

Abstract
Mitogen-activated protein kinase (MAPK) cascades are conserved signaling modules composed of three sequentially activated kinases (MAPKKK, MAPKK and MAPK). Because individual cells contain multiple MAPK cascades, mechanisms are required to ensure the fidelity of signal transmission. In yeast, external high osmolarity activates the HOG (high osmolarity glycerol) MAPK pathway, which consists of two upstream branches (SHO1 and SLN1) and common downstream elements including the Pbs2 MAPKK and the Hog1 MAPK. The Ssk2/Ssk22 MAPKKKs in the SLN1 branch, when activated, exclusively phosphorylate the Pbs2 MAPKK. We found that this was due to an Ssk2/Ssk22-specific docking site in the Pbs2 N-terminal region. The Pbs2 docking site constitutively bound the Ssk2/Ssk22 kinase domain. Docking site mutations drastically reduced the Pbs2–Ssk2/Ssk22 interaction and hampered Hog1 activation by the SLN1 branch. Fusion of the Pbs2 docking site to a different MAPKK, Ste7, allowed phosphorylation of Ste7 by Ssk2/Ssk22. Thus, the docking site contributes to both the efficiency and specificity of signaling. During these analyses, we also found a nuclear export signal and a possible nuclear localization signal in Pbs2.
Keywords: docking site/HOG pathway/MAP kinase cascade/Pbs2/signal transduction
Introduction
Mitogen-activated protein kinases (MAPKs) are a conserved family of protein kinases that serve major roles in intracellular signal transduction in eukaryotic cells (for a review see Chen et al., 2001). MAPK is activated through a cascade of three sequentially activated kinases: MAPK is phosphorylated and thus activated by a MAPK kinase (MAPKK), and MAPKK is activated by a MAPKK kinase (MAPKKK). Once activated in the cytoplasm, MAPK translocates to the nucleus where it regulates the expression of various effector genes by phosphorylating specific transcription factors (Ferrigno et al., 1998).
A number of distinct MAPKs have been described in eukaryotic organisms: five in Saccharomyces cerevisiae and >10 in mammals. Different external stimuli, such as mitogenic growth factors, pro-inflammatory cytokines, and osmotic and oxidative stresses, activate distinct subsets of MAPKs. There are also similarly large numbers of MAPKKs and MAPKKKs in each organism, potentially permitting an enormous number of MAPKKK–MAPKK–MAPK combinations. However, because proper responses to external stimuli are so critical to cells and organisms, several mechanisms exist to ensure highly selective recognition among homologous kinases. Specific substrate–enzyme interaction, i.e. recognition of the substrate phosphorylation site(s) by the kinase catalytic site, must obviously be important. However, substrate–active site interaction per se is not selective enough and other mechanisms exist that contribute to the specificity of the interaction. For example, in many cases, a scaffold protein tethers three kinases together, thus limiting the flow of signal (Whitemarsh and Davis, 1998; Elion, 2001). Some proteins, such as yeast Ste5 and mammalian JIP-1, are dedicated scaffolds, whereas others, such as yeast Pbs2, have both a scaffold and a kinase function (Choi et al., 1994; Marcus et al., 1994; Printen and Sprague, 1994; Posas and Saito, 1997). Furthermore, it recently became evident that MAPKs use a docking domain to facilitate specific interactions with their activators (MAPKKs), inhibitors (protein phosphatases) and substrates (transcription factors) (Sharrocks et al., 2000; Tanoue et al., 2000; Enslen and Davis, 2001). Both a docking domain and a scaffold protein, perhaps combined with the substrate site specificity, contribute in varying degree to the final specificity between the two components (Bardwell et al., 2001).
There are greater structural diversities among MAPKKKs than among MAPKKs and MAPKs, perhaps reflecting similarly diverse upstream stimuli (Chen et al., 2001). Thus, specific MAPKKK–MAPKK interaction is a potential fate-determining event in cellular responses to external stimuli. Nonetheless, not much is yet known about how MAPKKK–MAPKK specificity is determined. The relationships between MAPKKKs and MAPKKs are far from simple linear arrangements: more than one MAPKKK may activate a common MAPKK; and a MAPKKK may activate more than one MAPKK. Examples of these situations can be found in the yeast HOG (high osmolarity glycerol)– and mating pheromone–MAPK pathways (Figure 1A; for a review see Gustin et al., 1998).

Fig. 1. (A) Schematic diagram of the HOG osmoregulatory and the mating pheromone signal transduction pathways in yeast. For the sake of clarity, a simplified model of the pathways is shown. Although Ste20, Ste50 and Ste11 are shared by the two pathways, an osmostress signal emanating from Sho1 follows the black arrows, whereas a pheromone signal follows the white arrows. Ste11 can also be activated by nitrogen limitation (not shown). The arrows do not necessarily indicate direct interactions. (B) Summary of functional analyses of the Pbs2 N-terminal regulatory region. A series of pbs2 mutants with N-terminal deletions as indicated was constructed using a centromeric vector, YCplac22I′. The mutants were introduced into the osmosensitive strains TM260 (pbs2Δ) TM280 (pbs2Δ ssk2Δ ssk22Δ) or KT503 (pbs2Δ ste11Δ) and their ability to complement osmosensitivity was determined. The Pbs2 coding sequence is represented by the horizontal bars; the kinase catalytic domain is shown as a filled box, and three regulatory subdomains (RSD-I, -II and -III) are represented as I, II and III. Growth (+) or no growth (–) on YPD + 1.2 M NaCl is shown. (C) Complementation of the osmosensitivity of TM260, TM280 or KT503 by pbs2 mutants with various N-terminal deletions. Cells were streaked on YPD or YPD + 1.2 M NaCl plates, and incubated at 30°C.
In the yeast MAPK cascades, the Ste11 MAPKKK can be activated by at least three distinct stimuli: high osmotic stress, mating factor and nitrogen limitation. When activated, Ste11 can activate (i.e. phosphorylate) two different downstream MAPKKs, Ste7 and Pbs2. Surprisingly, however, when Ste11 is activated by mating factors or by nitrogen limitation, it only transduces signals to Ste7, whereas when Ste11 is stimulated by osmotic stress, it only activates Pbs2 (Posas and Saito, 1997). Specificity of Ste11 signaling appears to be regulated by the scaffold proteins Ste5 and Pbs2, which specifically tether multiple components of only one or the other pathway (Posas and Saito, 1997; Harris et al., 2001), although a role for the Hog1 MAPK in cross-talk suppression has also been reported (Hall et al., 1996; O’Rourke and Herskowitz, 1998; Davenport et al., 1999).
The Pbs2 MAPKK can also be activated by Ssk2 and Ssk22 MAPKKKs, which are activated only by osmotic stress (Maeda et al., 1995; Posas et al., 1996; Posas and Saito, 1998). Unlike Ste11, the Ssk2/Ssk22 MAPKKKs only activate Pbs2, and never activate Ste7 (Posas and Saito, 1997; this study). These observations could be explained if Ste7 cannot be recognized by Ssk2/Ssk22. Our results reported here, however, clearly indicate that the major specificity determinant of Pbs2–Ssk2/Ssk22 interaction is a novel type of docking site found in the N-terminus of Pbs2. Without the docking site, Pbs2 cannot interact with Ssk2/Ssk22 productively. Conversely, fusion of the Pbs2 docking site to Ste7 allows activation of Ste7 by Ssk2/Ssk22. Thus the specificity of Ssk2/Ssk22 substrate interactions is determined by the presence of a specific docking site in its substrate, Pbs2.
Results
The N-terminal ‘regulatory’ domain of the Pbs2 MAPKK contains at least three subdomains that govern its activity or specificity
The Pbs2 MAPKK has a long N-terminal non-kinase region that presumably contains a number of functional subdomains. In order to identify subdomains involved in the regulation of Pbs2 in response to osmotic stress, we constructed a series of pbs2 N-terminal deletion mutants. These mutants were tested for their ability to modulate osmosensitivity by introduction (carried on a centromeric plasmid) into three mutant hosts, namely TM260 (pbs2Δ), TM280 (pbs2Δ ssk2Δ ssk22Δ) and KT503 (pbs2Δ ste11Δ). These strains, being pbs2Δ, are defective in the HOG pathway, and therefore cannot grow on high osmolarity media. In addition, TM280 and KT503 are also defective, respectively, in the SLN1 or SHO1 branches of the HOG pathway (see Figure 1A). Because these two upstream branches are functionally redundant, the wild-type PBS2 rescues the osmosensitivity of all three host mutants (Figure 1C). In contrast, if a plasmid-borne pbs2 mutant is such that it can transduce the signal only from one of the upstream branches, it will rescue only certain of the three host strains. As summarized in Figure 1B, this analysis revealed that there are at least three subdomains, designated RSD (regulatory subdomain)-I, -II and -III, that are required for proper regulation of Pbs2.
The pbs2Δ(5–54) mutant, lacking RSD-I, could complement the osmosensitivity of the pbs2Δ or pbs2Δ ssk2Δ ssk22Δ cells, but not that of the pbs2Δ ste11Δ cells (Figure 1C). Because the deletion mutant, pbs2Δ(57–207) was able to complement the osmosensitivity of pbs2Δ ste11Δ, this delimited the RSD-I region to amino acids 5–56. More importantly, osmotic activation of the Hog1 MAPK (as probed by activation-specific Hog1 phosphorylation) was nearly abrogated by the pbs2Δ(5–54) mutation, when the SHO1 branch was inactivated by the ste11Δ mutation (Figure 2A). This indicates that RSD-I is required for signal transduction from the SLN1 branch through the redundant Ssk2/Ssk22 MAPKKKs, but not for signaling from the SHO1 branch via Ste11.

Fig. 2. (A and B) Effect of Pbs2 N-terminal deletions on Hog1 activation. (A) Centromeric plasmids carrying either no insert (Vector), wild-type PBS2 (wt) or the indicated pbs2 deletion mutations were introduced individually into TM260 (pbs2Δ), TM280 (pbs2Δ ssk2Δ ssk22Δ), or KT503 (pbs2Δ ste11Δ). Cells were collected before (–) and 5 min after (+) addition of 0.4 M NaCl, and Hog1 activation was examined by immunoblotting the total lysate with an anti-phosho-p38 antibody, which cross-reacts with activated Hog1. (B) Further characterization of the effect of pbs2Δ(5–54) and pbs2V54G mutations on Hog1 activation. Plasmids containing either the wild-type PBS2, or the pbs2Δ(5–54) or pbs2V54G mutants were introduced into TM260 or KT503. Cells were collected at the indicated times after addition of 0.4 M NaCl, and Hog1 activation was examined as in (A). (C and D) Identification of the sequence within the Pbs2 RSD-I region that is required for signal transduction from the SLNI branch. (C) Deletion analysis of the Pbs2 RSD-I region. The effect of a series of short deletions in the RSD-1 region on the ability of Pbs2 to complement osmosensitivity was investigated essentially as described in Figure 1B. The amino acid sequence of the minimally essential region, as well as the isolated missense mutations that showed the same phenotype as Δ(5–54), are shown at the bottom. (D) Osmosensitivity phenotypes of several mutants in RSD-I. the osmosensitivity of various RSD-I mutants was tested in three host strains, TM260, TM280 and KT503, as described in Figure 1C.
By the same line of reasoning, RSD-II is essential for signal transduction from the SHO1 branch, but not from the SLN1 branch (Figures 1C and 2A). Because pbs2Δ (108–206) showed essentially the same response as the wild-type, RSD-II is therefore confined to the interval 55–107. To dissect RSD-II further, we generated missense mutations in this interval using a PCR mutagenesis procedure and gap repair, and screened for mutants that have a similar phenotype to prototype pbs2Δ(57–207). Several mutants thus isolated harbored L95S (a mutation of leucine at amino acid 95 to serine), P96A, P96L or P96S. Previously, we reported that residues 91–101 (VNKPLPPLPVA) constitutively bind to the Sho1 SH3 domain, and that the P96S mutation (pbs2-13) disrupted this interaction (Maeda et al., 1995). Thus, it is likely that the newly isolated mutations at L95 and P96 also disrupt the same interaction, and that Sho1 binding accounts for the activities associated with RSD-II.
RSD-III (residues 283–353) is essential for Pbs2-mediated signal transduction. Deletion of RSD-III from Pbs2 resulted in a complete loss of its ability to transmit signals to Hog1, even in the presence of all three intact MAPKKKs (Ssk2, Ssk22 and Ste11) (Figures 1C and 2A). It is possible that RSD-III is crucial for the transmission of signals from Pbs2 to Hog1, which is common to both the SHO1 and the SLN1 branches, perhaps by serving as a docking site for Hog1. Indeed, a recent study on Wis1 (the fission yeast homolog of Pbs2) demonstrated that the homologous region contains a docking site for Sty1 (the fission yeast homolog of Hog1) (Nguyen et al., 2002). The function of the Pbs2 RSD-III is currently under study.
Regulatory subdomain I is required for Pbs2 to transduce an activating signal from Ssk2/Ssk22, but not from Ste11
To define further the region in RSD-I that is essential for the transduction of signals from the SLN1 branch, various pbs2 mutant genes with small deletions in the RSD-I region were constructed and examined as above (Figure 2C and D). Because deletion of either residues 4–45 or residues 57–207 did not prevent the mutant Pbs2 from transducing an activating signal either from the Ssk2/Ssk22 or from the Ste11 MAPKKK, the borders of RSD-1 were limited further to residues 46 and 56. As a second approach, we introduced randomly generated mutations within residues 1–80 of Pbs2 using a PCR-based mutagenesis procedure. Among ∼5000 clones screened, three mutants (V54E, A56P and a double mutant R53C/A56P) failed to rescue the osmosensitivity of the pbs2Δ ste11Δ cells, but still complemented the pbs2Δ or pbs2Δ ssk2Δ ssk22Δ cells. We introduced oligonucleotide-based site-directed mutations into each amino acid positions between 51 and 56 (L and P at H51; F, P, S, V and D at A52; H, L, P and C at R53; G and E at V54; Q at K55; and P at A56). Among these mutants, A52P, R53P, R53C, V54G, V54E and A56P failed to rescue the osmosensitivity of the pbs2Δ ste11Δ cells, but could complement the pbs2Δ and pbs2Δ ssk2Δ ssk22Δ cells (Figure 2C and D). The effect of two representative RSD-I mutations [pbs2Δ(5–54) and pbs2V54G] on Hog1 phosphorylation following osmotic stress was examined more closely (Figure 2B). Both mutations significantly reduced Hog1 phosphorylation at all time points tested, indicating that these mutations inhibited Hog1 signaling rather than merely delaying Hog1 activation.
Pbs2 RSD-I contains a specific docking site for Ssk2/Ssk22
Because the mutations in RSD-1 only affect signaling from the SLN1 branch, we considered it most likely that the RSD-1 region serves as a specific docking site for a component of the SLN1 branch. To test this hypothesis, residues 1–67 of Pbs2 were fused to the LexA DNA-binding domain. Full- or nearly full-length Ssk1, Ssk2 and Ssk22 were fused individually to the Gal4 activation (ACT) domain, and binding to Pbs2(1–67) was tested by the two-hybrid method. Ssk1, as expected, did not interact with Pbs2(1–67) (data not shown). While the full-length Ssk2 showed no significant binding to Pbs2(1–67), Ssk22(62–1331) did bind strongly. To localize the Pbs2-binding site in Ssk22, various deletion constructs of Ssk22 were examined for their binding to Pbs2(1–67). As shown in Figure 3A, the intact kinase domain of Ssk22 (residues 998–1331) was necessary and sufficient for its binding to Pbs2(1–67). Shorter constructs of Ssk2 that contain only its kinase domain also bound strongly to Pbs2(1–67) (Figure 3B). Results of quantitative β-galactosidase assays for representative constructs are shown in Table I. The failure of Ssk2(50–1579) to bind to Pbs2(1–67) in the two-hybrid system could be due to a steric hindrance by its N-terminal ACT domain sequence. Introduction of the V54G mutation into Pbs2(1–67) completely abolished its interaction with Ssk2 and Ssk22 (Table I). Thus, RSD-I contains a specific docking site for the Ssk2/Ssk22 kinase domains, and it is essential for activation of Pbs2 by Ssk2/Ssk22. It cannot be excluded, however, that other regions in Pbs2 have subsidiary contributions to the docking activity.

Fig. 3. Two-hybrid analysis of the binding of Ssk2/Ssk22 MAPKKKs to Pbs2 RSD-I. (A) Binding of Ssk22 to Pbs2 RSD-I. The indicated segments of Ssk22 were fused to the Gal4 activation domain. Their binding to the Pbs2(1–67) segment fused to the LexA DNA-binding domain [LexA-Pbs2(1–67)] was evaluated with a β-galactosidase filter assay. The precise amino acid positions of the Ssk22 fragments, and the presence (+) or absence (–) of binding are indicated on the right. The putative position of the Ssk1-binding domain, deduced from its sequence homology to that of Ssk2, is indicated by a broken box. (B) Binding of Ssk2 to Pbs2 RSD-I. The indicated segments of Ssk2 were fused to the Gal4 activation domain, and their binding to LexA-Pbs2(1–67) was tested as in (A).
Table I. Two-hybrid analysis demonstrating the binding of a Pbs2 docking site to the kinase domain of Ssk2 or Ssk22.
| Gal4 activationdomain construct | LexA DNA-binding domain construct |
|||
|---|---|---|---|---|
| RasV12 | Ssk1 | Pbs2(1–67) | Pbs2V54G(1–67) | |
| Raf | 6.2 | 1.4 | 0.3 | 0.3 |
| Ssk2(50–1579) | 0.3 | 248.6 | 0.5 | 0.5 |
| Ssk2(50–1148) | 0.4 | 203.7 | 0.7 | 0.4 |
| Ssk2(1128–1579) | 0.5 | 1.2 | 79.9 | 0.3 |
| Ssk22(62–1331) | 0.4 | 574.4 | 18.5 | 0.4 |
| Ssk22(998–1331) | 0.4 | 1.3 | 63.1 | 0.4 |
Values are arbitrary units of β-galactosidase activity (see Materials and methods), averaged from six assays (duplicate determinations with three independent transformants).
In vivo interaction between Pbs2 and Ssk2
So far, we have shown that an interaction between the kinase domain of Ssk2/Ssk22 and the RSD-I domain of Pbs2 is essential for the activation of Pbs2 by Ssk2/Ssk22. Insofar as Pbs2 is a substrate of Ssk2/Ssk22, however, the kinase domain of Pbs2 should also interact with the Ssk2/Ssk22 kinase domains. What, then, is the role of the docking site in RSD-I? To obtain further insight into the role of the RSD-I domain, the interaction between Pbs2 and Ssk2/Ssk22 was investigated further by in vivo co-precipitation experiments. To this end, Pbs2 was tagged at its C-terminus with the hemagglutinin (HA) epitope, and Ssk2 was tagged at its N-terminus with GST. In these experiments, we used the catalytically inactive Pbs2 K389M mutant to prevent toxicity of Pbs2 overexpression. The pbs2Δ mutant cells were co-transformed with Pbs2-HA (or its derivatives) together with GST–Ssk2 (or its derivatives). The GST–Ssk2 fusion proteins were affinity purified from cell extracts, followed by immunoblot analysis of co-precipitated HA-tagged proteins. The full-length Pbs2 (Pbs2-FL) bound both full-length Ssk2 (Ssk2-FL) and the Ssk2 kinase domain-only construct (Ssk2ΔN) (Figure 4A, lanes 1–3). Binding was unaffected by osmotic stress (data not shown). The V54G mutation greatly reduced the ability of Pbs2V54G-FL to bind to Ssk2-FL (compare lanes 1 and 4), indicating that the docking site in RSD-I is important for binding of Pbs2 to Ssk2 in vivo. Binding of Pbs2V54G-FL to Ssk2ΔN, however, was only moderately attenuated compared with wild-type Pbs2-FL (Figure 4A, lanes 2 and 5).

Fig. 4. (A) In vivo binding of Pbs2 to Ssk2. TM261 (pbs2Δ) was co-transformed with a plasmid expressing either full-length GST–Ssk2 (GST–Ssk2-FL), GST–Ssk2ΔN or GST alone, together with a second plasmid expressing either full-length HA-tagged Pbs2 (Pbs2-FL-HA) or its mutant or deleted derivatives, under the PGAL1 promoter. GST fusion proteins were precipitated from cell lysates using glutathione–Sepharose beads as described in Materials and methods. Co-precipitated Pbs2-HA (or its derivatives) was detected by immunoblotting using an anti-HA antibody as shown in the top row. The second row indicates the expression of Pbs2-HA in the total extract. The bottom two rows show the levels of GST–Ssk2-FL and GST–Ssk2ΔN, respectively, in the precipitates (GST alone was expressed at even higher levels; data not shown). (B) Direct binding of Pbs2 RSD-I to the Ssk2 kinase domain. The cell lysate from E.coli expressing MBP–Pbs2(1–67) fusion protein was incubated with purified GST–Ssk2ΔN, or GST, bound to glutathione–Sepharose beads. After incubation, beads were washed and subjected to immunoblot analysis with anti-MBP and anti-GST antibodies. (C) In vivo binding of Pbs2 to Ste11. TM261 (pbs2Δ) was co-transformed with a plasmid expressing either full-length GST–Ste11 (GST–Ste11-FL), GST–Ste11ΔN, GST–Ste11ΔC or GST alone, together with a second plasmid expressing either Pbs2-FL-HA or Pbs2V54G-FL-HA under the PGAL1 promoter. Precipitation of GST fusion proteins from cell lysates and detection of co-precipitated Pbs2-HA (or its mutant form) were performed as described in (A).
These results suggested that the docking site in RSD-I and the kinase domain of Pbs2 were two independent binding domains. To prove this hypothesis, we first examined the interaction of Ssk2 with the RSD-I domain alone, using Pbs2ΔC in which the Pbs2 C-terminal kinase domain is deleted. As shown in Figure 4A, lanes 7–12, Pbs2ΔC could bind to either Ssk2-FL or to the Ssk2 kinase domain (Ssk2ΔN), whereas the Pbs2V54GΔC mutant protein could not bind either. Thus, the Pbs2 N-terminus binds the Ssk2 kinase domain, independently of the Pbs2 kinase domain, and such binding is completely abrogated by the V54G mutation. We next tested if the Pbs2 kinase domain alone could bind Ssk2, using Pbs2ΔN (which lacks the N-terminal residues 5–300, but contains the entire kinase domain). Interestingly, Pbs2ΔN, just like Pbs2V54G-FL, bound to Ssk2ΔN, but not to Ssk2-FL (Figure 4A, lanes 13–15). These results corroborate the hypothesis that Pbs2 has two binding sites for Ssk2. In order to test if the observed Ssk2–Pbs2 binding is an indirect effect of another yeast protein that binds both Ssk2 and Pbs2, we tested binding of bacterially produced GST–Ssk2ΔN and maltose-binding protein (MBP)–Pbs2(1–67). As shown in Figure 4B, these bacterially produced proteins bind each other, proving that these proteins interact directly. Taken together, we conclude that Pbs2 constitutively binds to Ssk2 via its docking site, whereas Pbs2–Ssk2 kinase domain interaction can only occur when the Ssk2 kinase domain is released from its N-terminal autoinhibitory domain.
Previously, we reported that Pbs2 binds the Ste11 MAPKKK (Posas and Saito, 1997). While our functional data shown in Figures 1 and 2 imply that RSD-I is not required for specific interaction with Ste11, we tested if that is indeed the case. In Figure 4C, lane 2 shows that the wild-type Pbs2 binds the full-length Ste11, as previously reported, and lanes 3 and 4 show that the Pbs2 binding requires the Ste11 N-terminal half, i.e. its non-kinase domain. Furthermore, the Pbs2 V54G mutant protein could bind Ste11 just as well as the wild-type Pbs2 (Figure 4C, lanes 6–8), indicating that the Pbs2–Ste11 binding involves a different type of docking interaction from the Pbs2–Ssk2 interaction.
Pbs2 RSD-I, when fused to the Ste7 MAPKK, endows a new specificity to Ste7
A specific docking site could contribute either to specificity or to efficiency (or both) of signal transduction. To learn more about the role of the RSD-I docking site, we made use of the observation that Ssk2/Ssk22 never activates the Ste7 MAPKK. This could be either because the Ste7 kinase domain is incompatible with Ssk2/Ssk22, or because Ste7 lacks a proper docking site. If the latter is the case, then appending the Pbs2 RSD-I domain to Ste7 might allow activation of Ste7 by Ssk2/Ssk22. To distinguish between these possibilities, we fused a Pbs2 RSD-I segment (residues 1–67) to the N-terminus of Ste7 to generate a Pbs2–Ste7 fusion construct (Figure 5A), and determined its ability to be activated by Ssk2/Ssk22. Activation of Pbs2-Ste7 was monitored by the expression of its downstream reporter gene, FUS1-lacZ (McCaffrey et al., 1987; O’Rourke and Herskowitz, 1998).

Fig. 5. ‘Cross-talk’ activation of the mating pathway by Ssk2/Ssk22 via binding to a Pbs2–Ste7 fusion protein. (A) Schematic diagram of the Pbs2–Ste7 fusion protein used in this analysis. (B) Induction of FUS1-lacZ expression following activation of the Pbs2–Ste7 fusion protein by constitutively active Ssk2ΔN or Ssk22ΔN. The reporter strain KT007 (pbs2Δ FUS1::lacZ::LEU2) was co-transformed with either pYES2 (Vector), pYES2-Ssk2ΔN or pYES2-Ssk22ΔN together with either YCplac22′ (Vector), YCplac22′-Pbs2–Ste7 or YCplac22′-Pbs2V54G–Ste7. The cells were grown in SRaf medium, and were either harvested (–Gal) or incubated further for 2 h in the presence of 2.5% galactose (+Gal). FUS1-lacZ expression was measured by assaying the β-galactosidase activities in cell lysates as described in Materials and methods. For each combination of plasmids, three independent transformants were assayed in triplicate, and the average activity ± SD is shown. (C) FUS1-lacZ expression induced by osmotic stress via the Pbs2–Ste7 fusion protein. KT005 (pbs2Δ ste11Δ) was co-transformed with a FUS1-lacZ reporter plasmid, pSB231, and either YCplac22′ (Vector), YCplac22′-Pbs2–Ste7 or YCplac22′-Pbs2V54G–Ste7. The cells were grown in CAD and either harvested (–NaCl), or incubated further for 4 h following the addition of 0.4 M NaCl (+NaCl). (D) Restoration of pheromone-induced FUS1-lacZ expression in the ste7Δ mutant by expression of the Pbs2–Ste7 or Pbs2V54G–Ste7 hybrid protein. FP56 (ste7Δ) was co-transformed with pSB231, and either YCplac22′ (Vector), YCplac22′-Pbs2–Ste7 or YCplac22′-Pbs2V54G–Ste7. The cells were grown in CAD and either harvested (–αF), or incubated further for 2 h following the addition of 5 µM α-factor (+αF). FUS1-lacZ expression was measured as in (B).
First, we examined whether the constitutively active Ssk2ΔN or Ssk22ΔN could activate Pbs2–Ste7 through a ‘cross-talk’ signaling. Because expression of Ssk2ΔN or Ssk22ΔN in wild-type cells is toxic due to Pbs2–Hog1 hyperactivation (Maeda et al., 1995), we used a pbs2Δ host strain in which the FUS1-lacZ reporter is chromosomally integrated. When Ssk2ΔN or Ssk22ΔN was expressed from the inducible GAL1 promoter, no induction of FUS1-lacZ expression was observed in the control cells that carry only the empty vector (Figure 5B), confirming that neither Ssk2 nor Ssk22 can activate Ste7. In sharp contrast, significant induction of FUS1-lacZ expression occurred in the cells expressing the Pbs2–Ste7 fusion construct. This induction is due to the RSD-I sequence in Pbs2–Ste7, because in the cells expressing Pbs2V54G–Ste7, induction of the FUS1-lacZ reporter gene was of a similar level to that in cells carrying the empty vector alone.
We also investigated whether endogenous Ssk2/Ssk22, when activated by osmotic stress, could activate the Pbs2–Ste7 hybrid. In this experiment, both the PBS2 and STE11 genes were deleted in the host strain, in order to preclude any possible activation of Ste7 or Pbs2–Ste7 by osmotically activated Ste11. The pbs2Δ ste11Δ cells harboring a FUS1-lacZ reporter plasmid were transformed with a second plasmid expressing either Pbs2–Ste7, Pbs2V54G–Ste7 or a plasmid containing no insert. Expression of FUS1-lacZ was measured before (0 h) and after (4 h) continuous exposure to osmotic stress (0.4 M NaCl). In the control cells carrying the empty vector, no expression of FUS1-lacZ was observed (Figure 5C), confirming a previous report (O’Rourke and Herskowitz, 1998). In contrast, there was significant induction of FUS1-lacZ in the cells carrying the Pbs2–Ste7 plasmid. Importantly, no expression of FUS1-lacZ was detected with the cells carrying the Pbs2V54G–Ste7 plasmid, demonstrating that the cross-talk signal was due to RSD-I in the Pbs2–Ste7 hybrid.
To test whether Pbs2–Ste7 and Pbs2V54G–Ste7 could complement the Ste7 function in the mating pathway, we expressed these hybrid constructs in ste7Δ mutant cells, and examined the induction of FUS1-lacZ reporter gene by α-factor. Figure 5D shows that both constructs are equally functional in transmitting the mating signal, excluding the possibility that Pbs2V54G–Ste7 is non-functional for a reason other than its defect in specific Ssk2 docking interaction.
These results demonstrate that the kinase domain of Ste7 can, if forced, serve as a substrate of Ssk2/Ssk22. Nonetheless, in the absence of a docking interaction such as afforded by RSD-I, signaling between Ssk2/Ssk22 and Ste7 does not occur. We conclude, therefore, that the RSD-I docking site is a major determinant of the functional specificity between the Ssk2/Ssk22 MAPKKK and the Pbs2 MAPKK in vivo.
Pbs2 contains a nuclear export signal (NES) in RSD-I, and a possible nuclear localization signal (NLS) at its C-terminus
As part of the investigation of the function of RSD-I, we searched for proteins that might interact with RSD-I by two-hybrid screening using LexA-Pbs2(1–67) as bait. In this screen, two components of the nuclear pore complex (NPC), Nup57 and Nup100, were isolated as potential Pbs2 interacters. Because these NPC components contain the GLFG repeat motifs that bind transport cargo receptors (Allen et al., 2001), Pbs2(1–67) may interact indirectly with the Nup proteins via an importin or an exportin. As reported previously (Ferrigno et al., 1998; Reiser et al., 1999), Pbs2–green fluorescent protein (GFP) exclusively localizes to the cytoplasm regardless of osmotic stress (Figure 6A). In contrast, Pbs2Δ(5–54)–GFP predominantly localized to the nucleus (Figure 6B), suggesting that the RSD-I region contains a sequence required for either cytoplasmic retention or nuclear exclusion of Pbs2.

Fig. 6. Subcellular localization of the wild-type and mutant Pbs2 proteins. (A–G) TM261 (pbs2Δ) was transformed with either a plasmid expressing the wild-type Pbs2 fused to GFP (Pbs2–GFP) or its mutant derivatives, as indicated. Cells were grown to mid-log phase in CAD medium, and localization of the GFP fusion proteins was determined by fluorescence microscopy as described in Materials and methods. Localization of Pbs2–GFP and Pbs2Δ(4–18) was examined before (–NaCl) and 5 min after (+NaCl) addition of 0.4 M NaCl. Localization of the other proteins was observed in the absence of osmotic stress. The nuclear export signal (NES), within the first 20 amino acid residues of Pbs2, is shown in (E). DIC, differential interference contrast. (H) Analysis of Pbs2 C-terminal region deletion mutants. The Pbs2 C-terminal amino acid sequence, with the end of the kinase domain indicated by an arrow, is shown above. Below the sequence, C-terminal deletion mutations are indicated by horizontal bars. Thease deletion mutations were combined, individually, with the N-terminal NES mutation Δ(4–18), and the subcellular localizations of the double mutants were examined as in (F) and (G). ‘Wild-type’ refers to the Δ(4–18) mutation alone. Nuc, nuclear localization; Cyto, cytoplasmic localization.
We then considered whether a relationship exists between the Ssk2/Ssk22 binding activity and the nuclear exclusion activity of RSD-I. One possibility was that Ssk2/Ssk22 serves as a cytoplasmic anchor for the otherwise nuclear Pbs2 protein. To test this possibility, we determined the cellular localization of two additional deletion mutants, Pbs2Δ(31–54)–GFP and Pbs2Δ(4–18)–GFP. Pbs2Δ(31–54)–GFP, which lacks the Ssk2/Ssk22-binding site, nonetheless was localized predominantly to the cytoplasm (Figure 6D), whereas Pbs2Δ(4–18)–GFP localized to the nucleus (Figure 6C). These results clearly demonstrate that the domain that mediates nuclear exclusion is distinct from the docking site.
A close inspection of the Pbs2 sequence around residues 4–18 revealed that an NES-like sequence, with characteristic leucine residues, exists in this region (Figure 6E). Indeed, conversion of these leucine residues at residues 8 and 10 to alanine (L8A/L10A) had the same effect as Δ(5–54) or Δ(4–18), namely accumulation of mutant Pbs2–GFP in the nucleus (Figure 6E). Thus, it is likely that this is indeed a functional NES sequence. Although the Pbs2 mutants in the NES accumulated in the nucleus, those mutants could functionally complement the osmosensitivity of pbs2Δ strains (data not shown), probably because there are still sufficient amounts of the cytoplasmic Pbs2. They also did not affect the cellular localization of the Hog1 MAPK, regardless of the presence or absence of osmotic stress (data not shown).
Since the Pbs2–GFP molecule (∼100 kDa) is too large to diffuse passively though the nuclear pores (Rabut and Ellenberg, 2001), it probably uses an active nuclear transport process. Thus, we reasoned that Pbs2 either has an NLS or binds to a nuclear localizing partner. Interestingly, deletion of the Pbs2 C-terminal region (residues 636–668) reversed the nuclear accumulation of Pbs2Δ(5–54)–GFP, suggesting that this region facilitates nuclear import of Pbs2 (data not shown). Further analyses showed that deletion of 636–654, but not of 640–650, 645–668 or 654–668, prevented the nuclear accumulation (Figure 6F and G, and summarized in H), indicating that a sequence including the positions 636–639 (YITE) is required for the nuclear localization of Pbs2. Further analyses are needed to clarify whether this region contains an NLS or a binding site for a nuclear localizing partner.
Recently, it was reported that the Wis1 (Nguyen et al., 2002) and MKK6 (Hashimoto et al., 2000) proteins, fission yeast and carp homologs of Pbs2, respectively, also contain a NES sequence at their N-terminal region. These conserved patterns of MAPKK nuclear–cytoplasmic shuttling may be required for the activation or regulation of the MAPK pathway.
Discussion
In this report, we identified a docking site specific for Ssk2/Ssk22 MAPKKKs, an NES and a possible NLS within the Pbs2 MAPKK (summarized in Figure 7A). In recent years, specific interactions between MAPKs and their interacting partners have been intensely studied, culminating in the definition of the common docking (CD) domain in a number of MAPKs (Tanoue et al., 2000). The molecular determinants that underlie specific MAPKKK–MAPKK interaction, however, have been poorly understood. The N-terminal non-kinase domain of mammalian JNKK1 (MKK4) is known to be essential for its stable interaction with MEKK1 (Xia et al., 1998), but it is not known whether the JNKK1 N-terminal sequence alone binds MEKK1.
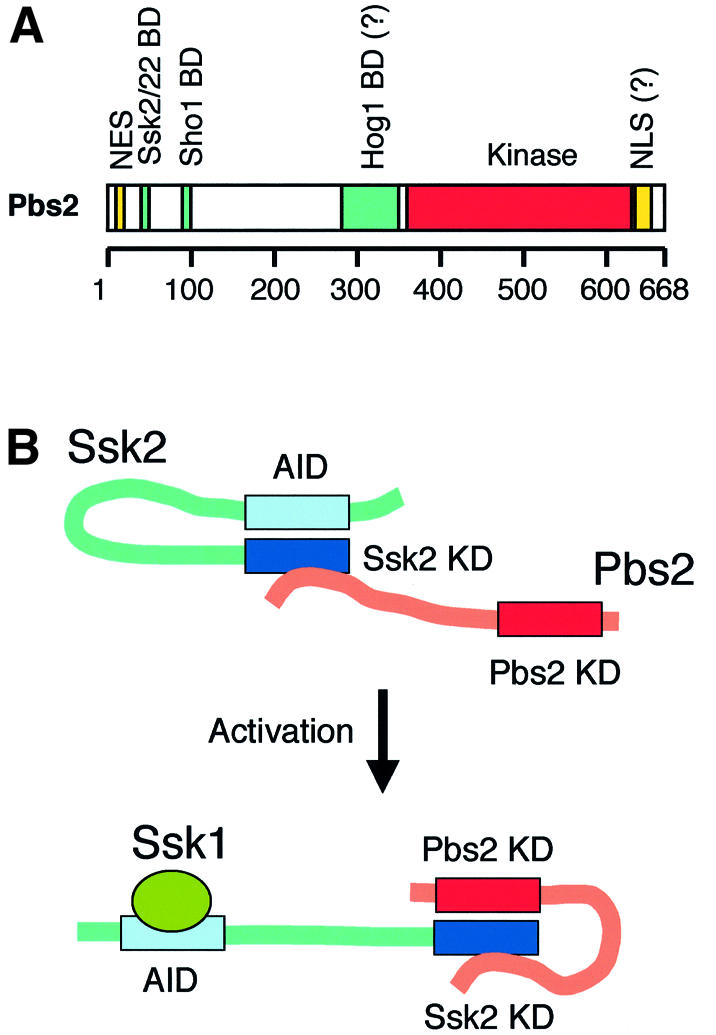
Fig. 7. (A) A schematic summary of the Pbs2 structure showing various functional domains. For details, see text. The horizontal bar represents the Pbs2 protein, and the scale below indicates amino acid positions. NES, nuclear export signal; BD, binding domain; NLS, nuclear localization signal. (B) A possible model of Pbs2 activation by Ssk2. In its inactive state, the Ssk2 N-terminal autoinhibitory domain (AID) binds and blocks the activity of its own kinase domain (KD). Pbs2 is constitutively bound, via its N-terminal docking domain, to Ssk2, but the Pbs2 kinase domain has no access to the blocked Ssk2 kinase domain. When the Ssk2 activator, Ssk1, binds to a region near the AID, the Ssk2 kinase domain becomes accessible to the Pbs2 kinase domain. Alternative models are also possible.
A specific docking site could contribute either to efficiency or to specificity (or both) in signal transduction. It is conceivable, for example, that RSD-I might increase signal efficiency by forming a pre-activation complex between Ssk2/Ssk22 and Pbs2. Thus, upon activation of Ssk2/Ssk22 by an upstream signal, the associated Pbs2 could be activated promptly. Indeed, there is evidence for very weak activation of Pbs2 by Ssk2/Ssk22 in the absence of the RSD-I docking site. In pbs2ΔRSD-I mutants, such as pbs2Δ(5–54), Hog1 phosphorylation was greatly reduced in the ste11Δ background, but not completely abolished. Signal strength was drastically enhanced when RSD-I was available for binding. Thus, the specific docking site for Ssk2/Ssk22 contributes to the efficient transmission of activating signals.
The weak but detectable signal in the pbs2ΔRSD-I mutants was due to direct interaction between the kinase domains of Ssk2/Ssk22 and Pbs2. Indeed, we have demonstrated that the Pbs2 kinase catalytic domain alone can interact with the catalytic domain of Ssk2/Ssk22. This raises the second possibility that the specificity of the interaction is encoded in the mutual compatibility of the MAPKKK and MAPKK kinase domains, while the docking domain serves an ancillary function. Our experiments using the Pbs2–Ste7 fusion construct, however, did not support this hypothesis. Activated Ssk2/Ssk22 can phosphorylate (and thus activate) the Ste7 kinase domain, if they are forced to interact with Ste7 via an artificially fused RSD-I. This indicates that the failure of Ssk2/Ssk22 to activate Ste7 under osmotic stress is not due to the incompatibility of the kinase domains. Rather, it is because Ste7 lacks a proper docking site that ensures a sufficient signaling efficiency. Similarly, it was reported that direct fusion of Ste11 to Pbs2 confined the signal to a particular pathway regardless of the type of stimulus (Harris et al., 2001). The absence of the Ssk2/Ssk22 docking site in Ste7, therefore, would effectively shield the mating pathway from inappropriate activation by Ssk2/Ssk22 under osmostress conditions. In other words, the enhancement of signaling efficiency by the docking interaction is so large that it practically determines the specificity of signal transmission.
Based on the results from our in vivo co-precipitation experiments, we propose the following molecular mechanism for the activation of Pbs2 by Ssk2/Ssk22 (Figure 7B). Pbs2 is constitutively bound to Ssk2/Ssk22 even under unstressed conditions. Although the Pbs2 kinase domain can also bind to the Ssk2/Ssk22 kinase domain, RSD-I is mainly responsible for the constitutive binding of Pbs2 to the full-length Ssk2/Ssk22, as evidenced by the drastically diminished binding of Pbs2V54G to Ssk2-FL. Direct binding between the kinase domains of Ssk2/Ssk22 and Pbs2, however, could occur if the Ssk2 N-terminal region was deleted. The Ssk2/Ssk22 N-terminal region presumably contains the kinase autoinhibitory domain (Maeda et al., 1995; Posas and Saito, 1998). MTK1, a human homolog of Ssk2/Ssk22, regulates its kinase activity using an analogous inhibitory machinery (Mita et al., 2002). Thus, Pbs2 activation by Ssk2/Ssk22 can be explained as follows. In the closed conformation, Ssk2/Ssk22 forms a pre-activation complex with Pbs2 via the RSD-I docking site. Upon osmotic activation, binding of Ssk1 to the N-terminal region of Ssk2/Ssk22 releases the autoinhibitory domain, resulting in conversion of Ssk2/Ssk22 to the open conformation, thereby allowing direct interaction between the Pbs2 and Ssk2/Ssk22 kinase domains. We could not reproducibly observe an enhanced interaction between Ssk2/Ssk22 and Pbs2 in the absence of the docking interaction after osmotic shock, further demonstrating the importance of the docking interaction for efficient signal transmission.
Materials and methods
Yeast strains
The following yeast strains were used: TM141 (MATa ura3 leu2 trp1 his3); TM231 (MATα ura3 leu2 his3 pbs2::URA3); TM260 (MATa ura3 leu2 trp1 pbs2::LEU2); TM261 (MATα ura3 leu2 his3 pbs2::LEU2); TM278 (MATa ura3 leu2 trp1 pbs2::URA3); TM280 (MATa ura3 leu2 trp1 pbs2::URA3 ssk2::LEU2 ssk22::LEU2); KT005 (MATa ura3 leu2 trp1 his3 pbs2::LEU2 ste11::HIS3); KT500 (MATα ura3 leu2 trp1 his3 pbs2::HIS3 ssk2::LEU2 ssk22::LEU2); KT503 (MATa ura3 leu2 trp1 his3 pbs2::URA3 ste11::HIS3); KT504 (MATa ura3 leu2 trp1 his3 pbs2::LEU2 sho1::HIS3); FP12 (MATa leu2 trp1 his3 LYS2::lexA-HIS3 URA3::lexA-lacZ pbs2::HIS3); and FP56 (MATa ura3 leu2 trp1 his3 ste7::HIS3). The FUS1-lacZ reporter strain KT007 (MATa ura3 leu2 trp1 his3 pbs2::HIS3 FUS1::lacZ::LEU2) was constructed using pFC23 (O’Rourke and Herskowitz, 1998).
Buffers and media
Standard yeast media and genetic procedures were as described previously (Rose et al., 1990). SRaf medium consists of 0.67% yeast nitrogen base and 2% raffinose with appropriate yeast synthetic drop-out medium supplement. CAD medium consists of 0.67% yeast nitrogen base, 2% glucose, 0.5% casamino acid and appropriate supplements. Buffer A is 50 mM Tris–HCl pH 7.5, 15 mM EDTA, 15 mM EGTA, 2 mM dithiothreitol, 0.2% Triton X-100, 1 mM phenylmethylsulfonyl fluoride (PMSF), 1 mM benzamidine, 5 µg/ml leupeptin, 50 mM NaF, 25 mM β-glycerophosphate and 150 mM NaCl. Z buffer is 50 mM Na2HPO4, 40 mM NaH2PO4, 10 mM KCl, 1 mM MgSO4, adjusted to pH 7.0. Buffer B is 50 mM Tris–HCl pH 7.5, 1 mM EDTA, 1% Triton X-100, 1 mM PMSF, 1 mM benzamidine, 5 µg/ml aprotinin and 150 mM NaCl.
Plasmids
Deletion mutants were constructed by a PCR-based strategy, and missense mutants by oligonucleotide-based mutagenesis. All mutations were confirmed by nucleotide sequence determination. pbs2 deletion and missense mutants were subcloned into a centromeric vector YCplac22′, a derivative of YCplac22 (Gietz and Sugino, 1988) in which the unique HindIII and EcoO109I sites were destroyed. HA-tagged Pbs2 and its derivatives were also constructed by a PCR strategy; a galactose-inducible PGAL1 promoter was placed upstream of the PBS2 open reading frame (ORF), and a sequence encoding the HA epitope was fused to the C-terminus. The fused constructs were subcloned into the multicopy vector pRS423 (Invitrogen). p423-Pbs2ΔC-HA and p423-Pbs2V54GΔC-HA contain a deletion of Pbs2 residues 355–648. p423-Pbs2ΔN-HA contains a deletion of residues 5–300. For the GST-fused Ssk2 constructs, the SSK2 ORF or its derivative, tagged with the HA-coding sequence at their C-terminus, was fused to the C-terminus of the GST ORF in the multicopy vector p426-TEG (Posas and Saito, 1998). p426-GST-Ssk2ΔN has residues 1–1215 of Ssk2 truncated. GST fusion proteins are constitutively expressed from the strong promoter PTEF1 in p426-TEG. GST–Ste11 fusion constructs were described previously (Posas et al., 1998). pSB231 was used for an episomal FUS1-lacZ reporter as described before (Posas and Saito, 1997). YCplac22′-Pbs2–Ste7 contains a fusion of Pbs2(1–67) with the intrinsic PBS2 promoter region to the N-terminus of full-length Ste7. pYES2-Ssk2ΔN and pYES2-Ssk22ΔN contain Ssk2 residues 1173–1579 and Ssk22 residues 951–1331, respectively, under the control of the PGAL1 promoter in pYES2 (Invitrogen). p416-Pbs2-GFP was described previously (Ferrigno et al., 1998).
Mutant screening
pbs2 mutants that were osmosensitive when the SHO1 branch was inactivated were isolated as follows. Briefly, a segment of PBS2 corresponding to its upstream promoter region and amino acids 1–80 was mutagenized by PCR in the presence of 0.1 mM MnCl2. The PCR products were co-transformed into KT504 (MATa pbs2Δ sho1Δ) together with a linearized PBS2 plasmid in which codons 1–56 had been removed to make a gap by restriction enzyme digestion. Transformants were replica-plated onto YPD plates containing 1.2 M NaCl to identify osmosensitive mutants. The osmosensitive transformants were individually crossed with TM231 (MATα pbs2Δ SHO1), to generate diploid TM231/KT504 (pbs2Δ/pbs2Δ SHO1/sho1Δ) cells. The pbs2 mutants which rendered TM231/KT504 osmoresistant were recovered and re-introduced into the three tester strains TM260, TM280 and KT503. The nucleotide sequence of pbs2 mutants that rendered TM260 and TM280, but not KT503, osmoresistant was determined. The pbs2 mutants that were osmosensitive when the SLN1 branch was inactivated were also isolated using a similar procedure, except that KT500 (MATα pbs2Δ ssk2Δ ssk22Δ) and TM278 (MATa pbs2Δ SSK2 SSK22) were used as the primary host and a mating partner, respectively.
Detection of phosphorylated Hog1
Cells were grown in CAD medium until an OD600 of ∼0.5 was attained. NaCl was added to a final concentration of 0.4 M, and cells were harvested at the indicated times, frozen in liquid nitrogen, suspended in SDS loading buffer, immediately boiled for 5 min, and subjected to SDS–PAGE. Activated Hog1, doubly phosphorylated at Thr174 and Tyr176, was detected by use of an anti-phospho-p38 antibody (Cell Signaling) together with the enhanced chemiluminescence (ECL) reagent (Amersham).
Two-hybrid analysis
Two-hybrid analysis was carried out essentially as previously described (Posas and Saito, 1998). pACTII-SSK2(50–1148), pACT-Raf, pLexA-SSK1 and pLexA-RasV12 were also described (Posas and Saito, 1998). The other constructs were generated by a PCR procedure. The FP12 yeast reporter strain was co-transformed with a pBTM116-based plasmid encoding either Pbs2(1–67), Pbs2V54G(1–67), Ssk1 or RasV12 together with a pACTII-based plasmid encoding either Raf or various Ssk2 or Ssk22 segments. Two-hybrid interactions were tested qualitatively using a colony-lift β-galactosidase filter assay. To assay β-galactosidase activity quantitatively, extracts of log-phase cells were made in Z buffer by four cycles of freezing and thawing. A 0.1 ml aliquot of cell extract was mixed with 0.7 ml of Z buffer containing 50 mM β-mercaptoethanol and 0.16 ml of 4 mg/ml o-nitrophenyl β-d-galactopyranoside (ONPG), and incubated at 37°C for the indicated times. Following quenching of the reaction with 0.4 ml of 1 M Na2CO3, samples were spun briefly in a microcentrifuge, and the OD420 of the supernatant was measured. β-galactosidase activity (Miller units) was calculated using the following formula: β-galactosidase units = 1000 × OD420/[incubation time (min) × volume of the culture (ml) × OD600 of culture] (Miller, 1972).
In vivo Ssk2-Pbs2 and Ste11-Pbs2 binding assay
Cells grown in SRaf were cultivated further for 4 h following the addition of 2% galactose. Cell extracts were prepared in buffer A using glass beads essentially as described previously (Posas et al., 1996). A 500 µg aliquot of protein was incubated with 50 µl of glutathione–Sepharose beads for 3 h at 4°C. Beads were washed extensively with buffer A, resuspended in SDS loading buffer and separated by SDS–PAGE. Immunoblotting was carried out with the 12CA5 anti-HA antibody (Roche) and detected with the ECL reagent.
In vitro Ssk2–Pbs2 binding assay
GST–Ssk2ΔN and GST proteins were expressed in Escherichia coli DH5α and purified using glutathione–Sepharose beads essentially as described previously (Posas et al., 1996). Approximately 5 µg of GST–Ssk2ΔN or an equimolar amount of GST bound to beads was incubated with 200 µg of cell lysate in buffer B prepared from E.coli expressing Pbs2(1–67) tagged with MBP at its N-terminus [MBP–Pbs2(1–67)] for 4 h at 4°C. Beads were washed extensively in buffer B, resuspended in SDS loading buffer and separated by SDS–PAGE. Immunobloting was performed using the anti-GST antibody (Amersham) or the 8G1 anti-MBP antibody (Cell Signaling) together with ECL reagent.
FUS1-lacZ reporter assay
To investigate the effects of Ssk2ΔN or Ssk22ΔN expression, KT007 (pbs2Δ FUS1::lacZ::LEU2) was co-transformed with either pYES2, pYES2-Ssk2ΔN or pYES2-Ssk22ΔN together with YCplac22′, YCplac22′-Pbs2–Ste7 or YCplac22′-Pbs2V54G–Ste7. The cells were grown in SRaf medium and were incubated further for 2 h following the addition of galactose (to a final concentration of 2.5%). β-galactosidase activity was assayed as described above. To study the effects of osmotic stress on the induction of Ste7 activation, KT005 (pbs2Δ ste11Δ) was co-transformed with a FUS1-lacZ reporter plasmid, pSB231, together with either YCplac22′, YCplac22′-Pbs2–Ste7 or YCplac22′-Pbs2V54G–Ste7. These cells were grown in the CAD medium and were incubated further for 4 h following the addition of NaCl (to a final concentration of 0.4 M). FUS1-lacZ expression was measured by the β-galactosidase liquid culture assay.
Microscopy
Yeast cells were observed without fixation using an Olympus BX51 fluorescence microscope. GFP signal was detected using a U-MNIBA filter (Olympus). Digital images were captured using an Orca-ER CCD camera with Aquacosmos Imaging software (Hamamatsu Photonics).
Acknowledgments
Acknowledgements
We thank I.Herskowitz and S.M.O’Rourke for the pFC23 plasmid, V.Reiser for technical advice, P.O’Grady for reading the manuscript, and S.Jibiki and E.Kasukawa for excellent technical assistance. This work was supported in part by Grants-in-Aid from the Ministry of Education, Culture, Sports, Science and Technology of Japan, a PRESTO grant from the Japan Science and Technology Corp., and a grant from the Asahi Glass Foundation.
References
- Allen N.P.C., Huang,L., Burlingame,A. and Rexach,M. (2001) Proteomic analysis of nucleoporin interacting proteins. J. Biol. Chem., 276, 29268–29274. [DOI] [PubMed] [Google Scholar]
- Bardwell A.J., Flatauer,L.J., Matsukuma,K., Thorner,J. and Bardwell,L. (2001) A conserved docking site in MEKs mediates high-affinity binding to MAP kinases and cooperates with a scaffold protein to enhance signal transmission. J. Biol. Chem., 276, 10374–10386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z., Gibson,T.B., Robinson,F., Silvestro,L., Pearson,G., Xu,B.E., Wright,A., Vanderbilt,C. and Cobb,M.H. (2001) MAP kinases. Chem. Rev., 101, 2449–2476. [DOI] [PubMed] [Google Scholar]
- Choi K.Y., Satterberg,B., Lyons,D.M. and Elion,E.A. (1994) Ste5 tethers multiple protein kinases in the MAP kinase cascade required for mating in S.cerevisiae. Cell, 78, 499–512. [DOI] [PubMed] [Google Scholar]
- Davenport K.D., Williams,K.E., Ullmann,B.D. and Gustin,M.C. (1999) Activation of the Sacchromyces cerevisiae filamentation/invasion pathways by osmotic stress in high-osmolarity glycogen pathway mutants. Genetics, 153, 1091–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elion A.E. (2001) The Ste5p scaffold. J. Cell Sci., 114, 3967–3978. [DOI] [PubMed] [Google Scholar]
- Enslen H. and Davis,R. (2001) Regulation of MAP kinases by docking domains. Biol. Cell, 93, 5–14. [DOI] [PubMed] [Google Scholar]
- Ferrigno P., Posas,F., koepp,D., Saito,H. and Silver,P.A. (1998) Regulated nucleo/cytoplasmic exchange of HOG1 MPAK requires the importin β homologs NMD5 and XPO1. EMBO J., 17, 5606–5614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gietz R.D. and Sugino,A. (1988) New yeast–Escherichia coli shuttle vectors constructed with in vitro mutagenized yeast genes lacking six-base pair restriction sites. Gene, 74, 527–534. [DOI] [PubMed] [Google Scholar]
- Gustin M.C., Albertyn,J., Alexander,M. and Davenport,K. (1998) MAP kinase pathways in the yeast Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev., 62, 1264–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall J.P., Cherkasova,V., Elion,E., Gustin,M.C. and Winter,E. (1996) The osmoregulatory pathway represses mating pathway activity in Saccharomyces cerevisiae: isolation of a FUS3 mutant that is insensitive ot the repression mechanism. Mol. Cell. Biol., 16, 6715–6723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris K., Lamson,R.E., Nelson,B., Hughes,T.R., Marton,M.J., Roberts,C.J., Boone,C. and Pryciak,P.M. (2001) Role of scaffolds in MAP kinase pathway specificity revealed by custom design of pathway-dedicated signaling proteins. Curr. Biol., 11, 1815–1824. [PubMed] [Google Scholar]
- Hashimoto H., Fukuda,M., Matsuo,Y., Yokoyama,Y., Nishida,E., Toyohara,H. and Sakaguchi,M. (2000) Identification of a nuclear export signal in MKK6, an activator of the carp p38 mitogen-activated protein kinases. Eur. J. Biochem., 267, 4362–4371. [DOI] [PubMed] [Google Scholar]
- Maeda T., Takekawa,M. and Saito,H. (1995) Activation of yeast PBS2 MAPKK by MAPKKKs or by binding of an SH3-containing osmosensor. Science, 269, 554–558. [DOI] [PubMed] [Google Scholar]
- Marcus S., Polverino,A., Barr,M. and Wigler,M. (1994) Complexes between STE5 and components of the pheromone-responsive mitogen-activated protein kinase module. Proc. Natl Acad. Sci. USA, 91, 7762–7766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCaffrey G., Clay,F.J., Kelsay,K. and Sprague,G.F. (1987) Identification and regulation of a gene required for cell fusion during mating of the yeast Saccharomyces cerevisiae. Mol. Cell. Biol., 7, 2680–2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller J.H. (1972) Experiments in Molecular Genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- Mita H., Tsutsui,J., Takekawa,M., Witten,E.A. and Saito,H. (2002) Regulation of MTK1/MEKK4 kinase activity by its N-terminal autoinhibitory domain and GADD45 binding. Mol. Cell. Biol., 22, 4544–4555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen A.N., Ikner,A.D., Shiozaki,M., Warren,S.M. and Shiozaki,K. (2002) Cytoplasmic localization of Wis1 MAPKK by nuclear export signal is important for nuclear targeting of Spc1/Sty1 MAPK in fission yeast. Mol. Biol. Cell, 13, 2651–2663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Rourke S.M. and Herskowitz,I. (1998) The Hog1 MAPK prevents cross talk between the HOG and pheromone response MAPK pathways in Saccharomyces cerevisiae. Genes Dev., 12, 2874–2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posas F. and Saito,H. (1997) Osmotic activation of the HOG MAPK pathway via Ste11p MAPKKK: scaffold role of Pbs2p MAPKK. Science, 276, 1702–1705. [DOI] [PubMed] [Google Scholar]
- Posas F. and Saito,H. (1998) Activation of the yeast SSK2 MAP kinase kinase kinase by the SSK1 two-component response regulator. EMBO J., 17, 1385–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posas F., Wurgler-Murphy,S.M., Maeda,T., Witten,E.A., Thai,T.C. and Saito,H. (1996) Yeast HOG1 MAP kinase cascade is regulated by a multistep phosphorelay mechanism in the SLN1–YPD1–SSk1 ‘two-component’ osmosensor. Cell, 86, 865–875. [DOI] [PubMed] [Google Scholar]
- Posas F., Witten,E.A. and Saito,H. (1998) Requirement of STE50 for osmostress-induced activation of the STE11 mitogen-activated protein kinase kinase kinase in the high-osmolarity glycerol response pathway. Mol. Cell. Biol., 18, 5788–5796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Printen J.A. and Sprague,G.F. (1994) Protein–protein interactions in the yeast pheromone response pathway: Ste5p interacts with all members of the MAP kinase cascade. Genetics, 138, 609–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabut G. and Ellenberg,J. (2001) Nucleocytoplasmic transport: diffusion channel or phase transition? Curr. Biol., 11, R551–R554. [DOI] [PubMed] [Google Scholar]
- Reiser V., Ruis,H. and Ammerer,G. (1999) Kinase activity-dependent nuclear export opposes stress-induced nuclear accumulation and retention of Hog1 mitogen-activated protein kinase in the budding yeast Saccharomyces cerevisiae. Mol. Biol. Cell, 10, 1147–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose M.D., Winston,F. and Hieter,P. (1990) Methods in Yeast Genetics: A Laboratory Course Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- Sharrocks A.D., Yang,S.H. and Galanis,A. (2000) Docking domains and substrate-specificity determination for MAP kinases. Trends Biochem. Sci., 25, 448–453. [DOI] [PubMed] [Google Scholar]
- Tanoue T., Adachi,M., Moriguchi,T. and Nishida,E. (2000) A conserved docking motif in MAP kinases common to substrates, activators and regulators. Nature Cell Biol., 2, 110–116. [DOI] [PubMed] [Google Scholar]
- Whitemarsh A.J. and Davis,R.J. (1998) Mammalian scaffold complex that selectively mediates MAP kinase activation. Science, 281, 1671–1674. [DOI] [PubMed] [Google Scholar]
- Xia Y., Wu,Z., Su,B., Murray,B. and Karin,M. (1998) JNKK1 organizes a MAP kinase module through specific and sequential interactions with upstream and downstream components mediated by its amino-terminal extension. Genes Dev., 12, 3369–3381. [DOI] [PMC free article] [PubMed] [Google Scholar]