

Abstract
In the eukaryotic cytosol, Hsp70 and Hsp90 cooperate with various co-chaperone proteins in the folding of a growing set of substrates, including the glucocorticoid receptor (GR). Here, we analyse the function of the co-chaperone Tpr2, which contains two chaperone-binding TPR domains and a DnaJ homologous J domain. In vivo, an increase or decrease in Tpr2 expression reduces GR activation, suggesting that Tpr2 is required at a narrowly defined expression level. As shown in vitro, Tpr2 recognizes both Hsp70 and Hsp90 through its TPR domains, and its J domain stimulates ATP hydrolysis and polypeptide binding by Hsp70. Furthermore, unlike other co-chaperones, Tpr2 induces ATP-independent dissociation of Hsp90 but not of Hsp70 from chaperone–substrate complexes. Excess Tpr2 inhibits the Hsp90-dependent folding of GR in cell lysates. We propose a novel mechanism in which Tpr2 mediates the retrograde transfer of substrates from Hsp90 onto Hsp70. At normal levels substoichiometric to Hsp90 and Hsp70, this activity optimizes the function of the multichaperone machinery.
Keywords: glucocorticoid receptor/Hsp70/Hsp90/molecular chaperones/protein folding
Introduction
Molecular chaperones aid in the folding of newly-synthesized polypeptides and the refolding of proteins after stress-induced denaturation. In the eukaryotic cytosol, the ATP-dependent chaperones Hsc70/Hsp70 (termed Hsp70 for simplicity) and Hsp90 often cooperate in the folding of a variety of substrate proteins. Hsp70 recognizes extended hydrophobic peptide sequences and generally acts at an early stage of polypeptide folding. In contrast, Hsp90 is thought to recognize the near-native conformations of a more restricted range of substrates, including several nuclear receptors (Bukau and Horwich, 1998; Buchner, 1999; Young et al., 2001; Hartl and Hayer-Hartl, 2002).
The function of the Hsp70/Hsp90 multichaperone machinery has been analysed in much detail for the progesterone receptor and glucocorticoid receptor (GR) (Pratt and Toft, 1997). After the initial binding of Hsp70, the co-chaperone Hop connects Hsp70 to Hsp90, which recognizes the ligand binding domain (LBD) of the receptor. Hsp70 and Hop are then replaced on the Hsp90–receptor complex by the immunophilin FKBP52 and the co-chaperone protein p23, and the receptor attains a state capable of binding the hormone ligand. In the presence of hormone, the receptor can proceed to activate transcription from specific steroid hormone response elements on the chromatin (Pratt and Toft, 1997; Chen and Smith, 1998; Johnson et al., 1998).
For both Hsp70 and Hsp90, polypeptide binding and release is regulated by an ATPase cycle. Hsp70 in its ATP-bound form has fast on- and off-rates for substrate polypeptides and ATP hydrolysis by Hsp70 is stimulated by the DnaJ homology domains (J domains) of Hsp40 and related co-chaperones. The ADP-bound state of Hsp70 binds substrates stably (Bukau and Horwich, 1998). Hsp90 also undergoes an ATP-regulated folding cycle, but unlike Hsp70, the substrate is held by the ATP-bound state of Hsp90 and released upon ATP hydrolysis. The co-chaperone p23 enhances the ATPase-dependent release of substrate (Young and Hartl, 2000). The nucleotide-free state of Hsp90 is transiently stabilized by Hop, permitting the loading of substrate onto Hsp90 from Hsp70 for another cycle (Prodromou et al., 1999).
The conserved C-terminal EEVD sequences of Hsp70 and Hsp90 mediate interactions with specialized tetratricopeptide repeat (TPR) domains in Hop and other related co-chaperones. In these TPR domains, a so-called ‘dicarboxylate clamp’ coordinates the terminal aspartate residue of the chaperones and specificity for Hsp70 or Hsp90 is determined by hydrophobic contacts with neighbouring residues. The N-terminal clamp domain of Hop is specific for Hsp70, whereas the central clamp domain is specific for Hsp90 (Scheufler et al., 2000; Brinker et al., 2002). Other TPR clamp co-chaperones recognize Hsp90 or Hsp70, or both chaperones (Young et al., 2001).
By yeast two-hybrid screening, we identified the cytosolic protein Tpr2 as an interacting partner of both Hsp90 and Hsp70. Tpr2 was originally found through interactions with the Ras-regulatory protein neurofibromin (Murthy et al., 1996), but the biological consequences of this interaction remain unresolved. Structure prediction revealed two putative TPR clamp domains and a J domain towards the C-terminus. In a Drosophila screen for the suppression of polyglutamine-induced eye degeneration, Tpr2 efficiently rescued the phenotype (Kazemi-Esfarjani and Benzer, 2000). However, the normal cellular function of Tpr2 has been unknown until now.
In this study, we demonstrate that Tpr2 functionally interacts with Hsp70 and Hsp90 in the folding of GR. Intracellular levels of Tpr2 are tuned for maximum efficiency of the chaperone system. The J domain of Tpr2 stimulates polypeptide binding by Hsp70, whereas the TPR domains cause nucleotide-independent dissociation of substrate polypeptide from Hsp90. Thus, Tpr2 may provide a mechanism for recycling substrates through the multichaperone machinery.
Results
Identification of human Tpr2
A yeast two-hybrid screen against a human brain cDNA library, using the 12 kDa C-terminal domain of human Hsp90 as bait, returned two positive isolates encoding Tpr2 (DDBJ/EMBL/GenBank accession No. U46571). Seven tetratricopeptide repeat motifs and a C-terminal J domain were predicted in the polypeptide sequence of Tpr2 (Figure 1A). A secondary two-hybrid screen using full-length Tpr2 as a bait yielded four positive isolates, three encoding segments of Hsp70 and the last encoding a segment of Hsp90. All of these isolates contained at least the complete C-termini of the chaperones (data not shown). This suggested that Tpr2 might bind Hsp70 and/or Hsp90 as a TPR domain co-chaperone, as well as interacting with Hsp70 through its J domain.

Fig. 1. Prediction of Tpr2 structural domains. (A) Domain prediction revealed seven tetratricopeptide motifs in human Tpr2 (motifs 1–7). Two sets of three motifs were predicted to form dicarboxylate clamp domains (T1 and T2). A DnaJ homology domain (J) near the C-terminus was also identified. The boundaries of the predicted domains are indicated as amino acid residue numbers. Introduced point mutations (dT1, dT2 and dJ) are listed with the respective amino acid exchange. (B) Sequences of the predicted T1 and T2 domains in Tpr2 were aligned against the Hsp70- and Hsp90-binding dicarboxylate clamp domains of Hop, TPR1 and TPR2A, respectively. Conserved residues that participate in the formation of the dicarboxylate clamp are underlined in boldface. Residues in boldface alone determine the specificity of chaperone binding. The arginine residues mutated to alanine in the dT1 and dT2 point mutants are marked with an asterisk. (C) The sequence of the predicted J domain in Tpr2 was aligned against the J domains of Hsp40 and Hdj-2. The functional HPD motif is underlined in boldface. Conserved residues are marked in boldface. The histidine residue mutated to alanine in the dJ point mutant is marked with an asterisk.
In the Hsp70- and Hsp90-binding TPR domains of Hop (TPR1 and TPR2A respectively), a cluster of three repeat motifs form a folded unit sufficient for binding the extended C-termini of the chaperones. The terminal aspartate residue of either chaperone is coordinated by five conserved ‘dicarboxylate clamp’ residues in the TPR domains (Scheufler et al., 2000). In Tpr2, the repeat motifs 1–3 and 5–7 could be aligned against the sequences of the Hop domains maintaining the correct position of the conserved clamp residues (Figure 1B, boldface and underlined) and we termed these predicted clamp domains T1 and T2 respectively (Figure 1A). Although the T1 domain contained an arginine instead of the usual lysine at the fourth position of the clamp, this conservative substitution may still permit binding to Hsp70 or Hsp90. However, residues of the Hop domains that determine the specificity of binding for Hsp70 or Hsp90 were divergent in the T1 and T2 domains of Tpr2, so it was unknown whether the chaperones would bind in a domain-specific manner. The predicted J domain of Tpr2 showed significant homology (∼45–48% identity) to the major human cytosolic J domain co-chaperones Hsp40 and Hdj2, and the functional HPD motif (Tsai and Douglas, 1996) was absolutely conserved (Figure 1C).
Modulation of Tpr2 expression reduces GR activation in vivo
The predicted structural features of Tpr2 and its interaction with Hsp70 and Hsp90 suggests that it functions in the multichaperone system involving both chaperones. This system has been extensively studied in the activation of GR. In live cells treated with dexamethasone, GR that is folded with the assistance of Hsp70 and Hsp90 can bind the hormone ligand and activate transcription from GR response elements (GRE) on the DNA. To measure this activity we transfected mouse N2A cells with a plasmid encoding a luciferase reporter gene downstream of a GRE, as well as a control plasmid encoding constitutively expressed β-galactosidase. Hormone treatment caused a strong activation of endogenous GR relative to untreated cells, as measured by luciferase expression normalized to β-galactosidase levels (Figure 2A, lanes 1 and 2). Co-transfection of myc-tagged Tpr2 strongly reduced hormone-dependent GR activation (Figure 2A, lane 4) to ∼40% of the control cells without exogenous Tpr2, at saturating levels of hormone. The myc-tagged Tpr2 did not affect basal transcription levels in the absence of hormone (Figure 2A, lane 3), nor did it change the abundance of GR protein (Figure 2C, lanes 1–2). Interestingly, even relatively low overexpression of Tpr2 over endogenous levels (Figure 2A, top, lane 4) significantly reduced GR activation. This suggested that the activity of the Hsp70/Hsp90 machinery in GR activation is highly sensitive to cellular levels of Tpr2. Overexpression of Hop also blocked GR activation (Figure 2A, lane 6), most probably through its known inhibition of Hsp90 ATPase activity (Prodromou et al., 1999). GR inhibition by overexpressed Hop was not additive with that of Tpr2 (Figure 2A, lane 8) and experiments described below suggest the two proteins have different mechanisms of action.

Fig. 2. Perturbation of Tpr2 expression levels reduce glucocorticoid receptor (GR) activation in vivo. Cells were transfected with a plasmid encoding a luciferase reporter gene downstream of a GR response element (GRE) and a control plasmid encoding β-galactosidase. Cells were treated for 24 h with 1 µM dexamethasone where indicated and harvested. Cell lysates were tested for luciferase activity and normalized against β-galactosidase activity. Samples were immunoblotted with antibodies against Tpr2, Hop or GR. In all figures, error bars show standard deviations from the mean of at least three independent experiments. (A) Empty vector (lanes 1–2) or vectors encoding myc-tagged Tpr2 (lanes 3–4), myc-tagged Hop (lanes 5–6) or both Tpr2 and Hop (lanes 7–8) were co-transfected into N2A cells together with the reporter and control plasmids. Top, immunoblot with antibodies against Tpr2 or Hop; transfected overexpressed myc-tagged proteins (o) are visible as bands above endogenous species (e). Bottom, GR-activated normalized luciferase expression in cells under conditions indicated. Columns correspond to the above immunoblot. (B) A double-stranded siRNA oligomer against the Tpr2 RNA was co-transfected into HeLa cells with the reporter and control plasmids (lanes 3–4). Control experiments included either an empty vector (lanes 1–2) or double-stranded RNA oligomers with a scrambled sequence (scRNA, lanes 5–6) or mutated sequence (mutRNA, lanes 7–8). Top, immunoblot against endogenous Tpr2 (e). Bottom, normalized GR-activated luciferase expression under indicated conditions. Columns correspond to the above immunoblot. (C) The indicated total cell lysates were resolved on SDS–PAGE and immunoblotted for GR, and for actin as a loading control. (D) Immunofluorescence of cells treated with siRNA against Tpr2 or control cells. Nuclei are stained with DAPI. Scale bar represents 100 µm. (E) Empty vector or vectors encoding Tpr2, GRΔLBD or both vectors were co-transfected with the reporter and control plasmids. Normalized GR-activated luciferase expression under indicated conditions are plotted.
Next we tested the effect of reducing Tpr2 expression on GR activity by using the small interfering RNA (siRNA) technique. Because siRNA knock-down of Tpr2 worked poorly in N2A cells, we used human HeLa cells in which siRNA had a clear and reproducible effect. GR activation in HeLa cells was also inhibited upon Tpr2 overexpression to ∼60% of the control (data not shown). The siRNA oligomer significantly diminished Tpr2 protein levels (Figure 2B, lanes 3–4) to <25% of control cells transfected with control oligomers having either a scrambled sequence or the same sequence except for three point mutations (Figure 2B, lanes 5–8). Remarkably, siRNA treatment also reduced the hormone-dependent activation of GR to ∼50% of the control (Figure 2B, lanes 3–4), similar to the effect caused by Tpr2 overexpression. The expression of GR itself was not affected by siRNA treatment or Tpr2 overexpression (Figure 2C, lanes 3–5). Immuno fluorescence microscopy confirmed that expression of Tpr2, which has a cytosolic distribution in control cells, was clearly reduced by siRNA in the majority of treated cells (Figure 2D).
These data suggest that Tpr2 may modulate the function of the Hsp70/Hsp90 machinery in the folding of GR. To confirm that Tpr2 interferes with this step and not downstream activation steps, the activation of a mutant GR, lacking the ligand binding domain (GRΔLBD) and thus independent of hormone and Hsp90 function, was tested (Hollenberg et al., 1987). In the absence of hormone, GRΔLBD transfection caused moderate expression from the GRE ∼5-fold over the basal level of the vector control. Overexpression of Tpr2 had no effect on the activity of GRΔLBD (Figure 2E), in agreement with an influence of the co-chaperone only on the hormone- and Hsp90-dependent activation steps.
Because both an increase and a decrease in cellular Tpr2 levels inhibited the chaperone-dependent activity of GR, the normal cellular levels of Tpr2 appear to be tuned for maximum efficiency of the chaperone machinery. Tpr2 has not been reported as a major component of chaperone–GR complexes and its abundance is ∼10-fold lower than that of Hop (data not shown). Thus, it is likely that Tpr2 acts as a substoichiometric regulator of the Hsp70/Hsp90 system.
Hsp90 and Hsp70 are the major interaction partners of Tpr2
Purified recombinant Tpr2 was used in vitro to investigate its mechanism of action. Point mutations introduced into conserved residues of purified Tpr2 (Figure 1A) were expected to disrupt the activity of the T1 (R91A, dT1), T2 (R323A, dT2) and J (H399A, dJ) domains, or different combinations thereof. All Tpr2 forms were soluble and monomeric (data not shown).
The specificity of chaperone binding to Tpr2 was tested in the absence of nucleotides to minimize binding between the J domain and Hsp70. The His-tagged Tpr2 proteins were incubated with rabbit reticulocyte lysate (RL), bound proteins were recovered with Ni-NTA agarose and chaperones were eluted with 500 mM NaCl, which is known to dissociate TPR clamp binding (Brinker et al., 2002). Hsp90 and Hsp70 were the major species bound to wild-type Tpr2 (Figure 3A, lane 1), as identified by immunoblotting (Figure 3B, lane 2). Mutation of both TPR clamp domains (dT12) strongly reduced the binding of Hsp90 and Hsp70 (Figure 3A, lane 2), indicating that the TPR domains contribute to chaperone binding. The dJ mutation alone had little influence on chaperone binding, but the triple mutant dT12J showed only background binding at the level of the control without His-tagged protein (Figure 3A, lanes 3–5). Hsp90 and Hsp70 therefore appear to be the major specifically-bound chaperone partners of Tpr2 in the cell lysate. When chaperone binding to Tpr2 was performed in RL containing an excess of the Hsp90 or Hsp70 C-terminal fragments, binding of both chaperones to Tpr2 was equally competed by either of the C-terminal fragments (Figure 3B, lanes 2–4). Thus, Hsp90 and Hsp70 compete for binding to the TPR domains of Tpr2.
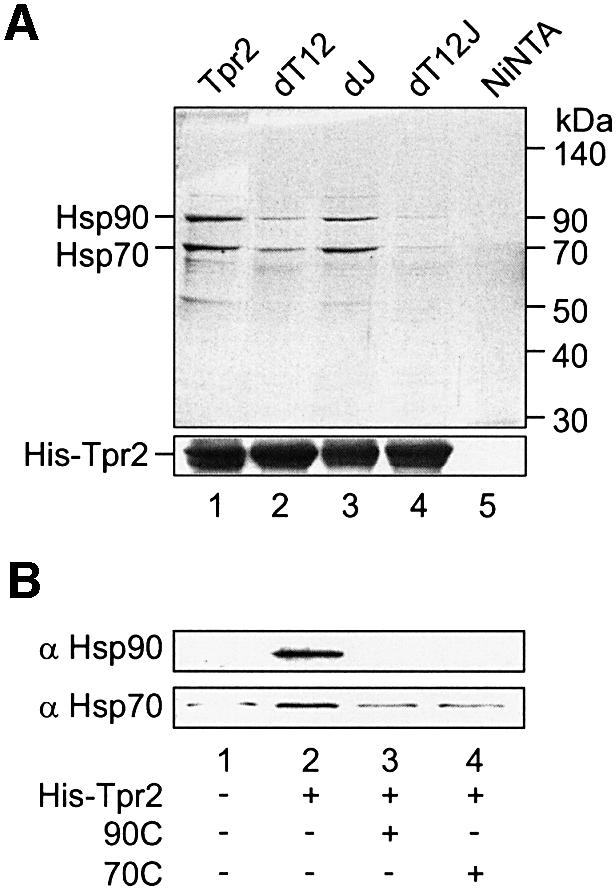
Fig. 3. Hsp90 and Hsp70 are the two major interaction partners of Tpr2. (A) Purified His-tagged wild-type (WT) Tpr2, Tpr2 point mutated in the TPR clamp domains (dT12) or J-domain (dJ), or with the combination of point mutations (dT12J) were tested for binding to reticulocyte lysate (RL) proteins. The indicated proteins at 10 µM concentration were incubated with RL, in parallel with a control reaction with no added protein (Ni-NTA). Complexes were recovered with Ni-NTA agarose and bound proteins eluted with 500 mM NaCl (top). Tpr2 proteins were re-eluted with SDS sample buffer containing 25 mM EDTA (bottom). Samples were resolved by SDS–PAGE and visualized by Coomassie blue staining. The two major bands eluting from WT Tpr2 were identified by immunoblotting as Hsp90 and Hsp70. The position of molecular weight standards is marked on the right. (B) 10 µM Tpr2 together with 50 µM of the C-terminal fragments of Hsp90 (90C, lane 3) or Hsp70 (70C, lane 4) were present during the binding reaction. After recovery with Ni-NTA agarose and elution with 500 mM NaCl, eluted proteins were resolved on SDS–PAGE and detected by immunoblotting against Hsp90 and Hsp70.
For a quantitative measure of the chaperone–Tpr2 interaction, we conducted surface plasmon resonance (SPR) experiments under nucleotide-free conditions. Because chip regeneration after each run required denaturing conditions, full-length Hsp90 and Hsp70, or domains thereof, could not be coupled to the sensor chip. Instead, 12mer peptides containing the C-terminal sequence of the respective chaperones (90C-12 and 70C-12), sufficient for domain-specific binding to the TPR domains of Hop (Scheufler et al., 2000; Brinker et al., 2002), were coupled to the chip. Binding affinities (KD) were calculated by assaying various concentrations of wild-type Tpr2 and the single dT1 (R91A) and dT2 (R323A) point mutants (Figure 4A; data not shown), and curve-fitting the plot of the relative equilibrium response units (Req) against the protein concentration. All proteins showed a fast on- and off-rate of binding to both the 90C-12 and 70C-12 peptides (Figure 4A, right panel; data not shown), similar to other chaperone–TPR domain interactions. Dissociation constants for wild-type Tpr2 were 2.7 µM for 90C-12 and 1.6 µM for 70C-12 (Table I) in a physiologically relevant range and slightly better than those measured with Hop (Brinker et al., 2002). The dT2 mutation affected binding more strongly than the dT1 mutation (Table I).

Fig. 4. Quantitative analysis of the Tpr2–chaperone interactions. 12mer peptides containing the C-terminal sequence of either Hsp70 or Hsp90 (70C-12 and 90C-12 respectively) were covalently coupled to a Biacore chip. Various concentrations of Tpr2 and its mutants were injected and the association and dissociation monitored by the surface plasmon resonance (SPR) signal. (A) Binding kinetics of Tpr2 in the concentration range of 0.1–30 µM were monitored (right panel) with immobilized 90C-12 or 70C-12. The relative response units during the equilibrium phase of binding to 90C-12 or 70C-12 were plotted against Tpr2 concentrations (left panel). (B) Binding efficiency to 70C-12 or 90C-12 of wild-type (WT) or indicated point mutants of Tpr2 at a constant protein concentration of 1 µM was tested. The relative response units during equilibrium binding were plotted. (C and D) Increasing concentrations (0.1–100 µM) of 70C-12 or 90C-12 in solution were used to compete for binding of Tpr2 to immobilized 70C-12 (C) or 90C-12 (D). A control peptide terminating in SKL, which is recognized by the TPR domain of Pex5p, but not by Hop, was also tested. Binding kinetics were monitored (right panels) and Tpr2 binding as a percentage of the control without soluble peptides was plotted against soluble peptide concentration (left panels).
Table I. Thermodynamic binding constants (KD) of wild-type and mutant Tpr2 to the Hsp70 and Hsp90 C-termini.
|
KD (µM) |
|||
|---|---|---|---|
| Tpr2 | dT1 | dT2 | |
| 70C-12 | 1.6 ± 0.2 | 9 ± 0.6 | 12.9 ± 0.3 |
| 90C-12 | 2.7 ± 0.4 | 9.3 ± 1.1 | 21.3 ± 3.5 |
The Tpr2 mutants were compared by measuring their equilibrium peptide binding at a constant protein concentration of 1 µM. Again, dT2 binding was slightly weaker than that of dT1, and mutations in both TPR domains (dT12) reduced binding further (Figure 4B). The dJ mutation had little effect on binding in either the wild-type or the dT12 context (Figure 4B). The binding of wild-type Tpr2 could also be competed by free peptides from either Hsp90 or Hsp70 (90C-12, 70C-12), independent of the immobilized binding partner (Figure 4C and D). A control peptide terminating in SKL could not compete Tpr2 binding (Figure 4C and D). This peptide is recognized by the tetratricopeptide repeat-containing protein Pex5p but not by dicarboxylate clamp TPR domains (Brinker et al., 2002). Thus, the TPR clamp domains in Tpr2 contribute independently to the binding of both Hsp90 and Hsp70.
The J domain of Tpr2 regulates Hsp70
J domain co-chaperones stimulate ATP hydrolysis by Hsp70 and thereby induce binding of Hsp70 to polypeptide substrates (Bukau and Horwich, 1998). The J domain of Tpr2 was tested for these characteristic functions. The steady-state ATPase rate of purified bovine Hsp70 was measured in the presence of wild-type and mutant Tpr2 and with the established J domain co-chaperone Hsp40. ATP hydrolysis by Hsp70 was similarly stimulated by both Hsp40 and Tpr2 (Figure 5A). As expected, the dJ mutation in Tpr2 largely abolished the ATPase stimulation of Hsp70, whereas the double clamp mutant (dT12) activated Hsp70 at a similar level to wild-type Tpr2 (Figure 5A). These data indicate that the J domain of Tpr2 is important for regulation of the Hsp70 ATPase cycle.

Fig. 5. The Tpr2 J domain regulates Hsp70. (A) 1 µM Hsc70/Hsp70, 2 µM Hsp40 and 2 µM wild-type or mutant Tpr2 in the indicated combinations were incubated at 30°C in the presence of [α-32P]ATP and 0.1 mM ATP. Aliquots of each reaction were stopped at different time points with 25 mM EDTA, resolved by thin layer chromatography and evaluated by PhosphorImager scanning. Steady-state ATPase rates were calculated from the linear range of the reactions. (B) Purified, partially-folded GR ligand binding domain (LBD) was bound to Ni-NTA agarose and beads were incubated for 10 min at room temperature with 5 µM Hsc70/Hsp70 and either no added protein (buffer), 5 µM wild-type (WT) or point mutated Tpr2, or Hsp40. Beads were recovered and bound proteins eluted with SDS sample buffer. Samples were resolved by SDS–PAGE and visualized by Coomassie blue staining. (C) Guanidine-denatured luciferase was diluted 100-fold into reactions containing 3% RL and ATP, supplemented with buffer or 1 µM Hsc70/Hsp70 with 2 µM Hsp40 or Tpr2 where indicated. Luciferase refolding at 30°C was monitored over time and plotted as a percentage of the activity of native luciferase.
The polypeptide binding activity of Hsp70 was next assayed using the purified ligand binding domain (LBD) of GR as a model substrate (Young and Hartl, 2000; Sondermann et al., 2001). The partially-folded myc-His-tagged LBD was incubated with Hsp70 in the presence of ATP, together with wild-type and mutant Tpr2, or Hsp40 as a control. Bound proteins were then recovered with Ni-NTA agarose. Hsp70 alone was unable to bind the polypeptide, but binding was clearly observed in the presence of Tpr2 (Figure 5B, lanes 1 and 2). As expected from the ATPase stimulation results, the dJ mutant of Tpr2 was defective in inducing Hsp70 binding (Figure 5B, lane 3). While mutations in the TPR clamp domains (dT12) appeared to have no effect, in combination with the dJ mutation, the induction of Hsp70 binding was completely abolished (Figure 5B, lanes 4 and 5). The level of Hsp70 binding induced by Tpr2 was the same as that caused by Hsp40 (Figure 5B, lane 6). Thus, the J domain of Tpr2 acts as predicted to trigger ATP hydrolysis and substrate binding by Hsp70. The TPR domains of Tpr2 may slightly stabilize Hsp70 binding to substrate in the absence of a functional J domain, but this stabilization is not required for substrate binding by Hsp70 (Figure 5B, lanes 3 and 4). Importantly, the association of Tpr2 itself with LBD was much weaker than that of Hsp70 and was abolished by mutation of the Hsp70-interacting TPR and J domains (Figure 5B, lanes 2 and 5). This suggests that Tpr2 does not bind to unfolded polypeptides directly, but only through Hsp70 or Hsp90, and that any effects of Tpr2 on polypeptide folding must be mediated by its interactions with the chaperones. In contrast, significant amounts of Hsp40 appeared in the recovered fractions (Figure 5B, lane 6), consistent with direct binding to the substrate (Minami et al., 1996).
J domain stimulation of Hsp70 ATPase is required for the refolding activity of the chaperone and we therefore asked whether Tpr2 could support the refolding of guanidine-denatured firefly luciferase by purified Hsp70. In the presence of ATP and 3% RL (Minami et al., 1996), refolding by Hsp70 and Hsp40 is enhanced by substoichiometric factors in the added lysate, but Hsp70 without Hsp40 refolds luciferase poorly (Figure 5C). Tpr2 was as effective as Hsp40 in the Hsp70-mediated refolding of luciferase, while Tpr2 alone had no effect (Figure 5C). Thus, activation of the Hsp70 ATPase by the Tpr2 J domain leads to the expected stable binding of polypeptides and Hsp70-dependent protein refolding.
Tpr2 dissociates Hsp90, but not Hsp70, from substrate polypeptide
To analyse the effects of Tpr2 on Hsp90 and Hsp70 simultaneously, we isolated complexes of the chaperones bound to the LBD of GR. In this previously established system (Young and Hartl, 2000; Sondermann et al., 2001), partially-folded myc-His-tagged LBD is incubated in RL to form chaperone–substrate complexes and the complexes are immune-isolated with anti-myc antibodies coupled to protein G–Sepharose. By radiolabelling either Hsp90 or Hsp70 in the RL, the dissociation of these chaperones from the isolated complexes in the presence or absence of Tpr2 can be quantitatively monitored.
As previously observed (Young and Hartl, 2000), incubation of the chaperone–LBD complexes with ATP induced some dissociation of radiolabelled Hsp90 (∼26%) compared with the control without ATP (12%) and the regulatory co-chaperone p23 enhanced Hsp90 release only in the presence of ATP (50% compared with 12% without ATP; Figure 6A). Intriguingly, Tpr2 in the absence of ATP caused significant dissociation of Hsp90 from complexes (36%; Figure 6A). Full-length Hop or its Hsp90-binding domain caused only a low level of Hsp90 release (15%) in the absence of ATP (Young and Hartl, 2000; data not shown), although their affinities for the Hsp90 C-terminus (KD, between 2 and 7 µM; Brinker et al., 2002) were similar to that of Tpr2 (KD, 2.7 µM; Table I). Thus, Tpr2 has an effect on Hsp90–substrate complexes distinct from that of p23 or Hop. The Hsp90 dissociation observed with Tpr2 and ATP together (53%; Figure 6A) is consistent with an additive effect of the apparently nucleotide-independent Tpr2-mediated release and of the release caused by ATPase cycling of Hsp90.

Fig. 6. Tpr2 dissociates Hsp90 but not Hsp70 from substrate polypeptide complexes. Partially-folded myc-His-tagged ligand binding domain (LBD) or guanidine-denatured myc-tagged luciferase were pre-bound to anti-myc antibodies covalently coupled to protein G–Sepharose. Radiolabelled wild-type Hsp90, or the Hsp90 D93N point mutant unable to bind ATP, or Hsp70 was generated by in vitro translation. The translation reactions were added to reticulocyte lysate (RL) containing ATP and chaperone–substrate complexes were formed for 10 min at 25°C and then immune-isolated. The dissociation of radiolabelled chaperones was assayed under different conditions. After 10 min of dissociation at 25°C, beads and supernatants were separated and proteins in the supernatants precipitated. Released and bound proteins were resolved by SDS–PAGE and quantified by PhosphorImager analysis. The fraction of radiolabelled chaperones released from the complexes was plotted as a percentage of the total. (A) Dissociation of wild-type Hsp90 from LBD was measured in the presence of buffer alone, or 5 µM p23 or Tpr2, without or with 2 mM ATP. (B) Dissociation of Hsp90 D93N from LBD was measured in the presence of buffer alone, or 5 µM p23 or Tpr2, without or with 2 mM ATP. (C) Dissociation of Hsp90 D93N from luciferase was measured in the presence of buffer alone, or 5 µM p23 or Tpr2, without or with 2 mM ATP. (D) Dissociation of Hsp90 D93N from LBD was measured in the presence of buffer alone, or 5 µM wild-type or point mutated Tpr2. (E) Dissociation of Hsp70 was measured in the presence of buffer alone, or 5 µM Bag-1, or 5 µM wild-type or point mutated Tpr2.
To ensure that the Hsp90–substrate dissociation caused by Tpr2 was not due to changes in Hsp90 ATPase activity, the steady-state ATP hydrolysis rate of yeast Hsp90 was measured and found to be identical in the presence and absence of Tpr2 (data not shown). We also tested a point mutant of Hsp90, D93N, which is unable to bind or hydrolyse ATP (Obermann et al., 1998). Radiolabelled Hsp90 D93N added to RL formed complexes with the LBD at a similar level to wild-type Hsp90, but as previously observed (Young and Hartl, 2000), could not be released from the complexes by the ATP-dependent activity of p23 (Figure 6B). In contrast, Tpr2 induced significant dissociation of Hsp90 D93N, at the same level either with or without ATP (30–33%; Figure 6B), similar to that observed with wild-type Hsp90 and Tpr2 in the absence of nucleotide (Figure 6A). Tpr2 therefore appears to dissociate Hsp90 from substrate polypeptide by a novel nucleotide-independent mechanism, unlike the ATP-dependent mechanism of p23.
Under conditions of stress, Hsp90 is thought to bind denatured proteins to maintain them in a soluble state competent for refolding by other chaperones, including Hsp70 (Buchner, 1999). This ‘holding’ function has been demonstrated in vitro for firefly luciferase, for which complexes have been isolated with Hsp90 and Hsp70 after dilution from guanidine into RL (Schneider et al., 1996). To examine the effect of Tpr2 on Hsp90 in this context, chaperone complexes with denatured myc-tagged luciferase were immune-isolated from RL and the dissociation of Hsp90 was tested. Radiolabelled Hsp90 D93N was included during complex formation to specifically examine ATP-independent dissociation. As observed for the Hsp90–LBD interaction, Tpr2 caused significant release of Hsp90 D93N from the complexes with luciferase, whereas p23 had no effect (Figure 6C). Thus, the substrate release activity of Tpr2 on Hsp90 appears to be a general mechanism independent of the bound substrate.
The contribution of the co-chaperone domains in Tpr2 to the ATP-independent dissociation of Hsp90 D93N from complexes with LBD was tested. Disruption of the TPR clamp domains (dT12) significantly reduced the complex-dissociation activity of Tpr2 compared with wild-type protein (Figure 6D). Mutation of the J domain (dJ) had no effect, whereas combined mutations of both TPR clamps and the J domain (dT12J) reduced complex dissociation to the level of the TPR clamp mutant (Figure 6D). These results suggest that the TPR clamp domains in Tpr2 act to induce the ATP-independent dissociation of Hsp90 from substrate.
The effect of Tpr2 on Hsp70–substrate interactions was similarly tested using radiolabelled Hsp70. ATP alone produced a low level of Hsp70 dissociation from the isolated LBD complexes, and as a positive control, Bag-1 in the presence of ATP induced significant release of Hsp70 (Figure 6E) by stimulating nucleotide exchange (Höhfeld and Jentsch, 1997; Sondermann et al., 2001). Tpr2 had no effect on Hsp70 dissociation compared with the buffer control, independent of ATP, and the various point mutants of Tpr2 behaved identically (Figure 6E). The complex-dissociation activity of Tpr2 is therefore specific for Hsp90 and is most likely mediated by binding of the TPR domains to that chaperone.
Different domains of Tpr2 cooperate to regulate Hsp90/Hsp70 machinery
To assess how the individual functions of the Tpr2 co-chaperone domains contribute to the activity of the whole protein in the multichaperone system, we tested mutants of Tpr2 for inhibition of GR activation in vivo. The dT12 double clamp mutant, which is defective in dissociating Hsp90 from substrate, was only partially active in inhibiting GR activation (Figure 7A). The dJ mutant, defective in Hsp70 activation, caused a similar reduced level of GR inhibition (Figure 7A) suggesting that the J and TPR domains are required for full activity of Tpr2. The dT12J mutant, disrupted in the three co-chaperone domains, was also partially inhibitory (Figure 7A). The partial function of this mutant could be due either to a residual affinity of the mutant for the chaperones in the highly crowded cellular environment or an additional activity of Tpr2. To clarify this question, we reconstituted the chaperone-dependent activation of GR in vitro using full-length recombinant GR added to RL and tested the Tpr2 mutants in this system.
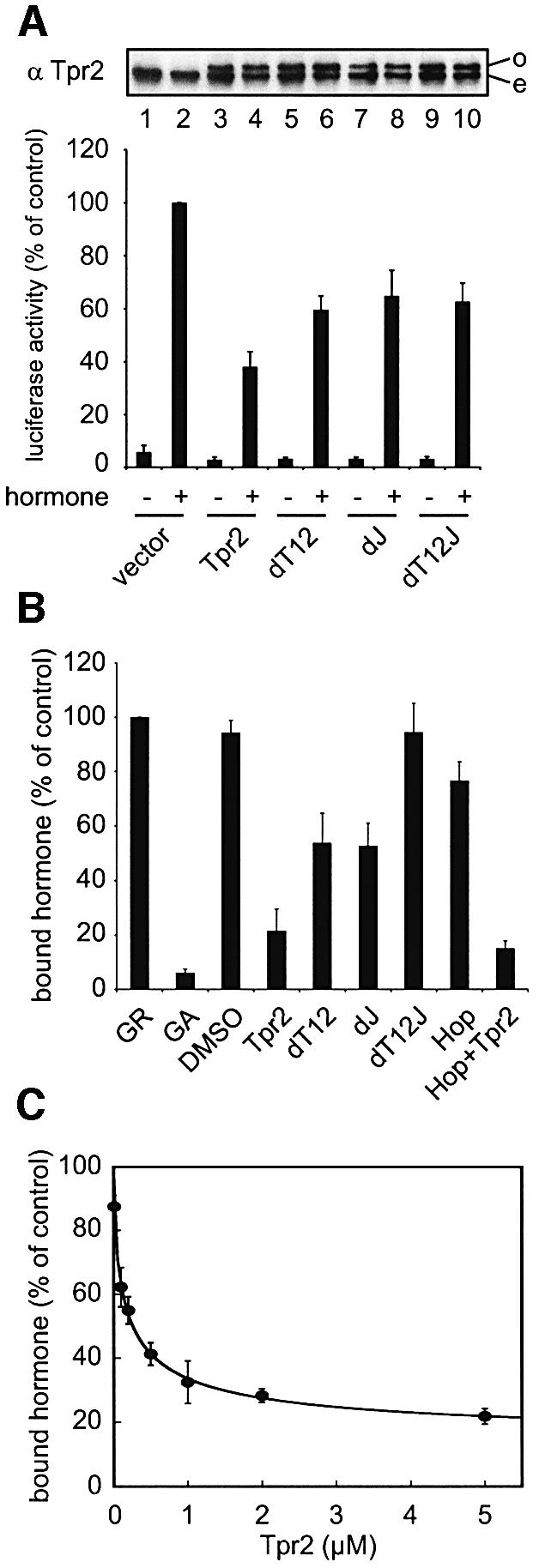
Fig. 7. The different domains of Tpr2 cooperate to regulate the Hsp70/Hsp90 machinery. (A) Glucocorticoid receptor (GR) activation in N2A cells with or without dexamethasone was determined as in Figure 2A. Empty vector, vectors encoding wild-type or mutant Tpr2 were co-transfected with the reporter and control plasmids. Top, immunoblot against overexpressed (o) and endogenous (e) Tpr2 forms. Bottom, normalized GR-activated luciferase expression under indicated conditions. (B) Full-length GR was added to reticulocyte lysate (RL) supplemented with various purified proteins and 2 mM ATP. Reactions were incubated at 42°C for 5 min to unfold the GR, then at 30°C for 15 min to allow chaperone-mediated refolding of the GR. [3H]Dexamethasone was allowed to bind the GR, unbound hormone was removed by fast gel filtration and bound hormone quantified by scintillation counting. Hormone binding by GR refolded in RL with no additions was set to 100%. As a negative control, RL was treated with 40 µM GA to inhibit Hsp90, or with an equivalent volume of DMSO as a solvent control. Other reactions were supplemented with 2 µM wild-type, mutant Tpr2 or Hop. (C) Hormone binding by GR after refolding in RL supplemented with increasing concentrations of wild-type Tpr2 was measured and plotted against the concentrations of Tpr2 added.
GR was partially unfolded in the lysate with a 42°C heat shock for 5 min and the reactions were returned to 30°C to allow refolding of the receptor by the RL chaperones in the presence of ATP. The amount of refolded GR was assayed by the binding of radiolabelled hormone. As a negative control, the RL was treated with geldanamycin (GA), a high-specificity and high-affinity inhibitor of Hsp90 which blocks GR folding in vivo (Whitesell and Cook, 1996). As expected, GA almost completely abolished the folding of GR, whereas the DMSO solvent control had no effect (Figure 7B). Purified Tpr2 added to the lysate strongly inhibited GR folding to ∼20% of the untreated control (Figure 7B). The in vitro experiments thus reproduce faithfully the inhibition of GR activation observed upon Tpr2 overexpression in vivo (Figure 2A). Excess Hop also moderately inhibited GR activation but this effect was not additive with the effect of Tpr2 (Figure 2A and 7B).
The mutants of Tpr2 were then used to analyse the functional contribution of its co-chaperone domains. Mutations in the TPR domains (dT12) or the J domain (dJ) reduced the ability of Tpr2 to inhibit GR activation, and combined mutation of the TPR clamps and the J domain (dT12J) practically abolished Tpr2 function in this assay (Figure 7B). Thus, in agreement with the in vivo results, both the Hsp90 dissociation and Hsp70 ATPase stimulation activities of Tpr2 appear to be necessary for full function of the co-chaperone. Moreover, there is no evidence for an additional activity of Tpr2. Because the effect of Tpr2 in vivo is apparently only on the chaperone-dependent folding of the GR and not directly on its transcriptional activation (Figure 2E), the partial effect of the dT12J mutant in vivo (Figure 7A) should also be on GR folding. In vitro, a low level of equilibrium binding between dT12J and the Hsp70 or Hsp90 C-termini is still observed (Figure 4B), with an estimated KD in the range of 50 µM. The binding of the mutant to chaperones will be increased in cells where the chaperones are in the 50 µM concentration range (Buchner, 1999), ∼20-fold higher than in the in vitro assays. Thus, the weak affinity of the dT12J mutant for the chaperones is most likely sufficient for its partial inhibition of GR activation in vivo.
Changes in Tpr2 expression in vivo strongly affected the activation of GR (Figure 2A) and this was confirmed in vitro by titrating the amount of exogenous Tpr2 added to the RL. As noted above, very little endogenous Tpr2 is present in cell lysates relative to Hop, which would mean less than micromolar concentrations in RL. Significant inhibition of GR folding was observed with very low levels of Tpr2 addition, with 50% inhibition achieved at 0.2 µM added Tpr2 (Figure 7C). At this concentration, total Tpr2 is still substoichiometric to Hsp90 and Hsp70 in RL, estimated at 2–5 µM each. These results support a function of Tpr2 as a regulatory factor of the Hsp70/Hsp90 chaperone system, rather than as a central component.
Discussion
The results presented here outline a novel role of the Tpr2 co-chaperone protein as a regulator of Hsp70/Hsp90 function. In the folding of steroid hormone receptors, the polypeptide is normally passed from Hsp70 to Hsp90 (Pratt and Toft, 1997). We have now shown that the TPR domains of Tpr2 work to disrupt Hsp90–substrate interactions, while the J domain induces ATP hydrolysis and substrate binding by Hsp70. In this manner, Tpr2 would transfer the substrate polypeptide from Hsp90 to Hsp70, i.e. ‘backwards’ on its usual folding pathway through the multichaperone machinery.
We propose that this function of Tpr2 helps the folding of a fraction of substrate polypeptides which are not yet properly folded after a single cycle through the Hsp70/Hsp90 system. Tpr2 would allow these polypeptides to re-enter the cycle immediately at the Hsp70 step. Importantly, release of the polypeptides into the bulk cytosol prior to re-entering the chaperone cycle would be prevented by direct substrate transfer from Hsp90 to Hsp70. Prematurely released polypeptides would run an increased risk of aggregation, which is non-productive or even harmful to the cell (Hartl and Hayer-Hartl, 2002). Thus, an optimal low level of Tpr2 would increase the efficiency of the Hsp70/Hsp90 system compared with its absence. Excess Tpr2, on the other hand, would inhibit the folding machinery by preventing Hsp90 from completing the folding of the substrates.
Imbalances in the expression of other co-chaperones of Hsp70 or Hsp90 are known to affect the GR activation pathway, in most cases negatively. The Hsp70 co-chaperone Bag-1 inhibits GR activation when overexpressed in cultured cells, presumably by interfering with Hsp70 during the folding of the receptor (Höhfeld and Jentsch, 1997; Takayama et al., 1997; Zeiner et al., 1997), but also by acting in the nucleus to specifically block GR-dependent transcription (Schneikert et al., 2000; Schmidt et al., 2002). The co-chaperone p23 regulates the Hsp90 ATPase cycle for GR folding (Dittmar et al., 1997; Young and Hartl, 2000) while mediating the disassembly of transcriptionally active receptor complexes, resulting in a modulation of receptor activity when overexpressed (Freeman and Yamamoto, 2002). Overexpressed Hop reduces GR activation (Figure 2A) probably by inhibiting the Hsp90 ATPase activity. By stabilizing the nucleotide-free state of Hsp90 (Prodromou et al., 1999), excess Hop would allow substrate binding by Hsp90, but would interfere with substrate folding which requires ATP binding by Hsp90 (Obermann et al., 1998). Tpr2, by uniquely moderating the overall flow of substrates from Hsp70 to Hsp90, would provide an additional important control point in this finely balanced system of regulatory co-chaperones. The ability of overexpressed Tpr2 to suppress polyglutamine-induced neurodegeneration in Drosophila eyes (Kazemi-Esfarjani and Benzer, 2000) may be caused in part by alterations in Hsp90-dependent signalling pathways.
The growing family of co-chaperones containing modular TPR clamp domains bind interchangeably to Hsp90 and/or Hsp70 to perform different cellular functions. For example, FKBP52 brings a peptidyl-prolyl isomerase activity to Hsp90–substrate complexes (Pratt and Toft, 1997), whereas Tom70 recognizes Hsp90 and Hsp70 for the purpose of mitochondrial targeting (Young et al., 2003). Unlike these, Hop (Prodromou et al., 1999) and Tpr2 appear to regulate the chaperones themselves. We expect that further analysis of these co-chaperones will continue to provide insight into the mechanisms of Hsp70 and Hsp90 function.
Materials and methods
Yeast two-hybrid screening
Yeast two hybrid experiments were performed as described (Young et al., 1998). Briefly, the sequence encoding residues 629–731 of human Hsp90α was inserted into the pAS2-1 vector and used as bait in a two-hybrid screen with a human brain cDNA library based on the pACT2 vector in Saccharomyces cerevisiae strains CG1945 or Y190 (Clontech, Palo Alto, CA). HIS+, β-galactosidase-expressing colonies were selected on synthetic media supplemented with 25 mM 3-amino-1,2,4-triazole. Isolates were re-tested in S.cerevisiae strain Y187 (Clontech). Rescreening using the Tpr2 coding sequence as bait was performed similarly. Predicted structural domains were analysed with the SMART algorithm at http://smart.embl-heidelberg.de
Protein preparation
DNA sequence encoding Tpr2 was inserted into pPROEXHTa (Invitrogen, Carlsbad, CA) in-frame with the N-terminal His-tag and point mutations were introduced stepwise by PCR. Proteins were expressed in E.coli BL21(DE3)pLysS cells grown in TB. Expression was induced by the addition of 1 mM IPTG for 5 h at 21°C. Soluble proteins were purified by Ni-NTA chromatography and, where needed, the His-tag was removed with TEV protease at 4°C (Scheufler et al., 2000). Bovine brain Hsp70 and Hsp90 and bacterially expressed C-terminal domains of human Hsp70 (C70, residues 382–641) and human Hsp90α (C90, residues 625–732), yeast Hsp90 (Hsp82), human p23, Hop and Bag-1, and rat GR LBD were purified as described previously (Young et al., 1998; Young and Hartl, 2000; Sondermann et al., 2001).
Cell culture
DNA sequences encoding Tpr2 or human Hop were inserted into pcDNA3.1 (Invitrogen) in-frame with the C-terminal myc-tag. The reporter vector provided GR-dependent luciferase expression (pGRE-luc, Clontech) and a vector expressing β-galactosidase (pSV-β-Gal; Promega, Madison, WI) was used as a control to normalize for transfection efficiency. Cells in 3 cm dishes were transfected with 0.5 µg pGRE-luc, 0.5 µg pSV-β-Gal and 1 µg of either pcDNA-Tpr2 (wild type or mutant), pcDNA-Hop or empty vector, using Lipofectamine PLUS (Invitrogen). After 24 h, cells were treated with 1 µM dexamethasone or an equal volume of EtOH solvent control for another 24 h and then harvested and lysed. The supernatants were tested with the β-galactosidase and luciferase enzyme assay systems (Promega). Where indicated, cells were co-transfected with 1 µg plasmid pGRΔLBD.
For siRNA experiments, a double-stranded synthetic RNA oligomer against the sequence 5′-TGCTCAGGCACAACAAGAGTT was used (Dharmacon). As a control, empty vector or RNA oligomers with the scrambled sequence 5′-ACTCTATCGCCAGCGTGACTT (Non-specific Control VII, Dharmacon) or the mutated sequence 5′-TGCCCAG GCACAGCAGGAGTT (mutations underlined) were used. A 7.5 µl aliquot of 20 µM siRNA oligomers per 3 cm dish were transfected with Lipofectamine PLUS (Invitrogen) in reactions separate from the plasmid transfections. Hormone stimulation and enzyme activity assays were carried out as above.
To monitor levels of individual proteins, equal amounts of cell lysate proteins were analysed by immunoblotting with an affinity-purified rabbit polyclonal antibody specific for Tpr2, a monoclonal antibody against Hop (SRA1500, Stressgen) and a polyclonal antibody against GR (H-300; Santa Cruz Biotechnology, Santa Cruz, CA). Cells were treated for immunofluorescence as described (Hille et al., 1992).
Chaperone binding
Binding reactions contained 50% desalted rabbit RL and 10 µM His-tagged Tpr2 or point mutants, in buffer B (25 mM HEPES–KOH pH 7.5, 100 mM KOAc, 5% glycerol) containing 30 mM imidazole and Ni-NTA agarose as described (Young et al., 2003). Bound proteins were eluted with 20 mM Tris–HCl pH 7.5, 500 mM NaCl and remaining proteins were eluted with SDS sample buffer containing 25 mM EDTA. All fractions were analysed by SDS–PAGE and Coomassie blue staining. For competition experiments, 50 µM C70 or C90 was present during the binding reaction. Complexes were treated as above and proteins were identified by immunoblotting with antibodies against Hsp70 (SPA820, Stressgen) and Hsp90 (rabbit polyclonal antiserum specific for the peptide NKTKPIWTRNPDDI sequence from human Hsp90α).
Surface plasmon resonance (SPR) analysis
SPR experiments were performed as described (Brinker et al., 2002) with some modifications. To introduce maleimido groups onto a B1 chip, 25 mM Sulfo-GMBS (Pierce, Rockford, IL) was injected for 5 min. Cysteine derivatized 12mer peptides of Hsp70 (C70-12, Ac-C-GSGSGPTIEEVD-OH) or Hsp90 (C90-12, Ac-C-GDDDTSRMEEVD-OH) were then injected for 5 min at 1 and 5 µM, respectively. The chip was regenerated by three 30 s washes with 6 M guanidinium hydrochloride. To assess the thermodynamic dissociation constant (KD), a concentration series (0.1–30 µM) of wild-type or mutant Tpr2 was injected. The KD was calculated with the BIAevaluation software (BIACORE), using a simple steady-state model for binding. To compare steady-state binding levels of Tpr2 mutants, proteins were injected at 1 µM concentration and the equilibrium response units of three independent experiments were averaged. For competition analysis, increasing amounts (0.1–100 µM) of 70C-12, 90C-12 or SKL (Ac-TKRRESKL-OH) peptides were pre-incubated for 5 min with 1 µM Tpr2 on ice before injection.
Activity assays and polypeptide binding of chaperones
The ATPase activities of Hsp70 and yeast Hsp90 (Hsp82) were determined as described (Young and Hartl, 2000; Sondermann et al., 2001). To measure luciferase refolding, firefly luciferase (Schneider et al., 1996) was denatured in 6 M guanidine hydrochloride and diluted 100-fold to a final concentration of 10 nM into buffer containing 50 mM KOAc, 20 mM HEPES–KOH pH 7.5, 5 mM MgOAc2, 2 mM ATP and 3% desalted RL (Minami et al., 1996). Reactions were supplemented with either buffer alone or purified Hsp40, Hsc70 and Tpr2 and incubated at 30°C. Luciferase activities at different time points were measured and compared with a native luciferase control.
For the stimulation of Hsp70 binding to substrate polypeptide, the myc-His-tagged LBD of rat GR was partially refolded in detergent (Young and Hartl, 2000) and bound to Ni-NTA agarose beads, which were incubated in buffer B containing 2 mM ATP, 5 mM MgOAc2 and the indicated amounts of proteins. After a 10 min incubation at room temperature, beads were recovered, washed with buffer B at 4°C and eluted with SDS sample buffer. Samples were analysed by SDS–PAGE and Coomassie blue staining. Binding of RL chaperones to detergent-refolded LBD or guanidine-denatured luciferase and analysis of the dissociation of chaperone–substrate complexes was performed as described (Schneider et al., 1996; Young and Hartl, 2000; Sondermann et al., 2001).
In vitro refolding of GR
The refolding of purified human recombinant GR (Panvera) was monitored by its binding to [1,2,4,5,7-3H]dexamethasone (Amersham, Arlington Heights, IL). Fifty per cent desalted RL in buffer B was supplemented with 2 mM ATP, 5 mM MgOAc2, 4.7 nM GR, 2 µM of the indicated proteins, 40 µM GA or an equal volume of the DMSO solvent control. Samples were heated to 42°C for 5 min to denature the GR. Reactions were transferred to 30°C for 5 min before adding 10 µM radiolabelled hormone, followed by a further incubation at 30°C for 10 min to allow refolding and hormone binding. Samples were then incubated at 4°C for 1 h to stabilize bound hormone. Unbound hormone was removed at 4°C using BioSpin 30 columns (BioRad) equilibrated in buffer B. GR-bound hormone in the eluate was detected by scintillation counting in a Packard Tri-Carb 1500 instrument.
Acknowledgments
Acknowledgements
This work was supported in part by the European Commission (QLK3-CT2000-00720) and by the Deutsche Forschungsgemeinschaft (SFB 284/Project Z3).
References
- Brinker A., Scheufler,C., Von Der Mulbe,F., Fleckenstein,B., Herrmann,C., Jung,G., Moarefi,I. and Hartl,F.U. (2002) Ligand discrimination by TPR domain. Relevance and selectivity of EEVD-recognition in Hsp70–Hop–Hsp90 complexes. J. Biol. Chem., 277, 19265–19275. [DOI] [PubMed] [Google Scholar]
- Buchner J. (1999) Hsp90 & Co.—a holding for folding. Trends Biochem. Sci., 24, 136–141. [DOI] [PubMed] [Google Scholar]
- Bukau B. and Horwich,A. (1998) The Hsp70 and Hsp60 chaperone machines. Cell, 92, 351–366. [DOI] [PubMed] [Google Scholar]
- Chen S. and Smith,D.F. (1998) Hop as an adaptor in the heat shock protein 70 (Hsp70) and Hsp90 chaperone machinery. J. Biol. Chem., 273, 35194–35200. [DOI] [PubMed] [Google Scholar]
- Dittmar K.D., Demady,D.R., Stancato,L.F., Krishna,P. and Pratt,W.B. (1997) Folding of the glucocorticoid receptor by the heat shock protein (hsp) 90-based chaperone machinery. The role of p23 is to stabilize receptor–hsp90 heterocomplexes formed by hsp90–p60–hsp70. J. Biol. Chem., 272, 21213–21220. [DOI] [PubMed] [Google Scholar]
- Freeman B.C. and Yamamoto,K.R. (2002) Disassembly of transcriptional regulatory complexes by molecular chaperones. Science, 296, 2232–2235. [DOI] [PubMed] [Google Scholar]
- Hartl F.U. and Hayer-Hartl,M. (2002) Molecular chaperones in the cytosol: from nascent chain to folded protein. Science, 295, 1852–1858. [DOI] [PubMed] [Google Scholar]
- Hille A., Klumperman,J., Geuze,H.J., Peters,C., Brodsky,F.M. and von Figura,K. (1992) Lysosomal acid phosphatase is internalized via clathrin-coated pits. Eur. J. Cell Biol., 59, 106–115. [PubMed] [Google Scholar]
- Höhfeld J. and Jentsch,S. (1997) GrpE-like regulation of the hsc70 chaperone by the anti-apoptotic protein BAG-1. EMBO J., 16, 6209–6216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollenberg S.M., Giguere,V., Segui,P. and Evans,R.M. (1987) Colocalization of DNA-binding and transcriptional activation functions in the human glucocorticoid receptor. Cell, 49, 39–46. [DOI] [PubMed] [Google Scholar]
- Johnson B.D., Schumacher,R.J., Ross,E.D. and Toft,D.O. (1998) Hop modulates Hsp70/Hsp90 interactions in protein folding. J. Biol. Chem., 273, 3679–3686. [DOI] [PubMed] [Google Scholar]
- Kazemi-Esfarjani P. and Benzer,S. (2000) Genetic suppression of polyglutamine toxicity in Drosophila. Science, 287, 1837–1840. [DOI] [PubMed] [Google Scholar]
- Minami Y., Höhfeld,J., Ohtsuka,K. and Hartl,F.U. (1996) Regulation of the heat-shock protein 70 reaction cycle by the mammalian DnaJ homolog, Hsp40. J. Biol. Chem., 271, 19617–19624. [DOI] [PubMed] [Google Scholar]
- Murthy A.E., Sohal,S.K., Carrington,M., Bishop,R.P. and Allsopp,B.A. (1996) Identification and characterization of two novel tetratricopeptide repeat-containing genes. DNA Cell Biol., 15, 727–735. [DOI] [PubMed] [Google Scholar]
- Obermann W.M., Sondermann,H., Russo,A.A., Pavletich,N.P. and Hartl,F.U. (1998) In vivo function of Hsp90 is dependent on ATP binding and ATP hydrolysis. J. Cell Biol., 143, 901–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratt W.B. and Toft,D.O. (1997) Steroid receptor interactions with heat shock protein and immunophilin chaperones. Endocr. Rev., 18, 306–360. [DOI] [PubMed] [Google Scholar]
- Prodromou C., Siligardi,G., O’Brien,R., Woolfson,D.N., Regan,L., Panaretou,B., Ladbury,J.E., Piper,P.W. and Pearl,L.H. (1999) Regulation of Hsp90 ATPase activity by tetratricopeptide repeat (TPR)-domain co-chaperones. EMBO J., 18, 754–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheufler C., Brinker,A., Bourenkov,G., Pegoraro,S., Moroder,L., Bartunik,H., Hartl,F.U. and Moarefi,I. (2000) Structure of TPR domain-peptide complexes: critical elements in the assembly of the Hsp70–Hsp90 multichaperone machine. Cell, 101, 621–632. [DOI] [PubMed] [Google Scholar]
- Schmidt U., Wochnik,G.M., Rosenhagen,M.C., Young,J.C., Hartl,F.U., Holsboer,F. and Rein,T. (2002) Essential role of the unusual DNA binding motif of BAG-1 for inhibition of the glucocorticoid receptor. J. Biol. Chem., 278, 4926–4931. [DOI] [PubMed] [Google Scholar]
- Schneider C., Sepp-Lorenzino,L., Nimmesgern,E., Ouerfelli,O., Danishefsky,S., Rosen,N. and Hartl,F.U. (1996) Pharmacologic shifting of a balance between protein refolding and degradation mediated by Hsp90. Proc. Natl Acad. Sci. USA, 93, 14536–14541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneikert J., Hübner,S., Langer,G., Petri,T., Jäättelä,M., Reed,J. and Cato,A.C.B. (2000) Hsp70–RAP46 interaction in downregulation of DNA binding by glucocorticoid receptor. EMBO J., 19, 6508–6516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sondermann H., Scheufler,C., Schneider,C., Hohfeld,J., Hartl,F.U. and Moarefi,I. (2001) Structure of a Bag/Hsc70 complex: convergent functional evolution of Hsp70 nucleotide exchange factors. Science, 291, 1553–1557. [DOI] [PubMed] [Google Scholar]
- Takayama S., Bimston,D.N., Matsuzawa,S., Freeman,B.C., Aime-Sempe,C., Xie,Z., Morimoto,R.I. and Reed,J.C. (1997) BAG-1 modulates the chaperone activity of Hsp70/Hsc70. EMBO J., 16, 4887–4896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai J. and Douglas,M.G. (1996) A conserved HPD sequence of the J-domain is necessary for YDJ1 stimulation of Hsp70 ATPase activity at a site distinct from substrate binding. J. Biol. Chem., 271, 9347–9354. [DOI] [PubMed] [Google Scholar]
- Whitesell L. and Cook,P. (1996) Stable and specific binding of heat shock protein 90 by geldanamycin disrupts glucocorticoid receptor function in intact cells. Mol. Endocrinol., 10, 705–712. [DOI] [PubMed] [Google Scholar]
- Young J.C. and Hartl,F.U. (2000) Polypeptide release by Hsp90 involves ATP hydrolysis and is enhanced by the co-chaperone p23. EMBO J., 19, 5930–5940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young J.C., Obermann,W.M. and Hartl,F.U. (1998) Specific binding of tetratricopeptide repeat proteins to the C-terminal 12-kDa domain of Hsp90. J. Biol. Chem., 273, 18007–18010. [DOI] [PubMed] [Google Scholar]
- Young J.C., Moarefi,I. and Hartl,F.U. (2001) Hsp90: a specialized but essential protein-folding tool. J. Cell Biol., 154, 267–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young J.C., Hoogenraad,N.J. and Hartl,F.U. (2003) Molecular chaperones Hsp90 and Hsp70 deliver preproteins to the mitochondrial import receptor Tom70. Cell, 112, 41–50. [DOI] [PubMed] [Google Scholar]
- Zeiner M., Gebauer,M. and Gehring,U. (1997) Mammalian protein RAP46: an interaction partner and modulator of 70 kDa heat shock proteins. EMBO J., 16, 5483–5490. [DOI] [PMC free article] [PubMed] [Google Scholar]