipped with an An60Ti rotor and double sector cells was used. Equilibrium sedimentation measurements were perfomed at 15 000 r.p.m. and 20°C for at least 40 h. Sedimentation velocity was analyzed at 40 000 r.p.m. and 20°C in the case of poly(A) polymerase and PABPN1, and at 60 000 r.p.m. and 4°C for the RNP and α-RNP variants. Poly(A) polymerase and PABPN1 were measured at 2 µM each in 50 mM Tris pH 8.0, 50 mM KCl, 10% glycerol. Oligo(A) was also used at 2 µM. α-RNP was analyzed at between 15 and 67 µM in 10 mM MOPS pH 6, 0.1 M NaCl. Data were collected at 230 and 260 nm and analyzed with the software provided by Beckman Instruments.
Assays
Specific polyadenylation assays (i.e. in the presence of Mg2+) were performed as described (Wahle, 1995). Protein concentrations and incubation times are indicated in the figure legends. Assays in which the incorporation of radiolabeled AMP into acid-precipitable material was measured were carried out under the same buffer conditions. For determination of kinetic constants, the mixture containing 20–200 fmol poly(A) polymerase was assembled in the absence of ATP, prewarmed for 2 min at 37°C, and the reaction started by the addition of 0.5 mM [α-32P]ATP (50–200 c.p.m./pmol). After 10–25 min, the reaction was stopped by the addition of trichloroacetic acid (TCA) and filtered (Wahle, 1991b). For EMSAs, RNA and proteins were incubated under specific polyadenylation conditions in the absence of ATP. Samples were analyzed in non-denaturing gels as described (Kühn et al., 2003).
Nitrocellulose filter-binding assays were carried out with 10 fmol 5′-end labeled A80 and 25–1000 fmol protein in filter-binding buffer (50 mM Tris–HCl pH 8.0, 100 mM NaCl, 1 mM EDTA, 0.5 mM DTT) in a final volume of 50 µl. After incubation at room temperature for 30 min, 45 µl of the mixture were applied to a nitrocellulose filter, which had been soaked in wash buffer (50 mM Tris–HCl pH 8.0, 100 mM NaCl, 1 mM EDTA) containing 5 µg/ml E.coli rRNA. The filter was washed with 5 ml of ice-cold wash buffer and bound radioactivity counted in a liquid scintillation counter.
GST pull-down assays were carried out as described (Kühn et al., 2003), including nuclease treatment of both the immobilized protein and the translation mixture. Approximately 2 µg of immobilized poly(A) polymerase were used per binding reaction.
Supplementary data
Supplementary data are available at The EMBO Journal Online.
Acknowledgments
Acknowledgements
We are grateful to Gudrun Scholz for skillful technical assistance; to Claudia Temme for the ΔN113 construct and initial experiments; to Kathrin Brunk for help in generating some of the transcription vectors; to Georges Martin (Biozentrum, University of Basel) for supplying purified poly(A) polymerase and expression clones; and to Sylke Meyer for critical reading of the manuscript. This work was supported by the Deutsche Forschungsgemeinschaft (to E.S., U.K. and E.W.) and the Fonds der Chemischen Industrie (to E.W.).
References
- Bienroth S., Wahle,E., Suter-Crazzolara,C. and Keller,W. (1991) Purification and characterisation of the cleavage and polyadenylation specificity factor involved in the 3′ processing of messenger RNA precursors. J. Biol. Chem., 266, 19768–19776. [PubMed] [Google Scholar]
- Bienroth S., Keller,W. and Wahle,E. (1993) Assembly of a processive messenger RNA polyadenylation complex. EMBO J., 12, 585–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Böhm G., Muhr,R. and Jaenicke,R. (1992) Quantitative analysis of protein far UV circular dichroism spectra by neural networks. Protein Eng., 5, 191–195. [DOI] [PubMed] [Google Scholar]
- Bradford M.M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem., 72, 248–254. [DOI] [PubMed] [Google Scholar]
- Chen Z., Li,Y. and Krug,R.M. (1999) Influenza A virus NS1 protein targets the poly(A) -binding protein II of the cellular 3′-end processing machinery. EMBO J., 18, 2273–2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christofori G. and Keller,W. (1989) Poly(A) polymerase purified from HeLa cell nuclear extract is required for both cleavage and polyadenylation of pre-mRNA in vitro. Mol. Cell. Biol., 9, 193–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edmonds M. (2002) A history of poly(A) sequences: from formation to factors to function. Progr. Nucleic Acids Res. Mol. Biol., 71, 285–389. [DOI] [PubMed] [Google Scholar]
- Fan X., Dion,P., Laganiere,J., Brais,B. and Rouleau,G.A. (2001) Oligomerization of polyalanine expanded PABPN1 facilitates nuclear protein aggregation that is associated with cell death. Hum. Mol. Genet., 10, 2341–2351. [DOI] [PubMed] [Google Scholar]
- Helmling S., Zhelkovsky,A. and Moore,C.L. (2001) Fip1 regulates the activity of poly(A) polymerase through multiple interactions. Mol. Cell. Biol., 21, 2026–2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller R.W., Kühn,U., Aragon,M., Bornikova,L., Wahle,E. and Bear,D.G. (2000) The nuclear poly(A) binding protein, PABP2, forms an oligomeric particle covering the length of the poly(A) tail. J. Mol. Biol., 297, 569–583. [DOI] [PubMed] [Google Scholar]
- Keller W., Bienroth,S., Lang,K.M. and Christofori,G. (1991) Cleavage and polyadenylation factor CPF specifically interacts with the pre-mRNA 3′ processing signal AAUAAA. EMBO J., 10, 4241–4249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kühn U., Nemeth,A., Meyer,S. and Wahle,E. (2003) The RNA binding domains of the nuclear poly(A) binding protein. J. Biol. Chem., 278, 16916–16925. [DOI] [PubMed] [Google Scholar]
- Manley J.L. (1983) Accurate and specific polyadenylation of mRNA precursors in a soluble whole-cell lysate. Cell, 33, 595–605. [DOI] [PubMed] [Google Scholar]
- Martin G. and Keller,W. (1996) Mutational analysis of mammalian poly(A) polymerase identifies a region for primer binding and a catalytic domain, homologous to the family X polymerases and to other nucleotidyl transferases. EMBO J., 15, 2593–2603. [PMC free article] [PubMed] [Google Scholar]
- Martin G., Keller,W. and Doublie,S. (2000) Crystal structure of mammalian poly(A) polymerase in complex with an analog of ATP. EMBO J., 19, 4193–4203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer S., Urbanke,C. and Wahle,E. (2002) Equilibrium studies on the association of the nuclear poly(A) binding protein with poly(A) of different lengths. Biochemistry, 41, 6082–6089. [DOI] [PubMed] [Google Scholar]
- Minvielle-Sebastia L. and Keller,W. (1999) mRNA polyadenylation and its coupling to other RNA processing reactions and to transcription. Curr. Opin. Cell Biol., 11, 352–357. [DOI] [PubMed] [Google Scholar]
- Moore C.L., Skolnik-David,H. and Sharp,P.A. (1986) Analysis of RNA cleavage at the adenovirus-2 L3 polyadenylation site. EMBO J., 5, 1929–1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murthy K.G.K. and Manley,J.L. (1995) The 160-kD subunit of human cleavage-polyadenylation specificity factor coordinates pre-mRNA 3′-end formation. Genes Dev., 9, 2672–2683. [DOI] [PubMed] [Google Scholar]
- Nemeth A., Krause,S., Blank,D., Jenny,A., Jenö,P., Lustig,A. and Wahle,E. (1995) Isolation of genomic and cDNA clones encoding bovine poly(A) binding protein II. Nucleic Acids Res., 23, 4034–4041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preker P.J., Lingner,J., Minvielle-Sebastia,L. and Keller,W. (1995) The FIP1 gene encodes a component of a yeast pre-mRNA polyadenylation factor that directly interacts with poly(A) polymerase. Cell, 81, 379–389. [DOI] [PubMed] [Google Scholar]
- Preker P.J., Ohnacker,M., Minvielle-Sebastia,L. and Keller,W. (1997) A multisubunit 3′-end processing factor from yeast containing poly(A) polymerase and homologues of the subunits of mammalian cleavage and polyadenylation specificity factor. EMBO J., 16, 4727–4737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith J.J., Rücknagel,K.P., Schierhorn,A., Tang,J., Nemeth,A., Linder,M., Herschman,H.R. and Wahle,E. (1999) Unusual sites of arginine methylation in poly(A)-binding protein II and in vitro methylation by protein arginine methyltransferases PRMT1 and PRMT3. J. Biol. Chem., 274, 13229–13234. [DOI] [PubMed] [Google Scholar]
- Wahle E. (1991a) A novel poly(A)-binding protein acts as a specificity factor in the second phase of messenger RNA polyadenylation. Cell, 66, 759–768. [DOI] [PubMed] [Google Scholar]
- Wahle E. (1991b) Purification and characterisation of a mammalian polyadenylate polymerase involved in the 3′ end processing of messenger RNA precursors. J. Biol. Chem., 226, 3131–3139. [PubMed] [Google Scholar]
- Wahle E. (1995) Poly(A) tail length control is caused by termination of processive synthesis. J. Biol. Chem., 270, 2800–2808. [DOI] [PubMed] [Google Scholar]
- Wahle E. and Rueegsegger,U. (1999) 3′-end processing of pre-mRNA in eukaryotes. FEMS Microbiol. Rev., 23, 277–295. [DOI] [PubMed] [Google Scholar]
- Wahle E., Lustig,A., Jenö,P. and Maurer,P. (1993) Mammalian poly(A) binding protein II. J. Biol. Chem., 268, 2937–2945. [PubMed] [Google Scholar]
- Zarkower D., Stephenson,P., Sheets,M. and Wickens,M. (1986) The AAUAAA sequence is required both for cleavage and for polyadenylation of simian virus 40 pre-mRNA in vitro. Mol. Cell. Biol., 6, 2317–2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J., Hyman,L. and Moore,C. (1999) Formation of mRNA 3′ ends in eukaryotes: mechanism, regulation and interrelationship with other steps in mRNA synthesis. Microbiol. Mol. Rev., 63, 405–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhelkovsky A., Helmling,S. and Moore,C. (1998) Processivity of the Saccharomyces cerevisiae poly(A) polymerase requires interactions at the carboxyl-terminal RNA binding domain. Mol. Cell. Biol., 18, 5942–5951. [DOI] [PMC free article] [PubMed] [Google Scholar]