

Abstract
The cardiac isoform of 6-phosphofructo-2-kinase/ fructose-2,6-bisphosphatase (PFK-2), regulator of the glycolysis-stimulating fructose-2,6-bisphosphate, was among human HeLa cell proteins that were eluted from a 14-3-3 affinity column using the phosphopeptide ARAApSAPA. Tryptic mass fingerprinting and phospho-specific antibodies showed that Ser466 and Ser483 of 14-3-3-affinity-purified PFK-2 were phosphorylated. 14-3-3 binding was abolished by selectively dephosphorylating Ser483, and 14-3-3 binding was restored when both Ser466 and Ser483 were phosphorylated with PKB, but not when Ser466 alone was phosphorylated by AMPK. Furthermore, the phosphopeptide RNYpS483VGS blocked binding of PFK-2 to 14-3-3s. These data indicate that 14-3-3s bind to phosphorylated Ser483. When HeLa cells expressing HA-tagged PFK-2 were co-transfected with active PKB or stimulated with IGF-1, HA-PFK-2 was phosphorylated and bound to 14-3-3s. The response to IGF-1 was abolished by PI 3-kinase inhibitors. In addition, IGF-1 promoted the binding of endogenous PFK-2 to 14-3-3s. When cells were transduced with penetratin-linked AARAApSAPA, we found that this reagent bound specifically to 14-3-3s, blocked the IGF-1-induced binding of HA-PFK-2 to 14-3-3s, and completely inhibited the IGF-1-induced increase in cellular fructose-2,6-bisphosphate. These findings suggest that PKB-dependent binding of 14-3-3s to phospho-Ser483 of cardiac PFK-2 mediates the stimulation of glycolysis by growth factor.
Keywords: glycolysis/growth factors/Pasteur effect/penetratin/Warburg effect
Introduction
Fructose-2,6-bisphosphate (fru-2,6-P2) is a signalling metabolite (Stitt 1990; Dihazi et al 2001; Okar et al., 2001) which, in mammalian cells, activates 6-phosphofructo-1-kinase in glycolysis and inhibits fructose-1,6-bisphosphatase in gluconeogenesis. This makes fru-2,6-P2 pivotal in balancing cellular responses to demands for glucose for energy, against the need for glucose for biosynthesis, storage and/or export (Pilkis et al., 1995). Both the synthesis and degradation of fru-2,6-P2 are catalysed by isoforms of the bifunctional 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase (PFK-2/FBPase-2) (El-Maghrabi et al., 2001). Isoforms were named according to where they were first discovered (liver, skeletal muscle, foetal, cardiac, testis, brain and placental/ ubiquitous/inducible forms), but their distributions may be wider than these names indicate (Marsin et al., 2002). At least four genes encode PFK-2/FBPase-2 proteins, PFKFB1-4, with additional diversity generated by expression from different promoters and differential mRNA splicing (Darville et al., 1991; El-Maghrabi et al., 2001; Okar et al., 2001; Watanabe and Furuya, 2001).
How fru-2,6-P2 levels respond to extracellular and metabolic signals depends on the cellular complement of PFK-2/FBPase-2 isoforms, whose distinctive kinase/phosphatase activity ratios and different N- or C-terminal regulatory domains are tailored to suit the functions of different organs (El-Maghrabi et al., 2001). In the liver, glycolysis mainly provides precursors for lipogenesis and is inhibited when gluconeogenesis and glucose export predominate. Glucagon activates the cAMP-dependent protein kinase (PKA), which phosphorylates Ser32 of the major liver PFK-2/FBPase-2 isoform, thereby inhibiting its PFK-2 activity while activating FBPase-2 activity (Pilkis et al., 1980; El-Maghrabi et al., 2001). Hence, the fru-2,6-P2 concentration decreases, gluconeogenesis is promoted and glycolysis inhibited. Insulin activates liver PFK-2 by mechanisms that may involve sugar metabolites and dephosphorylation of PFK-2/FBPase-2 (Nishimura and Uyeda, 1995; El-Maghrabi et al., 2001), and increasing tissue fru-2,6-P2 has been reported to overcome hepatic insulin resistance in type 2 diabetes (Wu et al., 2002).
In contrast, glycolysis in heart muscle is geared towards energy production, and adapts rapidly to availability of lipids, glucose or alternate fuels, hormones, oxygen and workload (Depré et al., 1998a,b). When the heart is deprived of oxygen, the TCA cycle is compromised and there is a strong shift to anaerobic glycolysis (Pasteur Effect) (Pasteur, 1866; Depré et al., 1998a). One mechanism underlying the Pasteur Effect is that lack of oxygen increases the cellular AMP/ATP ratio, activating the AMP-activated protein kinase (AMPK), which phosphorylates Ser466 and increases the Vmax of cardiac PFK-2 (Marsin et al., 2000). Insulin and IGF-1 enhance the ability of cardiac myocytes to survive ischaemia by several mechanisms, including phosphophatidylinositol-3 kinase (PI 3-kinase)-dependent activation of cardiac PFK-2, and stimulation of glycolysis (Lefebrvre et al., 1996). Protein kinase B (PKB, also known as Akt), which is activated downstream of PI 3-kinase, phosphorylates Ser466 and Ser483 of cardiac PFK-2 in vitro, thereby activating the enzyme by increasing both its Vmax and the affinity for one of its substrates, fructose-6-phosphate (Deprez et al., 1997). However, a dominant-negative PKB construct failed to block insulin regulation of cardiac PFK-2 (Bertrand et al., 1999), and a partially-characterized enzyme, termed WISK, was proposed to be the true insulin-stimulated PFK-2 kinase (Deprez et al., 2000). Cardiac PFK-2 has also been reported to be activated in response to adrenalin via phosphorylation of Ser466 and Ser483 by PKA (Deprez et al., 1997, 1998a). None of these stimuli has any reported effects on the FBPase-2 activity of the cardiac enzyme, which is very low (Depré et al., 1998a,b).
Here, we identified cardiac PFK-2 during a project to find novel human 14-3-3-binding proteins (Milne et al., 2002). 14-3-3 proteins play central regulatory roles in eukaryotic cells by binding to phosphopeptide motifs in diverse target proteins, thereby modulating the function of the binding partner (Tzivion and Avruch, 2002). In a number of cases, binding of 14-3-3s to phosphorylated targets promotes events that support cell survival (Masters et al., 2002), and apoptosis is triggered by expression of a protein construct that binds 14-3-3s and blocks their binding to cellular targets (Masters and Fu, 2001). Two optimal 14-3-3-binding consensus sequences RSXpSXP (mode I) and RXXXpSXP (mode II) have been selected by screening peptide libraries (Rittinger et al., 1999), although several proteins bind to 14-3-3s via unconventional phosphopeptide binding sequences (Jelich-Ottmann et al., 2001; J.E.Harthill, A.Kulma, M.Pozuelo Rubio, B.H.C.Wong and C.MacKintosh, unpublished). Here we show that binding of cardiac PFK-2 to 14-3-3s is dependent on phosphorylation by PKB at Ser483 (RNYpSVGS), which does not conform precisely to a canonical 14-3-3-binding motif. The effects of transducing cells with a penetratin-linked 14-3-3-binding peptide indicate that 14-3-3s are essential mediators of the growth factor-induced increase in cellular fru-2,6-P2 by PFK-2.
Results
Isolation of cardiac isoform of PFK-2 by 14-3-3 affinity chromatography of HeLa cell extracts
Human HeLa cell extract was mixed with 14-3-3–Sepharose (Moorhead et al., 1999; Milne et al., 2002), which was washed extensively with 0.5 M NaCl and mock-eluted with control peptides. Proteins that bound via the phosphopeptide binding site on the 14-3-3s were specifically eluted by competition with a synthetic peptide, ARAApSAPA, which binds 14-3-3s (Moorhead et al., 1999). Comparison of protein profiles and 14-3-3 overlays of fractions from the column showed that the ARAApSAPA elution pool was highly enriched in 14-3-3 binding proteins (compare Figure 1A with B).
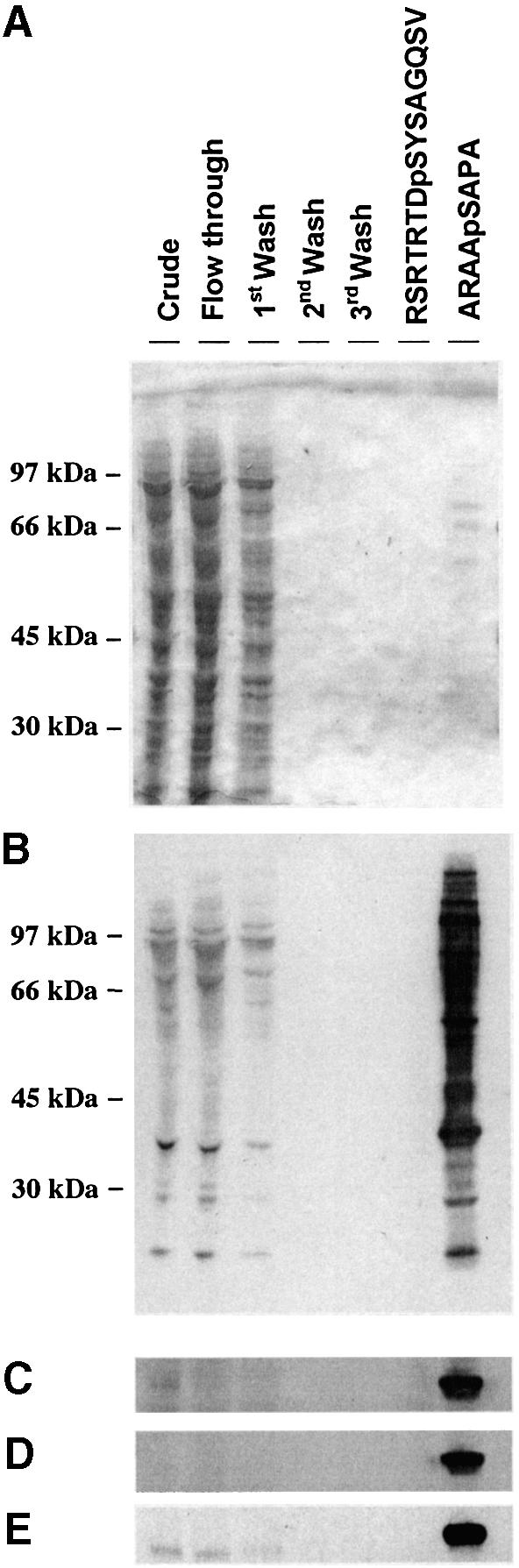

Fig. 1. Phosphorylated cardiac PFK-2 is highly enriched in the specific phosphopeptide elution pool from a 14-3-3 column. An extract of serum-grown HeLa cells was chromatographed on 14-3-3–Sepharose (see Materials and methods). The following amounts of protein were run on 10% Bis-Tris SDS–PAGE and transferred to nitrocellulose: extract, flow through, and start of salt wash (30 µg); middle and end of salt wash (protein undetectable); control peptide mock elution pool (<1 µg); and ARAApSAPA pool (2 µg). Blots were analysed for protein with Ponceau S (A) and DIG-14-3-3 overlay (B). Western blotting was performed with an antibody recognizing both the seryl-phosphorylated and unphosphorylated forms of the peptide RRNSFTP, corresponding to residues 463–469 of human cardiac PFK-2 (C), phospho-specific cardiac PFK-2 (pSer466) antibody (D), and phospho-specific cardiac PFK-2 (pSer483) antibody (E). DIG-14-3-3 overlays were performed as for western blots, except that DIG-labelled 14-3-3 was used instead of primary antibody, followed by anti-DIG horseradish peroxidase-secondary antibody (Moorhead et al., 1999). (F) Different amounts of synthetic peptides were tested by dot-blot using affinity-purified antibodies that bound to both phosphorylated and unphosphorylated RRN(p)S466TFP peptides (top panel), phospho-specific antibodies against RRNpS466FTP (pSer466 antibody, middle panel), and phospho-specific antibodies against RNYpS483VGS (pSer483 antibody, bottom panel). (G) Fractions from a 14-3-3–Sepharose column where 1 mM RNYS483VGS was used as the control peptide, and 1 mM RNYpS483VGS (corresponding to the pSer483 phosphorylation site on PFK-2) was used to elute proteins. PFK-2 was identified using the same antibody as in (C). (H) Untransfected HeLa cells were serum-starved for 12 h, then stimulated for 20 min with or without 100 ng/ml IGF-1, as indicated. Cell extracts (3 mg of protein) were incubated for 1 h with 50 µl 14-3-3–Sepharose, and washed pellets were extracted with SDS sample buffer, run on SDS–PAGE and probed with the same anti-PFK-2 antibody used in (C).
The pool of 14-3-3-binding proteins was fractionated further by anion-exchange chromatography, and fractions were analysed for 14-3-3-binding proteins using the DIG-14-3-3 overlay assay. A 58-kDa protein in a 300–400 mM NaCl Mono Q fraction was identified by tryptic mass fingerprinting as the cardiac isoform of 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase (PFK-2) (Figure 2; and Supplementary figure 9, available at The EMBO Journal Online). The cardiac PFK-2 aligned with a DIG-14-3-3-binding signal (Figure 2), indicating that cardiac PFK-2 could bind directly to 14-3-3s. Masses corresponding to mono-phosphorylated forms of the peptides Arg463-Arg476 and Asn480-Ala495 were present (Supplementary figure 9). Finding phosphopeptides in MALDI-TOF spectra of complex mixtures is unusual; perhaps the basic nature of the phosphorylated Arg463-Arg476 and Asn480-Ala495 peptides promoted their positive ionization and explains their prominence in the spectrum.
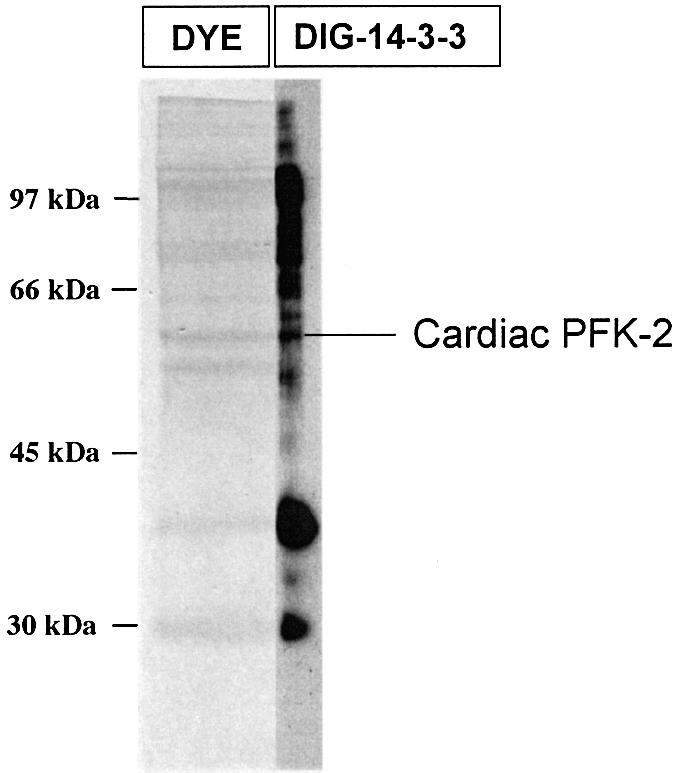
Fig. 2. Cardiac PFK-2 is among 14-3-3 affinity-purified HeLa proteins. 14-3-3 affinity-purified proteins (200 µg) were fractionated further by Mono Q anion-exchange chromatography. Fractions that were eluted between 300 and 400 mM NaCl were run on a 10% Bis-Tris SDS–polyacrylamide gel, blotted onto a Fluorotrans membrane, and stained with sulphorhodamine B. A narrow slice of the stained lane was processed in an overlay (labelled DIG-14-3-3) to identify 14-3-3-binding proteins. The remainder of the protein bands were analysed by MALDI-TOF tryptic mass fingerprinting (see Supplementary data).
Both Ser466 and Ser483 of cardiac PFK-2 can be phosphorylated by several protein kinases, including PKB/WISK (Bertrand et al., 1999; Deprez et al., 2000), while Ser466 is phosphorylated by the AMPK (Marsin et al., 2000). Antibodies were raised against the phosphopeptides RRNpS466FTP and RNYpS483VGS (where pS corresponds to phosphorylated Ser466 and Ser483 of cardiac PFK-2, respectively), which are the same antigens as those used by Marsin et al. (2000). Antibodies were isolated that recognize both the seryl-phosphorylated and unphosphorylated forms of the peptide, RRNSFTP, corresponding to residues 463 to 469 (Figure 1F, top panel). In addition, we isolated antibodies specific for the phosphorylated peptides RRNpSFTP (phospho-specific Ser466 antibody, Figure 1F, middle panel) and RNYpSVGS (phospho-specific Ser483 antibody, Figure 1F, bottom panel). All three antibodies detected the cardiac PFK-2 weakly in the crude HeLa cell extract, but very strongly in the ARAApSAPA eluate of the 14-3-3 column (Figure 1C–E). In addition, the specific activity of PFK-2 was higher in the ARAApSAPA phosphopeptide elution pool from the 14-3-3 column than in the crude extract (data not shown). These findings confirmed the identity of the purified protein as cardiac PFK-2, the specificity of elution of PFK-2 by the 14-3-3-binding phosphopeptide, and the phosphorylation of Ser466 and Ser483 on 14-3-3 affinity-purified PFK-2. Two forms of cardiac PFK-2 (54 and 58 kDa) have been described, which originate from the B gene by alternative splicing (Rider et al., 1992; Deprez et al., 1997). However, only the 58 kDa cardiac PFK-2 protein was apparent in the 14-3-3 column elution pool.
14-3-3s bind directly to phosphorylated cardiac PFK-2
Generally, but with a few exceptions (Masters et al., 1999), 14-3-3s bind to phosphorylated serine or threonine residues in target proteins (Rittinger et al., 1999; Tzivion and Avruch, 2002). Therefore, the phosphorylation dependence of the PFK-2/14-3-3 interaction was tested by dephosphorylating the 14-3-3 affinity-purified proteins. With 50 mU/ml protein serine/threonine phosphatase 2A (PP2A), both Ser466 and Ser483 on the 14-3-3 affinity-purified PFK-2 were dephosphorylated, and the binding of the PFK-2 to DIG-14-3-3s was abolished (Figure 3A). In the control, PFK-2 remained phosphorylated on Ser466 and Ser483, and still bound to DIG-14-3-3s after incubation with a mixture of PP2A and its inhibitor microcystin-LR (Figure 3A). When the dephosphorylated preparation was incubated with MgATP and PKB, the PFK-2 became phosphorylated on both Ser466 and Ser483, and its ability to bind DIG-14-3-3s was restored (Figure 3A). In contrast, when the dephosphorylated preparation was incubated with MgATP alone, Ser466 of PFK-2, but not Ser483, was phosphorylated and DIG-14-3-3 binding was not restored (Figure 3A). When a lower concentration of PP2A (10 mU/ml) was used in a similar experiment, Ser483 was selectively dephosphorylated. The resulting PFK-2 that was still phosphorylated on Ser466 did not bind 14-3-3s (Figure 3B).
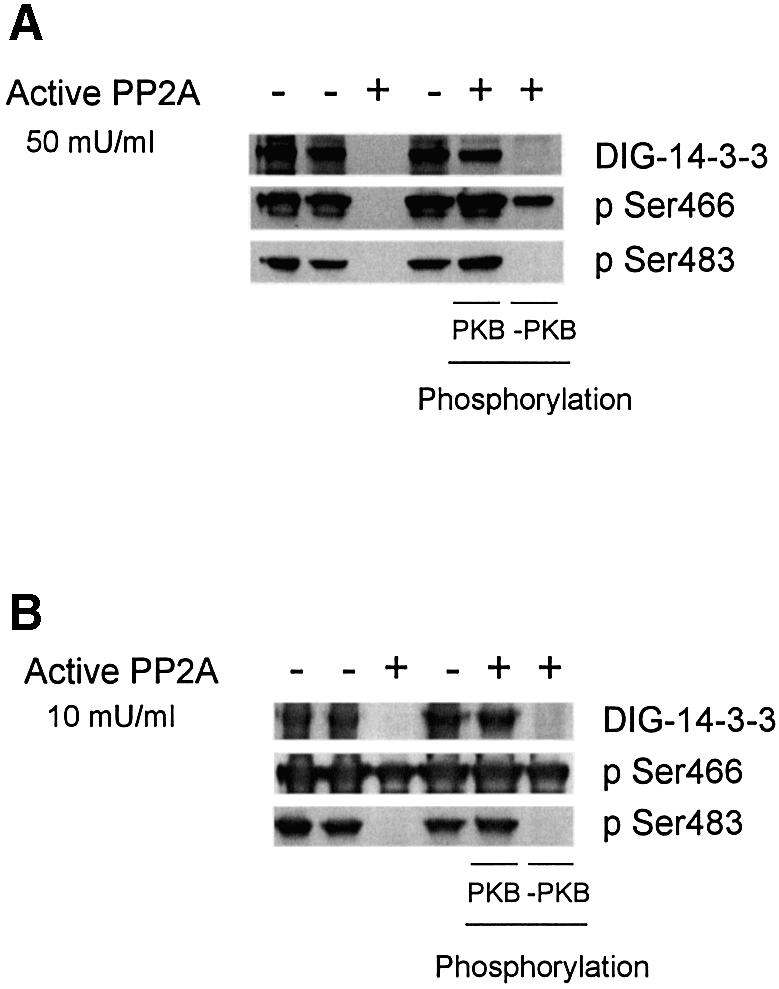
Fig. 3. Effects of dephosphorylation and rephosphorylation of cardiac PFK-2 in the 14-3-3 column elution pool. (A) Lanes 1 and 2 contain 2 µg of 14-3-3-binding proteins eluted with ARAApSAPA from a 14-3-3 affinity column [plus or minus 5 µM microcystin-LR (MC-LR), respectively]. For lanes 3, 5 and 6, 14-3-3-binding proteins (2 µg) were dephosphorylated with 50 mU/ml PP2A for 30 min at 30°C, and dephosphorylation was stopped with 5 µM MC-LR. Lane 4 is a control where the MC-LR and PP2A had been pre-mixed. In lanes 5 and 6, the dephosphorylated protein was rephosphorylated with 100 µM ATP/10 mM MgCl2, with (lane 5) or without (lane 6) 1 U/ml PKB for 30 min at 30°C, and the reaction was stopped with sample buffer. Samples were analysed by DIG-14-3-3 overlay and western blotting using phospho-specific cardiac PFK-2 (pSer466) and phospho-specific cardiac PFK-2 (pSer483) antibodies as indicated. (B) Identical experiment to (A), except that PP2A was used at 10 mU/ml.
These findings demonstrated that binding of 14-3-3s depended on phosphorylation of the cardiac PFK-2, and implicated phosphoSer483 as the binding site. The endogenous protein kinase(s) that phosphorylated Ser466, but not Ser483 (Figure 3A and B, last lane), might be the AMPK and/or p70 S6 kinase, both of which were found in the 14-3-3 column eluate (see Supplementary data).
14-3-3s bind directly to cardiac PFK-2 phosphorylated by PKB, but not by AMPK
For further characterization of the interaction between 14-3-3s and cardiac PFK-2, we generated a glutathione S-transferase (GST)–PFK-2 fusion protein. The GST–PFK-2 was phosphorylated on both Ser466 and Ser483 by PKB (Figure 4A), but only on Ser466 by AMPK (Figure 4B), as expected from previous studies (Bertrand et al., 1999; Deprez et al., 2000; Marsin et al., 2000). Consistent with the experiment shown in Figure 3, DIG-14-3-3s in the overlay assay bound directly to GST–PFK-2 that had been phosphorylated by PKB (Figure 4A), but there was no 14-3-3-binding signal for GST–PFK-2 that had been phosphorylated on Ser466 by the AMPK (Figure 4B). Similarly, PFK-2 that was phosphorylated on Ser466, but not Ser483, by PKA did not bind to 14-3-3s (not shown). Furthermore, a synthetic phosphopeptide corresponding to the pSer483 site of PFK-2 (RNYpS483VGS), but not the unphosphorylated version, was able to competitively elute PFK-2 from the 14-3-3–Sepharose column (Figure 1G). Taken together, these data implicate phosphoSer483 as the 14-3-3 binding site.
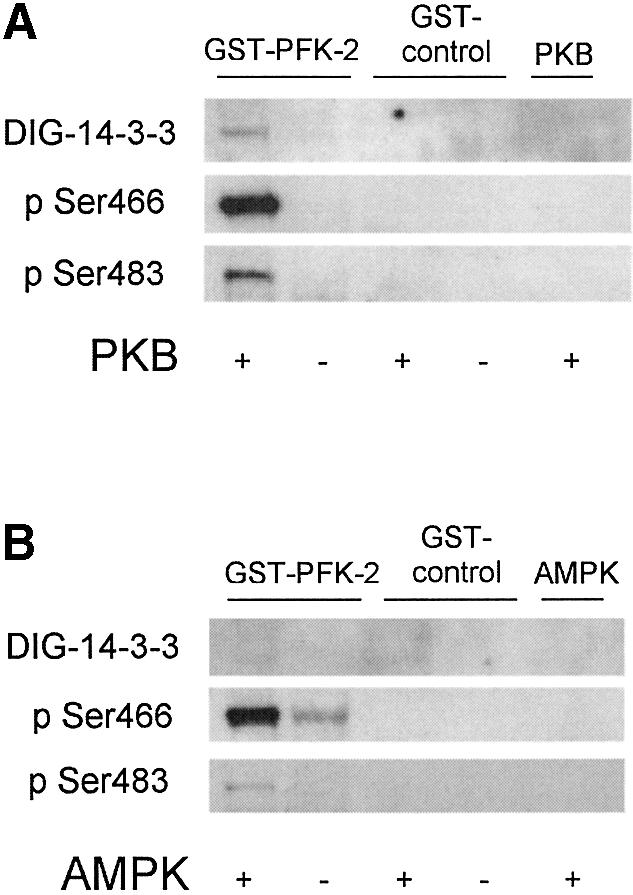
Fig. 4. Direct binding to 14-3-3s after phosphorylation of GST-tagged cardiac PFK-2 with PKB, but not after phosphorylation with AMPK. (A) Purified GST–PFK2, GST control or no substrate were incubated in the presence and absence of PKB (1 U/ml) with MgATP for 30 min at 30°C, and analysed by DIG-14-3-3 overlays, phospho-specific cardiac PFK-2 (pSer466) antibody, and phospho-specific cardiac PFK-2 (pSer483) antibody, as indicated. (B) As for A, expect that AMPK (10 U/ml) was used in place of PKB.
IGF-1-, PI 3-kinase- and PKB-dependent binding of 14-3-3s to HA-PFK-2 and endogenous PFK-2
Consistent with the possibility that Ser466 and Ser483 of the 14-3-3 affinity-purified PFK-2 (Figure 1) had been phosphorylated in vivo by PKB, the extracts used for these experiments were from cells grown in the presence of serum, and PKB activity was >3-fold higher than the basal level in serum-starved cell extracts (not shown).
The physiological regulation of 14-3-3 binding to PFK-2 was tested formally in cells transfected with a construct expressing HA-PFK-2. In HeLa cells, Ser473 of PKB was maximally phosphorylated and PKB maximally activated within a few minutes of stimulation with IGF-1 (Figure 5A; data not shown). IGF-1 also stimulated the phosphorylation of both Ser466 and Ser483 of HA-PFK-2, and these phosphorylations were blocked by the PI 3-kinase inhibitor LY 294002, but not by the mTOR inhibitor rapamycin, or UO126, which inhibits the activation of MAPK (Lefebrvre et al., 1996; Davies et al., 2000) (Figure 5A).
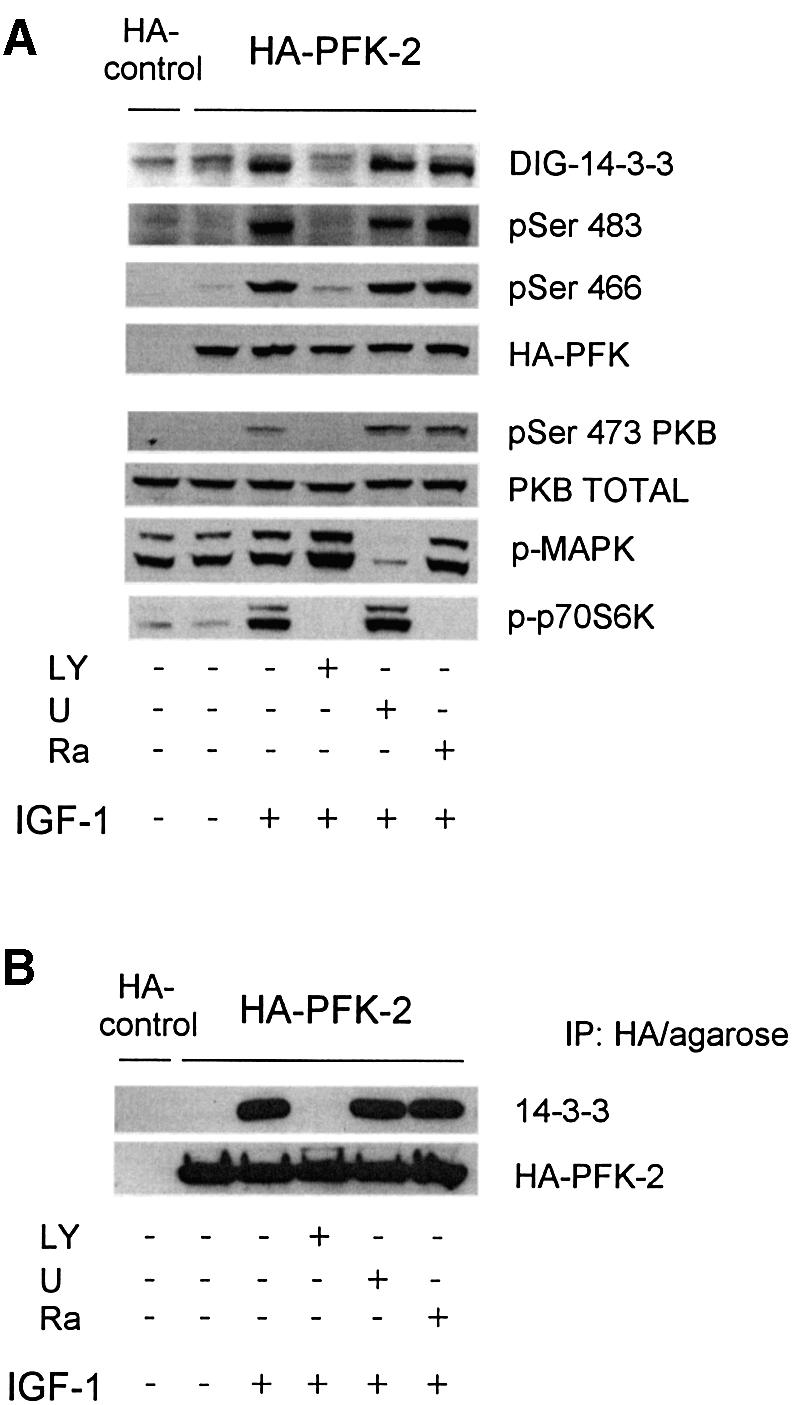

Fig. 5. Binding of 14-3-3s to HA-PFK-2 in IGF-1-treated HeLa cells. HeLa cells were transfected with a plasmid expressing HA-PFK-2. After 16 h, cells were serum-starved for a further 12 h, then stimulated for 20 min with or without 100 ng/ml IGF-1. Where indicated, cells were incubated with LY294002 (LY; 100 µM for 1 h), U0126 (U; 10 µM for 1 h) or rapamycin (Ra; 100 nM for 30 min) prior to stimulation with IGF-1. (A) Cell extracts (30 µg) were probed for binding to DIG-14-3-3s (14-3-3 overlay), phospho-specific antibodies that recognize pSer483 and pSer466 on cardiac PFK-2, anti-HA antibodies, phospho-specific antibodies against pSer473 on PKBα, and anti-PKB-total. Only the part of the 14-3-3 overlay in the region of HA-PFK-2 is shown. (B) HA-PFK-2 and associated proteins were precipitated from cell extracts (500 µg of lysate protein) with 20 µl of anti-HA affinity matrix (Roche). The washed immunoprecipitates were resolved using SDS–PAGE, transferred to nitrocellulose and probed for binding to the K19 antibodies that recognize all seven human 14-3-3 isoforms and anti-HA antibodies. (C) The washed immunoprecipitates were probed for binding to antibodies specific for individual 14-3-3 isoforms (see Materials and methods) and anti-HA antibodies.
IGF-1 also induced the binding of 14-3-3 proteins to HA-PFK-2 in HeLa cells, as assessed by the ability of HA-PFK-2 to bind 14-3-3s in the overlay assay (Figure 5A) and co-precipitation of HeLa cell 14-3-3s proteins recognized by the pan-14-3-3 K-19 antibody (Figure 5B). Consistent with the IGF-1-induced binding of 14-3-3s and PFK-2 being mediated by PKB, binding was prevented by the PI 3-kinase inhibitor LY 294002 (Figure 5). When untransfected cells were stimulated with IGF-1, the amount of PFK-2 in the HeLa cell extracts that could bind 14-3-3–Sepharose was much higher than that in extracts from unstimulated cells (Figure 1H). Thus, the binding of both HA-PFK-2 (Figure 5A and B) and endogenous PFK-2 (Figure 1H) to 14-3-3s was promoted by growth factors. Furthermore, when HeLa cell extract was passed through an anti-14-3-3 antibody column, PFK-2 bound and was specifically eluted by the 14-3-3-binding phosphopeptide ARAApSAPA (Figure 6), demonstrating that endogenous PFK-2 had been bound to the phosphopeptide binding site of 14-3-3s in the HeLa cell extract.
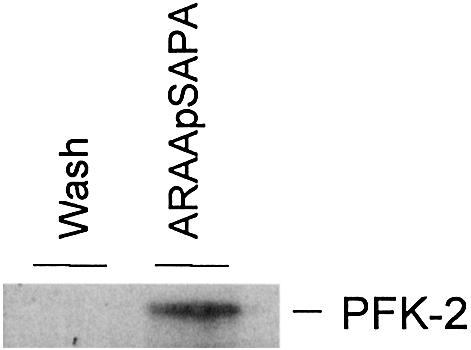
Fig. 6. Binding of endogenous 14-3-3s to endogenous PFK-2. An extract of serum-grown HeLa cells (500 mg) was passed through anti-14-3-3 antibody column (1 mg K-19 antibody bound to agarose). After washing with 500 column volumes of 0.5 M NaCl, 14-3-3-binding proteins were eluted with 1 mM ARAApSAPA. Samples of salt wash and the ARAApSAPA elution pool were concentrated, run on SDS–PAGE and transferred to nitrocellulose. Western blotting (same antibody as in Figure 1C) was used to identify endogenous PFK-2.
Using 14-3-3 isoform-specific antibodies, we found that the zeta (ζ) and theta (θ) 14-3-3 isoforms co-precipitated with HA-PFK-2 from extracts of IGF-1-stimulated HeLa cells (Figure 5C, IP lanes). The proportion of total 14-3-3ζ and 14-3-3θ that bound to the HA-PFK-2 was low, which would be expected because HA-PFK-2 is only one of many other proteins that bind to 14-3-3s (Harthill et al., 2002) (Figure 1). Whether other 14-3-3 isoforms also bind PFK-2 is not yet clear.
The possibility that phosphorylation by PKB promotes 14-3-3 binding to PFK-2 was examined further in cells expressing constitutively active PKB. Targeting PKB to cell membranes through a myristylation consensus is sufficient to promote its phosphorylation on Thr308 and Ser473 (Figure 7A), and also activation (Andjelkovic et al., 1997). In HeLa cells containing active myristylated PKB, GFP–HA-PFK-2 was phosphorylated on both Ser483 and Ser466, and was capable of binding to 14-3-3s in an overlay assay (Figure 7A), and to 14-3-3s in the HeLa cell extracts, as demonstrated by co-precipitation with anti-GFP antibodies (Figure 7B). Similarly, a constitutively active PKB in which the Thr308 and Ser473 activating phosphorylation sites are changed to aspartate residues (PKB-DD) (Lawlor and Alessi, 2001) promoted the phosphorylation of Ser483 and Ser466 of HA-PFK-2, and its binding to 14-3-3s (not shown).

Fig. 7. Binding of 14-3-3s to HA-PFK-2 in myristylated PKB-transfected HeLa cells. HeLa cells were transfected with plasmid pCMV5-PFK-2 to express HA-PFK-2 (A), or pGFPPFK2 to express GFP–HA-PFK-2 (B). Where indicated, cells were co-transfected with a plasmid expressing PKB with a myristylation consensus site (pCMV5-PKBmyr). (A) Cell extracts (30 µg) were analysed for binding to DIG-14-3-3s (14-3-3 overlay), phospho-specific antibodies that recognize pSer483 and pSer466 on cardiac PFK-2 and anti-HA antibodies, and phospho-specific antibodies against pSer473 on PKBα and anti-PKB. (B) GFP–HA-PFK-2 and associated proteins were precipitated from cell extracts (500 µg) with 3 µg anti-GFP antibodies (20 µl of protein G–Sepharose). Washed anti-GFP immunoprecipitates were subjected to SDS–PAGE, transferred to nitrocellulose and blotted with K19 antibodies that recognize all seven human 14-3-3 isoforms and anti-GFP antibodies.
Effects of disrupting 14-3-3 interactions in vivo on fru-2, 6-P2 levels and HA-PFK-2 activity
The activity of 14-3-3 affinity-purified PFK-2 in the standard assay (see Materials and methods) was unaffected by 14-3-3s and/or the ARAApSAPA peptide in vitro (not shown), although we cannot rule out the possibility that 14-3-3s affect the kinetic properties of the enzyme. We therefore aimed to determine whether disrupting 14-3-3 binding to PFK-2 inside cells had any functional effect. A 14-3-3-binding phosphopeptide and unphosphorylated control were synthesized attached to both an N-terminal penetratin sequence to make them cell-permeable, and a fluorescein label for visualization of their uptake into cells. In agreement with Richard et al. (2003), fluorescence microscopy of living cells indicated that endocytosis may play a role in the cellular internalization of the penetratin conjugates (Figure 8A). We also made the penetratin peptides with biotin tags so that they could be extracted from cell lysates with streptavidin. When HeLa cells were incubated in 30 µM biotin-penetratin-AARAApSAPA, washed and extracted, 14-3-3 proteins were found in the streptavidin–Sepharose precipitates (Figure 8B). In contrast, 14-3-3s from extracts of cells incubated with biotin-penetratin-AARAAGAPA did not bind streptavidin (Figure 8B).



Fig. 8. Use of penetratin-ARAApSAPA to test the effects of disrupting 14-3-3 binding to cellular PFK-2. (A) HeLa cells were incubated with 30 µM of fluorescein-penetratin-AARAASAPA (dP) or 30 µM of fluorescein-penetratin-AARAApSAPA (P) for 1 h. Live cells were observed by fluorescence microscopy. Arrows indicate examples of green fluorescent spots in cells. (B–D) HeLa cells were transfected with the plasmid expressing HA-PFK-2, and after 16 h were serum-starved for a further 12 h, then stimulated for 20 min, 1 h or 2 h with 100 ng/ml IGF-1. Where indicated, the cells were incubated with 100 µg/ml (30 µM) biotin-penetratin-AARAApSAPA or biotin-penetratin-AAR AAGAPA for 1 h prior to stimulation with IGF-1. (B) Biotinylated peptides and associated proteins were precipitated from cell extracts (500 µg of lysate protein) with 20 µl streptavidin-agarose (Amersham-Pharmacia Biotech). Washed pellets were extracted in SDS sample buffer, subjected to SDS–PAGE, transferred to nitrocellulose and blotted with the K19 pan-14-3-3 antibodies. (C) HA-PFK-2 and associated proteins were precipitated from cell extracts (500 µg of lysate protein) with 20 µl anti-HA-agarose. Washed precipitates were subjected to SDS–PAGE, transferred to nitrocellulose and blotted with the K19 pan-14-3-3 and anti-HA antibodies (upper two panels). Lysates from each set of cells (30 µg of protein) were probed with phospho-specific antibodies that recognize pSer483 and pSer466 on cardiac PFK-2 and anti-HA antibodies (lower three panels). (D) Effect of penetratin-AARAApSAPA and penetratin-AARAAGAPA on the IGF-1-induced increase in cellular fru-2,6-P2 levels. Cells were washed, extracted and assayed for fru-2,6-P2 at the times indicated. Results represent means ± standard deviations for three separate experiments, with each assayed in duplicate.
14-3-3s were bound to HA-PFK-2 that was extracted from IGF-1-stimulated cells in the absence or presence of biotin-penetratin-AARAAGAPA (Figure 8C). However, biotin-penetratin-AARAApSAPA selectively blocked the co-precipitation of 14-3-3s with HA-PFK-2 (Figure 8C). Thus, the biotin-penetratin-AARAApSAPA could bind specifically to 14-3-3s and disrupt their binding to cellular targets such as PFK-2.
The activity of HA-PFK-2 extracted from IGF-1-stimulated cells was ∼1.3-fold higher than unstimulated cells (not shown), compared with the 2-fold increase reported previously (Deprez et al., 1997). Neither the biotin-penetratin-AARAApSAPA nor control peptides had any obvious effect on the activity of extracted HA-PFK-2 (not shown). However, despite its apparent lack of effect on HA-PFK-2 activity, the biotin-penetratin-AARAApSAPA peptide, but not the control, completely blocked the IGF-1-induced increase in cellular fru-2,6-P2 levels (Figure 8D). Within 1 h of cell stimulation, fru- 2,6-P2 concentrations increased 2-fold above the levels in unstimulated cells. Pretreatment with penetratin-AARAApSAPA, but not penetratin-AARAAGAPA, completely suppressed the IGF-1-induced increase in fru-2,6-P2 (Figure 8D). The suppressive effect of penetratin-AARAApSAPA on cellular fru-2,6-P2 concentration continued for at least 2 h (Figure 8D). Most of the measured fru-2,6-P2 was generated by the HA-PFK-2, but the lower levels of this metabolite in untransfected cells were also regulated by IGF-1 and penetratin-AARAApSAPA (not shown).
The penetratin-linked AARAApSAPA peptide did not block the IGF-1-induced phosphorylation of Ser466 and Ser483 of HA-PFK-2 (Figure 8C).
Discussion
Control of energy supplies, metabolism and cell survival must be interdependent, although our understanding of the cellular integration of these processes is rudimentary. Therefore, we were intrigued to find an interaction between an enzyme of metabolism, cardiac PFK-2, that functions to support survival and continued beating of heart muscle (Depré et al., 1998a) and the 14-3-3s that have been implicated in promoting cell survival (Masters et al., 2002). In order to bind 14-3-3s, the PFK-2 had to be phosphorylated on Ser483 by PKB in vitro (Figures 1, 3 and 4), or in cells that were stimulated with IGF-1 or transfected with active forms of PKB (Figures 5 and 7). Endogenous PFK-2 and 14-3-3s bound to each other in HeLa cell extracts (Figure 6), and the ability of endogenous PFK-2 to bind 14-3-3–Sepharose was promoted by IGF-1 stimulation of cells (Figure 1H), indicating that the HA-PFK-2 was a valid reporter of physiological regulation of the endogenous enzyme. PKB is a central mediator of cell survival and metabolic regulation. PKB was first identified as the v-Akt retroviral oncogene product, PKB isoforms are overexpressed in several cancers, and the enzyme behaves experimentally as a strong promoter of cell survival and inhibitor of apoptosis (Lawlor and Alessi, 2001). Interestingly, several cellular proteins bind 14-3-3s after phosphorylation by PKB, including the transcription factor FKHR (e.g. Rena et al., 2001), and the insulin- and growth factor-regulated protein p39 (Harthill et al., 2002).
We found that a cell-permeable 14-3-3-binding phosphopeptide bound selectively to 14-3-3s, disrupted their binding to HA-PFK-2 (Figure 8) and other targets (not shown), and inhibited IGF-1-induced increases in cellular fru-2,6-P2 levels (Figure 8C). Upstream components of IGF-1 signalling, including the IGF-1 receptor (Parvaresch et al., 2002), interact with 14-3-3s in a phosphorylation-dependent manner. However, we found that the penetratin-AARAApSAPA did not affect the IGF-1 stimulated phosphorylation of PFK-2 (Figure 8B). These data suggest that 14-3-3 binding to cardiac PFK-2 is essential for mediating the IGF-1-induced increase in cellular fru-2,6-P2. Our findings indicate that penetratin-AARAApSAPA has exciting potential for dissecting the regulation of other cellular processes by 14-3-3s.
The only case where activation of an enzyme by 14-3-3s is understood at the structural level is serotonin N-acetyltransferase (AANAT). Binding of a 14-3-3 dimer restricts the movement of a floppy loop in AANAT, forcing the enzyme into the conformation that is most favourable for substrate binding (Obsil et al., 2001). For PFK-2, there was no obvious effect of the penetratin-AARAApSAPA on the IGF-1-induced increase in extractable HA-PFK-2 activity (not shown), so the precise mechanism of fru-2,6-P2 regulation by 14-3-3s is still puzzling. Perhaps 14-3-3s affect the kinetic properties or subcellular location of the PFK-2.
PhosphoSer483 is clearly implicated as the 14-3-3-binding site on PFK-2, but the surrounding sequence (RNYpS483VGS) lacks the proline residue that is in the +2 position of the canonical 14-3-3-binding phosphopeptides (Rittinger et al., 1999) and the binding motif of AANAT (Obsil et al., 2001). In AANAT, this proline causes a sharp bend that allows the protein backbone to exit the binding groove of the 14-3-3 protein. Perhaps the PFK-2 structure does not sweep away from the binding site so sharply.
One obvious question surrounds the significance of finding the cardiac PFK-2 in extracts of the carcinoma-derived HeLa cell line. Cancer cells are characterized by the Warburg Effect, meaning that they display high rates of glycolysis, including generation of lactate, and respire poorly, not only in the anaerobic environment within a tumour, but even when O2 is not limiting (Warburg, 1930). Why glycolysis should be favoured over the TCA cycle in cancers is puzzling because glycolysis generates less ATP for each molecule of glucose. Recently, however, it was discovered that the glycolytic intermediate fru-1,6-P2 feeds into the pentose phosphate pathway to provide nucleotides for DNA replication in proliferating cancer cells (Chesney et al., 1999). In dissecting the mechanisms for promoting glycolysis, high levels of fru-2,6-P2 have been found in many cancer cells (Mojena et al., 1992), and the inducible PFK-2 (iPFK-2), a splice variant of the PFKFB3 gene, was found to be expressed in tumours (Chesney et al., 1999) and induced under control of the hypoxia-inducible factor-1 (Minchenko et al., 2002). Inhibiting iPFK-2 decreased the levels of pentose phosphate pathway intermediates (Chesney et al., 1999). Our findings suggest the additional possibility that PKB-dependent binding of 14-3-3s to cardiac PFK-2 may contribute to regulation of fru-2,6-P2 levels in cancer cells.
Finally, while this work was in progress, we found that Arabidopsis thaliana fructose-6 phosphate, 2-kinase/ fructose-2,6-bisphosphatase (termed F2KP in plants) binds 14-3-3s after phosphorylation in vitro or in vivo (A.Kulma, D.Villadsen, D.G.Campbell, S.E.M.Meek, J.E.Harthill, T.H.Nielsen and C.MacKintosh, manuscript in preparation). This means that 14-3-3s interact with at least four enzymes that contain both synthetase- and phosphohydrolase-like domains, namely sucrose- phosphate synthase, a trehalose-phosphate synthase (Moorhead et al., 1999), F2KP from plants (A.Kulma et al., in preparation) and cardiac PFK-2 (this study). Perhaps there is a general significance in 14-3-3s interacting with bifunctional synthetase/phosphatase enzymes.
Materials and methods
Materials
Synthetic peptides were from Graham Bloomberg (University of Bristol), oligonucleotides from MWG-Biotech, microcystin-LR from Linda Lawton (Robert Gordon’s University, Aberdeen), frozen human HeLa cell pellets from 4c Biotech (Ghent, Belgium), Vivaspin concentrators from Vivascience, tissue culture reagents from Gibco-BRL, sequencing grade trypsin from Roche Molecular Biochemicals, and precast 10% Bis-Tris SDS–polyacrylamide gels from Invitrogen. All chromatographic columns were from Amersham-Pharmacia Biotech. All other chemicals were from BDH Chemicals or Sigma-Aldrich.
14-3-3 affinity chromatography and anion exchange chromatography of human HeLa cell extracts
HeLa cells (5 × 109) were thawed and extracted in 20 ml lysis buffer comprising 50 mM Tris pH 7.5, 1 mM EDTA, 1 mM EGTA, 1% (v/v) Triton X-100, 10 mM β-glycerophosphate, 50 mM NaF, 1 mM sodium orthovanadate, 5 mM sodium pyrophosphate, 0.27 M sucrose, 1 mM benzamidine, 0.2 mM phenylmethylsulfonyl fluoride, 10 µg/ml leupeptin and 0.1% (v/v) 2-mercaptoethanol. The broken cells were centrifuged at 15 000 g for 60 min, and the supernatant was collected and filtered. The clarified solution was mixed end-over-end for 1 h at 4°C with 5 ml Sepharose linked to BMH1/BMH2 (the Saccharomyces cerevisiae 14-3-3 isoforms) (Moorhead et al., 1996, 1999). The mixture was poured into a column, flow-through collected, and the column washed with 600 column volumes of 50 mM Tris–HCl pH 7.5, 500 mM NaCl, 1 mM DTT (buffer A) and then 12 ml of a control synthetic phosphopeptide that does not bind 14-3-3s (1 mM RSRTRTDpSYSAGQSV in buffer A). Proteins that bind to the phosphopeptide binding site of 14-3-3s were eluted with 12 ml of 1 mM ARAApSAPA phosphopeptide in buffer A. Samples (12 ml) from the middle and end of the 0.5 NaCl wash, the control peptide mock-elution pool (12 ml) and the ARAApSAPA elution pool (12 ml) were each concentrated to ∼400 µl, desalted in Vivaspin concentrators, and analysed as indicated in Figure 1.
A sample (200 µg) from an ARAApSAPA elution pool was filtered through a 0.22 µm spin filter (Costar), and fractionated further by anion exchange chromatography (Mono Q PC 1.6/5). A gradient of salt (0 to 1 M NaCl) in buffer (25 mM Tris–HCl pH 7.5, 1 mM DTT) was applied to the column, and fractions were collected.
Tryptic mass fingerprinting
Proteins were resolved in 10% Bis-Tris SDS–PAGE, transferred to a nitrocellulose membrane and stained with Sulphorhodamine B. Narrow strips of the protein-containing lanes were cut off and incubated with DIG-14-3-3 to identify proteins that bound directly to 14-3-3s. The remainder of the protein bands was excised, alkylated and digested with trypsin, and masses of tryptic peptides were determined by matrix-assisted laser desorption/ionization (MALDI-TOF) mass spectrometry in an Elite STR mass spectrometer (PerSeptive Biosystems, CA) in reflectron mode (Campbell and Morrice, 2002).
Antibodies
Antibodies specific for phosphoSer466 of human cardiac PFK-2 were raised against the phosphopeptide RRNpS466FTP (where pS denotes phosphoserine), and antibodies specific for phosphoSer483 were raised against the peptide RNYpS483VGS. The peptides were conjugated separately to bovine serum albumin and keyhole limpet haemocyanin, mixed and injected into sheep at Diagnostics Scotland (Penicuik, UK) The antisera were affinity-purified by Jane Leitch [Division of Signal Transduction Therapy (DSTT), University of Dundee], first by binding to the phosphorylated peptides coupled to N-hydroxysuccinimide-activated CH–Sepharose, which bound both phospho- and non-phosphospecific antibodies. The eluate was then passed through a column of unphosphorylated peptide, and the flow-through contained phosphospecific antibodies.
The following antibodies against protein kinases were used for western blotting: sheep anti p70S6K (used at 1 µg/ml) and sheep anti-PKB (1 µg/ml) from DSTT; sheep anti-AMPKα1 and AMPKα2 (isoforms of the catalytic subunit) from Grahame Hardie (University of Dundee); and rabbit anti-JNK (c-Jun N-terminal kinase) and rabbit anti-SAPK2 (p38) from Cell Signaling Technology.
Antibodies specific for the zeta (ζ, sc-1019) and theta (θ, sc-732) 14-3-3 isoforms, the K-19 pan-14-3-3 antibody (sc-629) and the K-19 pan-14-3-3 antibody bound to agarose (sc-629 AC) were from Santa Cruz.
cDNA cloning and sequencing
The cardiac PFK-2 cDNA was cloned by RT–PCR from total HeLa-cell RNA. Primers MP251 (ATGTCTGGGGCATCTTCCTCAGAACAG) and MP266 (ggatccgccaccatgtacccatacgatgtgccagattacgcctctggggcatcttcctcagaacagaac) were used to amplify the open reading frame and add an HA-tag at the 5′-terminus. The resulting 1515-bp cDNA fragment was ligated into pCR2.1-TOPO (Invitrogen). The insert was identical to the DDBJ/EMBL/GenBank sequence CAA06605, apart from an inserted C between K27 and S28. The plasmid was digested with BamH1 and ligated into the same site in pGEX6P-1 (Pharmacia) to produce pGEXPFK2 (encoding GST–HA-cardiac PFK-2, termed GST–PFK-2), pCMV5 (Clontech) to produce pCMVHAPFK2 (encoding HA-tagged PFK2) and EGFP-C1 (Clontech) to produce pGFPPFK2 (encoding GFP–HA-PFK-2). Sequences of all DNA constructs were checked using The Sequencing Service, managed by Nick Helps (University of Dundee; www.dnaseq.co.uk). For transfection of HeLa cells, Fugene 6 (24 µl; Roche) and 3–6 µg DNA was added to each 10-cm diameter dish of cells.
Production and use of GST–cardiac PFK-2
Expression of GST–PFK-2 fusion protein was induced with isopropyl-β-d-thiogalactopyranoside (IPTG) in Escherichia coli BL21 cells (Life Technologies), containing the pGEXPFK2 plasmid. The GST–PFK-2 protein was purified by glutathione Sepharose chromatography (Amersham Pharmacia Biotech). The purified protein was dialysed and stored in 50% (v/v) glycerol at –80°C. The authenticity of the protein was confirmed by tryptic mass fingerprinting (Campbell and Morrice, 2002) and western blotting.
Protein kinases and protein phosphatase 2A
AMPK was purified from rat liver to 4000 U/mg using SAMS peptide substrate (Dale et al., 1995), by Kevin Green and colleagues in Grahame Hardie’s group (University of Dundee). Recombinant PKBα lacking the pleckstrin homology domain (ΔPH-PKBα-S473D) was activated to 30 U/ml by phosphorylation with PDK1, and the PDK1 was subsequently removed (Walker et al., 1998), by Carla Clark (DSTT). The catalytic subunit of PKA was from Calbiochem. One unit transfers 1 pmol of phosphate to histone H1 in 1 min at 30°C. PP2A was purified from bovine heart by Jean Harthill and Julie Diplexcito (MRC Unit, Dundee), and 1 mU dephosphorylates 1 nmol phosphorylase a/min in a standard assay (MacKintosh and Moorhead, 1999).
6-phosphofructo-2-kinase activity and cellular fru-2,6-P2 assays
For 6-phosphofructo-2-kinase assays, enzyme was incubated for 30 min at 30°C with 5 mM KPi, 1 mM fru 6-P, 2 mM ATP and 3 mM glu 6-P in 50 mM MOPS–KOH pH 7.3, 5 mM MgCl2, 1 mM EDTA and 10% (v/v) ethylene glycol. The reaction was stopped with KOH. Fru-2,6-P2 in cell extracts, or produced by PFK-2 in vitro, was measured by following the activation of pyrophosphate fructose 6-P phosphotransferase (PFP) in converting fru-6-P to fru-1,6-P2 (Nielsen, 1992). PFP was extracted from potato tubers by Dorthe Villadsen, a visitor to the MRC Unit, Dundee.
Supplementary data
Supplementary data are available at The EMBO Journal Online.
Acknowledgments
Acknowledgements
We thank Agnieszka Kieloch and Richard Grier for culturing cells, colleagues at the DSTT for supplies, Maria Deak for PKB constructs, Grahame Hardie for AMPK, and Alan Prescott for help with cell imaging. We also thank the European Community programme ‘Quality of Life and Management of Living Resources’ for a Marie Curie Fellowship to M.P.R. under contract No. QLK1-CT-2000-51184, the Wellcome Trust for a studentship to B.W., the BBSRC and MRC for grants to C.M., and the pharmaceutical companies that support DSTT (AstraZeneca, Boehringer Ingelheim, GlaxoSmithKline, Novo-Nordisk and Pfizer).
References
- Andjelkovic M. et al. (1997) Role of translocation in the activation and function of protein kinase B. J. Biol. Chem., 272, 31515–31524. [DOI] [PubMed] [Google Scholar]
- Bertrand L., Alessi,D.R., Deprez,J., Deak,M., Viaene,E., Rider,M.H. and Hue,L. (1999) Heart 6-phosphofructo-2-kinase activation by insulin results from Ser-466 and Ser-483 phosphorylation and requires 3-phosphoinositide-dependent kinase-1, but not protein kinase B. J. Biol. Chem., 274, 30927–30933. [DOI] [PubMed] [Google Scholar]
- Campbell D.G. and Morrice,N.A. (2002) Identification of protein phosphorylation sites by a combination of mass spectrometry and solid phase Edman sequencing. J. Biomol. Technol., 13, 119–130. [PMC free article] [PubMed] [Google Scholar]
- Chesney J., Mitchell,R., Benigni,F., Bacher,M., Spiegel,L., Al-Abed,Y., Han,J.H., Metz,C. and Bucala,R. (1999) An inducible gene product for 6-phosphofructo-2-kinase with an AU-rich instability element: role in tumor cell glycolysis and the Warburg effect. Proc. Natl Acad. Sci. USA, 96, 3047–3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dale S., Wilson,W.A., Edelman,A.M. and Hardie,D.G. (1995) Similar substrate recognition motifs for mammalian AMP-activated protein kinase, higher plant HMG-CoA reductase kinase-A, yeast SNF1 and mammalian calmodulin-dependent protein kinase I. FEBS Lett., 361, 191–195. [DOI] [PubMed] [Google Scholar]
- Darville M.I., Chikri,M., Lebeau,E., Hue,L. and Rousseau,G.G. (1991) A rat gene encoding heart 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase. FEBS Lett., 288, 91–94. [DOI] [PubMed] [Google Scholar]
- Davies S.P., Reddy,H., Caivano,M. and Cohen,P. (2000) Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem. J., 351, 95–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depré C., Rider,M.H., Hue,L. (1998a) Mechanisms of control of heart glycolysis. Eur. J. Biochem., 258, 277–290. [DOI] [PubMed] [Google Scholar]
- Depré C., Ponchaut,S., Deprez,J., Maisin,L. and Hue,L. (1998b) Cyclic AMP suppresses the inhibition of glycolysis by alternative oxidizable substrates in the heart. J. Clin. Invest., 101, 390–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deprez J., Vertommen,D., Alessi,D.R., Hue,L. and Rider,M.H. (1997) Phosphorylation and activation of heart 6-phosphofructo-2-kinase by protein kinase B and other protein kinases of the insulin signalling cascades. J. Biol. Chem., 272, 17269–17275. [DOI] [PubMed] [Google Scholar]
- Deprez J., Bertrand,L., Alessi,D.R., Krause,U., Hue,L. and Rider,M.H. (2000) Partial purification and characterization of a wortmannin-sensitive and insulin-stimulated protein kinase that activates heart 6-phosphofructo-2-kinase. Biochem. J., 347, 305–312. [PMC free article] [PubMed] [Google Scholar]
- Dihazi H., Kessler,R. and Eschrich,K. (2001) Phosphorylation and inactivation of yeast 6-phosphofructo-2-kinase contribute to the regulation of glycolysis under hypotonic stress. Biochemistry, 40, 14669–14678. [DOI] [PubMed] [Google Scholar]
- El-Maghrabi M.R., Noto,F., Wu,N. and Manes,N. (2001) 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase: suiting structure to need, in a family of tissue-specific enzymes. Curr. Opin. Clin. Nutr. Metab. Care, 4, 407–410. [DOI] [PubMed] [Google Scholar]
- Harthill J.E., Pozuelo Rubio,M., Milne,F.C. and MacKintosh,C. (2002) Regulation of the 14-3-3-binding protein p39 by growth factors and nutrients in rat PC12 pheochromocytoma cells. Biochem. J., 368, 565–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jelich-Ottmann C., Weiler,E.W. and Oecking,C. (2001) Binding of regulatory 14-3-3 proteins to the C terminus of the plant plasma membrane H+-ATPase involves part of its autoinhibitory region. J. Biol. Chem., 276, 39852–39857. [DOI] [PubMed] [Google Scholar]
- Lawlor M.A. and Alessi,D.R. (2001) PKB/Akt: a key mediator of cell proliferation, survival and insulin responses? J. Cell Sci., 114, 2903–2910. [DOI] [PubMed] [Google Scholar]
- Lefebrvre L., Mechin,M.C., Louckx,M.P., Rider,M.H. and Hue,L. (1996) Signaling pathways involved in the activation of heart 6-phosphofructo-2-kinase by insulin. J. Biol. Chem., 271, 22289–22292. [DOI] [PubMed] [Google Scholar]
- MacKintosh C. and Moorhead,G. (1999) Assay and purification of protein (serine/threonine) phosphatases. In Hardie,D.G. (ed.), Protein Phosphorylation: A Practical Approach, 2nd edition. Oxford University Press, Oxford, UK, pp. 153–181. [Google Scholar]
- Marsin A.S., Bertrand,L., Rider,M.H., Deprez,J., Beauloye,C., Vincent,M.F., Van den Berghe,G., Carling,D. and Hue,L. (2000) Phosphorylation and activation of heart PFK-2 by AMPK has a role in the stimulation of glycolysis during ischaemia. Curr. Biol., 10, 1247–1255. [DOI] [PubMed] [Google Scholar]
- Marsin A.S., Bouzin,C., Bertrand,L. and Hue,L. (2002) The stimulation of glycolysis by hypoxia in activated monocytes is mediated by AMP-activated protein kinase and inducible 6-phosphofructo-2-kinase. J. Biol. Chem., 277, 30778–3078. [DOI] [PubMed] [Google Scholar]
- Masters S.C. and Fu,H. (2001) 14-3-3 proteins mediate an essential anti-apoptotic signal. J. Biol. Chem., 276, 45193–45200. [DOI] [PubMed] [Google Scholar]
- Masters S.C., Pederson,K.J., Zhang,L., Barbieri,J.T. and Fu,H. (1999) Interaction of 14-3-3 with a nonphosphorylated protein ligand, exoenzyme S of Pseudomonas aeruginosa. Biochemistry, 38, 5216–5221. [DOI] [PubMed] [Google Scholar]
- Masters S.C., Subramanian,R.R., Truong,A., Yang,H., Fujii,K., Zhang,H. and Fu,H. (2002) Survival-promoting functions of 14-3-3 proteins. Biochem. Soc. Trans, 30, 360–365. [DOI] [PubMed] [Google Scholar]
- Milne F.C., Moorhead,G., Pozuelo Rubio,M., Wong,B., Kulma,A., Harthill,J.E., Villadsen,D., Cotelle,V. and MacKintosh C. (2002) Affinity purification of diverse plant and human 14-3-3-binding partners. Biochem. Soc. Trans, 30, 379–381. [DOI] [PubMed] [Google Scholar]
- Minchenko A., Leshchinsky,I., Opentanova,I., Sang,N., Srinivas,V., Armstead,V. and Caro,J. (2002) Hypoxia-inducible factor-1-mediated expression of the 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase-3 (PFKFB3) gene. Its possible role in the Warburg effect. J. Biol. Chem., 277, 6183–6187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mojena M., Bosca,L., Rider,M.H., Rousseau,G.G. and Hue,L. (1992) Inhibition of 6-phosphofructo-2-kinase activity by mercaptopurines. Biochem. Pharmacol., 43, 671–676. [DOI] [PubMed] [Google Scholar]
- Moorhead G., Douglas,P., Morrice,N., Scarabel,M., Aitken,A. and MacKintosh,C. (1996) Phosphorylated nitrate reductase from spinach leaves is inhibited by 14-3-3 proteins and activated by fusicoccin. Curr. Biol., 6, 1104–1113. [DOI] [PubMed] [Google Scholar]
- Moorhead G. et al. (1999) Phosphorylation-dependent interactions between enzymes of plant metabolism and 14-3-3 proteins. Plant J., 18, 1–12. [DOI] [PubMed] [Google Scholar]
- Nielsen T.H. (1992) Difference in fructose-2,6-biphosphate metabolism between sections of developing barley leaves. Physiol. Plant, 84, 577–583. [Google Scholar]
- Nishimura M. and Uyeda,K. (1995) Purification and characterisation of a novel xylulose 5-phosphate-activated protein phosphatase catalysing dephosphorylation of fructose-6-phosphate, 2-kinase: fructose-2,6-bisphosphatase. J. Biol. Chem., 270, 26341–26346. [DOI] [PubMed] [Google Scholar]
- Obsil T., Ghirlando,R., Klein,D.C., Ganguly,S. and Dyda,F. (2001) Crystal structure of the 14-3-3ζ:serotonin N-acetyltransferase complex: a role for scaffolding in enzyme regulation. Cell, 105, 257–267. [DOI] [PubMed] [Google Scholar]
- Okar D.A., Manzano,A., Navarro-Sabate,A. Riera,L., Bartrons,R. and Lange,A.J. (2001) PFK-2/FBPase-2: maker and breaker of the essential biofactor fructose-2,6-bisphosphate. Trends Biochem. Sci., 26, 30–35. [DOI] [PubMed] [Google Scholar]
- Parvaresch S., Yesilkaya,T., Baer,K., Al-Hasani,H. and Klein,H.W. (2002) 14-3-3 binding to the IGF-1 receptor is mediated by serine autophosphorylation. FEBS Lett., 532, 357–362. [DOI] [PubMed] [Google Scholar]
- Pasteur L. (1866) Etudes sur le Vin. Monograph. Editors, Masson et cie, Paris, France. [Google Scholar]
- Pilkis S.J. et al. (1980) Phosphorylation of rat hepatic fructose-1,6-bisphosphatase and pyruvate kinase. J. Biol. Chem., 255, 2770–2775. [PubMed] [Google Scholar]
- Pilkis S.J., Claus,T.H., Kurland,I.J. and Lange,A.J. (1995) Annu. Rev. Biochem., 64, 799–835. [DOI] [PubMed] [Google Scholar]
- Rena G., Prescott,A.R., Guo,S., Cohen,P. and Unterman,T.G. (2001) Roles of the forkhead in rhabdomyosarcoma (FKHR) phosphorylation sites in regulating 14-3-3 binding, transactivation and nuclear targetting. Biochem. J., 354, 605–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard J.P., Melikov,K., Vives,E., Ramos,C., Verbeure,B., Gait,M.J., Chernomordik,L.V. and Lebleu,B. (2003) Cell-penetrating peptides. A reevaluation of the mechanism of cellular uptake. J. Biol. Chem., 278, 585–590. [DOI] [PubMed] [Google Scholar]
- Rider M.H., Vandamme,J., Lebeau,E., Vertommen,D., Vidal,H., Rousseau,G.G., Vandekerckove,J. and Hue,L. (1992) The two forms of bovine heart 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase result from alternative splicing. Biochem. J., 285, 405–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rittinger K., Budman,J., Xu,J., Volinia,S., Cantley,L.C., Smerdon,S.J., Gamblin,S.J. and Yaffe,M.B. (1999) Structural analysis of 14-3-3 phosphopeptide complexes identifies a dual role for the nuclear export signal of 14-3-3 in ligand binding. Mol. Cell, 4, 153–166. [DOI] [PubMed] [Google Scholar]
- Stitt M. (1990) Fructose-2, 6-bisphosphate as a regulatory molecule in plants. Annu. Rev. Plant Physiol. Plant Mol. Biol., 41, 153–185. [Google Scholar]
- Tzivion G. and Avruch,J. (2002) 14-3-3 proteins: active cofactors in cellular regulation by serine/threonine phosphorylation. J. Biol. Chem., 277, 3061–3064. [DOI] [PubMed] [Google Scholar]
- Walker K.S., Deak,M., Paterson,A., Hudson,K., Cohen,P. and Alessi,D.R. (1998) Activation of protein kinase B beta and gamma isoforms by insulin in vivo and by 3-phosphoinositide-dependent protein kinase-1 in vitro: comparison with protein kinase B alpha.Biochem. J., 331, 299–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburg O. (1930) The Metabolism of Tumour. Vol II. Constable, London, UK. [Google Scholar]
- Watanabe F. and Furuya,E. (2001) Alternative splicing of novel exons of rat heart-type fructose-6-phosphate 2-kinase/fructose-2,6-bisphos phatase gene. Biochem. Biophys. Res. Commun., 282, 803–810. [DOI] [PubMed] [Google Scholar]
- Wu C., Okar,D.A., Newgard,C.B. and Lange,A.J. (2002) Increasing fructose-2,6-bisphosphate overcomes hepatic insulin resistance of type 2 diabetes. Am. J. Physiol. Endocrinol. Metab., 282, E38–E45. [DOI] [PubMed] [Google Scholar]