

Abstract
The AP-1 transcription factor c-Jun is a prototypical nuclear effector of the JNK signal transduction pathway. The integrity of JNK phosphorylation sites at serines 63/73 and at threonines 91/93 in c-Jun is essential for signal-dependent target gene activation. We show that c-Jun phosphorylation mediates dissociation of an inhibitory complex, which is associated with histone deacetylase 3 (HDAC3). The subsequent events that ultimately cause increased mRNA synthesis are independent of c-Jun phosphorylation and its interaction with JNK. These findings provide an ‘activation by de-repression’ model as an explanation for the stimulatory function of JNK on c-Jun.
Keywords: c-Jun transcription factor/HDAC/MAP kinase/phosphorylation/repression of transcription/signaling
Introduction
Whereas it is well established that the interaction of MAP kinases with responsive transcription factors and their subsequent phosphorylation often coincides with changed transcriptional output, the mechanistic basis for such processes is not well understood (Treisman, 1996; Chang and Karin, 2001). We have chosen the Jun-N-terminal-kinase (JNK)/c-Jun module to study mechanisms of transcription factor activation in eukaryotic cells. JNK is a MAPK that phosphorylates and activates c-Jun in response to a range of extracellular stimuli (Leppä and Bohmann, 1999; Davis, 2000). JNK-to-c-Jun signaling has been implicated in cellular proliferation, differentiation and apoptosis. Loss of either JNK or c-Jun activity is incompatible with embryonic development and impairs proliferation of mouse embryonic fibroblasts in tissue culture (Jochum et al., 2001). Jun proteins can also contribute to oncogenesis in different settings. c-Jun cooperates with Ras and is required for Ras to transform fibroblasts. Moreover, c-Jun is essential for the development of chemically induced tumors in mice (Eferl et al., 2003). v-jun is a retrovirally transduced allele of c-jun that can cause fibrosarcoma in chickens as well as wound-induced tumors in v-jun-expressing transgenic mice (reviewed in Vogt, 2001). While the molecular biology underlying the role of Jun in cell transformation and tumorigenesis is not completely understood at present, it is believed to involve the signaling and transcriptional properties of the protein.
JNK docks to c-Jun on a sequence referred to as the δ-domain (Dai et al., 1995; Kallunki et al., 1996). The relatively stable interaction between JNK and c-Jun facilitates phosphorylation of the latter when JNK is activated by its upstream kinase, JNKK. The relevant MAPK–substrate sites on c-Jun are serines 63 and 73 (Binetruy et al., 1991; Smeal et al., 1991) and either threonine 91 or 93 or both (Hibi et al., 1993; Derijard et al., 1994, Papavassiliou et al., 1995). Current models assume that phosphorylation facilitates the interaction of signal responsive transcription factors with the basal transcriptional machinery or with transcriptional co-activators, including histone acetyl transferases (HATs; Treisman, 1996; Mayr and Montminy, 2001). Examples for such scenarios include the transcription factors CREB and Smad3 where increased binding to the HAT CBP following phosphorylation seems to mediate signal-dependent gene activation (Janknecht et al., 1998; Mayr and Montminy, 2001). While CBP also appears to be required for transcriptional activation by AP-1 (Mayr and Montminy, 2001) and can bind to the N-terminal region of c-Jun in vitro, there is no evidence that this interaction is regulated by or dependent on phosphorylation (Bannister et al., 1995). More generally, no co-activators or components of the basal transcriptional machinery have yet been identified that preferentially bind to phosphorylated c-Jun.
As an alternative to the phosphorylation-induced binding of a co-activator, release of transcriptional inhibitors upon phosphorylation might also explain the signal-dependent activation of transcription factors. Examples for such inhibitory activities include prominently histone deacetylases (HDACs; Khochbin et al., 2001). HDACs antagonize the stimulatory effect of HATs on chromatin remodeling. Indeed, several transcription factors, notably nuclear receptors, recruit HDACs to promoters and thereby repress transcription in the absence of appropriate signals (Glass and Rosenfeld, 2000). Once such a transcription factor is activated, HDAC-associated protein complexes are exchanged for HAT-containing complexes, resulting in increased target gene expression.
Thus, the activity of a transcription factor in a given cellular context may be regulated at the level of co-activator, or repressor interaction, or both. Here we show that in the absence of JNK signaling a repressor activity that is associated with HDAC3 inhibits c-Jun. Phosphorylation of c-Jun by JNK relieves this repression, thereby enhancing the transcriptional activity of c-Jun. Our data indicate that JNK phosphorylation causes dissociation of the HDAC3-containing repressor complex, releasing c-Jun from a repressed state. The subsequent steps which lead to enhanced transactivation by c-Jun appear to be independent of JNK, as unphosphorylated c-Jun can stimulate transcription efficiently if the repressor complex is dissociated. Experiments with JNK-deficient cells show that inactive JNK cannot be an essential component of the repressor complex and that c-Jun can activate transcription in the absence of its kinase, provided that the repressor function is abrogated. Based on these findings, we propose an explanation for the increased transcriptional and transforming potential of v-Jun, which acts as an activated version of c-Jun even though it cannot be phosphorylated by JNK.
Results
c-Jun contains an inhibitory domain which mediates JNK responsiveness
To gain insight into the mechanism by which JNK stimulates c-Jun target gene transcription, we employed different types of reporter assays in cultured cells. We intended to restrict the analysis to the transcriptional function of c-Jun itself independent of heterodimerization with other AP-1 family members. To this end, we constructed fusion proteins that replaced the basic region/leucine zipper (bZIP) domain of c-Jun with the heterologous DNA-binding domain of the yeast transcription factor GAL4 (GAL-DBD, Figure 1A). In GAL–Jun1–256 all of c-Jun, except the bZIP domain, is fused to the GAL-DBD. As a reporter we stably integrated a Gal4-responsive luciferase gene into the genome of 293 cells. We chose stably integrated reporters because, presumably due to their packaging into chromatin, they acted more reliably and displayed less variable baseline activity than comparable reporters that were transiently transfected.
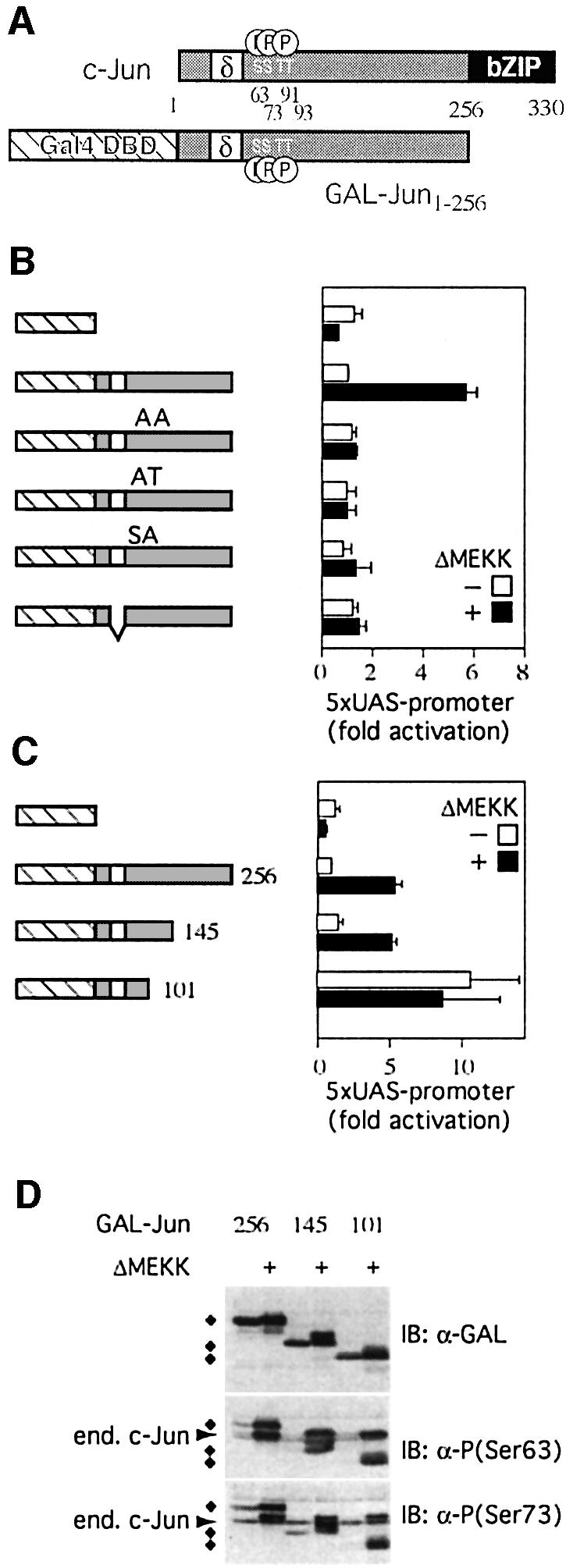
Fig. 1. Features of c-Jun that confer JNK responsiveness. (A) Schematic structure of c-Jun and GAL–Jun: the DNA-binding and dimerization domains of c-Jun (bZIP, black box) and GAL–Jun (GAL-DBD, cross-hatched box), the δ-domain (white box) and the JNK phosphorylation sites (serines 63, 73 and threonines 91, 93) are shown. Numbers indicate amino acid positions. (B) The transcriptional activity of GAL–Jun1–256 MAPK phosphorylation site mutants (AA: serines 63, 73 and threonines 91, 93 were replaced by alanines; AT: serines 63, 73 were replaced by alanines; SA: threonines 91, 93 were replaced by alanines) or a δ- domain deletion mutant were measured in reporter assays with or without activation of the JNK pathway by ΔMEKK. The graph displays the fold increase of normalized reporter activity after transfection with plasmids coding for the indicated fusion proteins relative to GAL– Jun1–256 which was set to 1 (means ± SD of 3–5 independent experiments). (C) Activities of GAL–Jun1–256 and different deletion mutants were determined as in (B). Note that Gal–Jun1–101 is constitutively active. (D) 293 cells were transiently transfected with the indicated expression constructs together with an empty expression vector or an expression plasmid encoding ΔMEKK. Lysates of transfected cells were analyzed by immunoblotting (IB) using either antibodies specific for the GAL-DBD or phospho-c-Jun-specific antibodies against the phosphorylated serine 63 or serine 73 (diamonds indicate fusion proteins, cross-reactivity with endogenous c-Jun is indicated by an arrow).
At moderate levels of expression, comparable to endogenous c-Jun, GAL–Jun1–256 did not stimulate the reporter when compared with GAL-DBD alone. However, when JNK activity in the transfected cells was stimulated by expression of a constitutively active form of the JNK kinase kinase MEKK1 (ΔMEKK), reporter activity increased 6-fold (Figure 1B). Thus, the assay system recapitulates the activation of c-Jun by JNK signaling. Replacement of all potential JNK phosphorylation sites, serines 63 and 73 and threonines 91 and 93, by alanines (Figure 1B, AA) abrogated responsiveness to JNK. Furthermore, substitution mutants in which only subsets of the JNK phosphorylation sites have been mutated (Figure 1B, AT and SA) show that both the serines and the threonines are required for signal-dependent regulation of c-Jun. Similar to the mutation of the phosphorylation sites, deletion of the δ-domain, which serves as the JNK docking site, impaired JNK-dependent transcriptional activation (Figure 1B).
Next, we examined which features of c-Jun in addition to the phosphorylation sites and the δ-domain are important for JNK responsiveness. A series of truncations of the GAL–Jun1–256 protein was generated and tested in reporter assays (Figure 1C). Similar to GAL–Jun1–256, a Gal4 fusion protein containing amino acids 1–145 of c-Jun (GAL–Jun1–145) was sufficient to recapitulate JNK responsiveness (Figure 1C).
In contrast, a shorter form including only residues 1–101 (GAL–Jun1–101) was already transcriptionally active in the absence of a JNK signal and activation of JNK did not increase this constitutive activity further (Figure 1C). This increased signal-independent activity is not caused by a higher expression level or constitutive phosphorylation of GAL–Jun1–101, as confirmed by western blot experiments (Figure 1D). Interestingly, phosphorylation of GAL–Jun1–101 was still signal-dependent and increased upon JNK activation by MEKK co-expression, evidently without enhancing transcriptional activity. Thus, phosphorylation of c-Jun and its transcriptional activity do not absolutely correlate and can be uncoupled from each other.
The findings described above suggest that GAL– Jun1–145, but not GAL–Jun1–101, contains a negatively acting domain that suppresses transactivation of the reporter gene. JNK signaling appears to neutralize this repressing function, permitting target gene activation. A conceivable mechanistic explanation for these observations is that JNK phosphorylation displaces a repressor activity that precludes transcription activation by c-Jun. Consistent with these results, a titratable repressor of c-Jun has been postulated by Baichwal and colleagues more than a decade ago (Baichwal and Tjian, 1990; Baichwal et al., 1992). The described activity acted via the so-called ε-domain (amino acids 101–128 of c-Jun), which resides in the region that we now also find to be important for JNK signaling to c-Jun.
Analogous to the experiments by Baichwal et al., we performed transactivation assays using different GAL–Jun fusion proteins and co-expressed them with an excess of full-length c-Jun, which acts as a competitor for the repressor as it cannot bind, and therefore not activate, the GAL-reporter (Figure 2A). Overexpression of full-length c-Jun increases the transactivation potential of GAL– Jun1–256 in our experimental system (Figure 2B). Furthermore, we find that a c-Jun-derived competitor lacking the bZIP region (ΔbZIP), which cannot bind to DNA, was sufficient to enhance GAL–Jun1–256 activity (Figure 2B). This rules out that the stimulatory effect of full-length c-Jun is caused indirectly by c-Jun target gene expression. The GAL–Jun1–101 truncation mutant, as well as a mutated version of GAL–Jun1–256 that lacks the ε-domain could not be further activated by co-expression of full-length c-Jun (Figure 2B). Taken together, these data are most readily explained by a repressor, which requires amino acids 101–145 of c-Jun for a stable interaction with the transcription factor. This repressor, or an essential subunit of it can be dissociated from c-Jun by titration with an excess of competitor. Interestingly, co-expressed full-length c-Jun reduces the activity of a GAL–Jun mutant devoid of the ε-domain. This squelching effect supports the notion that in addition to the above-described inhibitor, c-Jun also binds to titratable co-activators. However their effect only becomes apparent when the influence of the inhibitor is eliminated, in the case of our experiment by the removal of the ε-domain.

Fig. 2. A titratable repressor inhibits c-Jun transcriptional activity via the same region that mediates JNK responsiveness. (A) Schematic model to illustrate repressor function on c-Jun transcriptional activity. See Figure 1 for a legend. In its inactive state c-Jun is bound by a repressor (R). This interaction is stabilized by the ε-domain (shaded box) and the δ-domain (white box). (B) The transcriptional activity of GAL–Jun1–256, ‘the activator’, can be enhanced by co-expression of full-length c-Jun, ‘the competitor’, due to competition for the repressor. 293 cells with a stably integrated 5× UAS luciferase reporter were transiently transfected with expression constructs encoding various derivatives of GAL–Jun as activators. The different activators were GAL–Jun1–256, GAL–Jun1–101 or an ε-domain deletion mutant. In addition, an empty expression vector (white bars) or an expression plasmid encoding either full-length c-Jun (black bars) or a c-Jun deletion mutant lacking the bZIP region (gray bar) was co-transfected as ‘competitor’. The fold activation compared to GAL–Jun is represented (means ± SD of 3–5 independent experiments). The activity of GAL–Jun1–256 alone was set to 1. (C) The activation of the human collagenase I promoter by c-Jun is inhibited by a titratable repressor. 293 cells were transiently transfected with a luciferase reporter gene under the control of the human collagenase I promoter (region –517 to +63) together with an empty expression vector or an expression plasmid encoding full-length c-Jun (gray bars). For competition assays, a full-length c-Jun expression plasmid, as activator, was co-transfected with a GAL–Jun1–256 expression plasmid as the competitor. Activities were determined as in (B). The fold activation compared with the basal activity of the collagenase promoter, which was set to 1, is shown (average ± average deviation of two independent experiments).
Next, we examined whether the repressor also regulates transactivation of a natural target gene by c-Jun via its own DBD. The collagenase 1 promoter can be moderately activated by expression of full-length c-Jun (Figure 2C). Parallel over-expression of GAL–Jun1–256 increased c-Jun-dependent transcription from the collagenase 1 promoter significantly. This indicates that the competition for the repressor activity between c-Jun and Gal–Jun is reciprocal.
Phosphorylation of c-Jun by JNK destabilizes the interaction with repressor
Based on the data described above, we proposed the hypothesis that JNK might activate c-Jun by decreasing its affinity for the ε-domain-specific repressor or one of its components (Figure 3A). To test this idea, we measured transactivation by GAL–Jun1–256 (‘the activator’ in this experimental setting) when expressed either together with wild-type c-Jun (‘the competitor’) or with a phosphorylation point mutant, in which all JNK substrate sites have been replaced with alanines (JunAla). Compared with wild-type c-Jun, c-JunAla more potently stimulated GAL– Jun1–256 activity when co-expressed as a competitor (Figure 3B, upper part). Upon increased JNK signaling (+ΔMEKK) the difference between the phosphorylatable and non-phosphorylatable competitor to stimulate the activator was even more pronounced. This indicates that the distribution of repressor factors between the activator and the competitor is influenced by their respective phosphorylation states (Figure 3B, right panel). If wild-type c-Jun is used as a competitor, it reduces the amount of repressor bound to the activator, resulting in enhanced transcription of the reporter. In the presence of JNK activation, the affinities of both, the activator and the competitor, are decreased by phosphorylation. The net effect is no further enhancement of activator-mediated transcription by JNK under these conditions. However, competition by non-phosphorylatable c-Jun is more effective, due to its higher affinity to the repressor compared with wild-type c-Jun phosphorylated at basal levels. If the activator’s phosphorylation is increased by JNK signaling, non-phosphorylatable competitor leads to a substantial shift of the distribution manifested in a super-enhancement of transcription.

Fig. 3. c-Jun phosphorylation and repressor binding. (A) Hypothesis: phosphorylation of c-Jun by JNK appears to reduce the affinity to the repressor thereby enhancing c-Jun’s transcriptional activity. (B) Phosphorylated c-Jun competes less efficiently for repressor than non-phosphorylated c-Jun. Left panel: 293 cells with a stably integrated 5× UAS luciferase reporter were transiently transfected with expression constructs encoding GAL–Jun1–256 or GAL–Jun1–256Ala as activators and the indicated c-Jun derivatives as competitors. To stimulate phosphorylation of c-Jun, ΔMEKK was co-transfected (indicated with +). The normalized activity of GAL–Jun1–256 alone was set to 1 (means ± SD of 3–5 independent experiments). Note broken scale for values >50 (//). Right panel: model to explain the results shown in the graph on the left (for further details see text). (C) In the absence of the repression domain GAL–Jun1–101 activates transcription independently of its MAPK phosphorylation sites. 293 cells with a stably integrated 5× UAS luciferase reporter were transiently transfected with expression constructs encoding GAL–Jun or deletion mutants with, or without, MAPK phosphorylation sites (GAL–Jun1–101 and GAL–Jun1–101Ala, respectively). Activities were determined as in (A). Fold activation (average ± average deviation of two independent experiments) relative to GAL–Jun1–256, which was set to 1, is shown. (D) Phosphorylation of GAL–Jun1–256 is not increased by co-expression of full-length c-Jun. Lysates of 293 cells transfected as indicated were analyzed by immunoblotting (IB) using either antibodies specific for the HA epitope tag or antibodies against the phosphorylated serine 63 or serine 73 of c-Jun. Note that both Gal–Jun1–256 and c-Jun are HA-tagged. Arrows demarcate hypo- and hyperphosphorylated forms of GAL–Jun1–256; diamonds indicate endogenous hypo- (open diamonds) and hyperphosphorylated c-Jun (black diamonds). (E) The δ-domain of c-Jun stabilizes the interaction with the repressor. 293 cells with a stably integrated 5× UAS luciferase reporter were transiently transfected with expression constructs encoding GAL–Jun or a mutant, GAL–Junδ–, which lacks the δ- domain, as activator, together with an expression plasmid encoding either full-length c-Jun or a δ-domain deletion mutant, c-Junδ– as competitor. Activities were determined as in (A). Percent activation compared with the activity of GAL–Jun + c-Jun which was set to 100% is shown (means ± SD of 3–5 independent experiments).
To further corroborate the model that phosphorylation weakens the repressor/c-Jun interaction, we used a non-phoshorylatable activator (GAL–Jun1–256Ala) and co- expressed it, in the absence or presence of JNK signaling, with wild-type c-Jun as a competitor (Figure 3B, lower part). Without JNK signaling the wild-type-Jun competitor causes increased transactivation by GAL–Jun1–256Ala. However, increased phosphorylation of the competitor (+ΔMEKK) abolished this effect, presumably because it no longer efficiently titrates the repressor. The conclusion that the negative effect of JNK on repressor binding and titration is due to the phosphorylation of the competitor is shown when c-JunAla is used as a competitor. In this experiment the inhibition of the activator can also be relieved under conditions of active JNK signaling.
These experiments show that after removal of the repressor function even non-phosphorylatable c-Jun can efficiently enhance transcription. Accordingly, in a c-Jun mutant that cannot bind the repressor or essential components of it (Figure 3C, GAL–Jun1–101) all phosphorylation sites can be replaced by alanines without affecting its activity (Figure 3C, GAL–Jun1–101Ala, AA). Once c-Jun is de-repressed, either by deleting the repressor-binding domain or by sequestering the repressor activity, it does not have to be phosphorylated to transactivate and, presumably, to interact with co-activators. Consistently, although GAL–Jun efficiently enhances transcription when the inhibitor is dissociated by co-expressed c-Jun, its phosphorylation does not increase under these conditions (Figure 3D).
The above experiments show that phosphorylation of c-Jun regulates repressor binding and not vice versa. Interestingly, whereas low amounts of transfected GAL–Jun1–256 or GAL–Jun1–256Ala expression plasmid do not activate the reporter in comparison to GAL-DBD alone, higher amounts do this even in the absence of JNK signaling (data not shown). Presumably under these conditions the concentration of activator exceeds that of the repressor, precluding efficient inhibition. In conclusion, we have identified a plausible mechanism for signal-dependent enhancement of c-Jun transactivation, which relies on phosphorylation-dependent de-repression of c-Jun.
JNK1 and JNK2 are not essential components of the repressor and are dispensable for c-Jun to activate transcription
It has previously been reported that the ε-domain is essential for binding a repressor activity, and that the δ-domain stabilizes this interaction (Baichwal and Tjian, 1990; Baichwal et al., 1992). The δ-domain serves as a docking site for a stable interaction between c-Jun and JNK, which can be detected even in the absence of signaling (Dai et al., 1995; C.Weiss, unpublished data). For these reasons, it has been suggested that JNK in its inactive form might constitute the repressor or contribute to repressor function (Dai et al., 1995; Vogt, 2001). This hypothesis gained credibility with the finding that the yeast MAPK Kss1 in the inactive state can bind to and thereby inhibit its target transcription factor STE12 (Madhani et al., 1997). In agreement with earlier data, we find that a mutant of c-Jun that lacks the δ-domain can compete less efficiently for the repressor activity than wild-type c-Jun. Conversely, a δ-domain deletion mutant when it is acting as a transactivator, is more easily relieved of repression (Figure 3E).
To test the role of JNK in c-Jun repression directly, we performed experiments in JNK-deficient 3T3 cell lines, which were derived from mice in which the genes encoding both JNK1 and JNK2 have been disrupted (Ouwens et al., 2002). In such jnk1–/–, jnk2–/– cells c-Jun phosphorylation in response to inducers of JNK signaling is completely absent (Tournier et al., 2000 and data not shown). The presence of the repressor activity in wild-type 3T3 cells was evident because increasing amounts of a wild-type c-Jun competitor enhanced the activity of the GAL–Jun activator as shown above for 293 cells. Strikingly, a similar result was obtained in jnk1–/–, jnk2–/– double knock-out cells, indicating that these JNK-deficient cells have repressor activity and demonstrating that JNK is not required for this function (Figure 4A).

Fig. 4. JNK is not the repressor and is dispensable for transcription activation by c-Jun. (A) wt-3T3 or jnk1–/–, jnk2–/– 3T3 cells were transiently transfected with a 5× UAS luciferase reporter and expression vectors for activator and competitor proteins as indicated. Fold activation compared with GAL–Jun1–256 (activity set to 1, means ± SD of three independent experiments) is shown. (B) Phosphorylation of c-Jun by JNK weakens the interaction with the inhibitor. Left panel: wt-3T3 or jnk1–/–, jnk2–/– 3T3 cells were transiently transfected with a 5× UAS luciferase reporter and the GAL–Jun1–256Ala expression construct as activator, together with full-length c-Jun expression plasmid as competitor. Where indicated, ΔMEKK, was co-transfected. Activities were determined as in (A). The relative reporter gene activities (means ± SD of three independent experiments) are shown. The activity of GAL–Jun1–256Ala in the presence of c-Jun competitor was set to 100%. Right panel: model to explain the results shown in the graph on the left (for further details see text). (C) The δ-domain of c-Jun stabilizes the interaction with the repressor in the absence of JNK. wt-3T3 or jnk1–/–, jnk2–/–3T3 cells were transiently transfected with a 5× UAS luciferase reporter and expression constructs as indicated. Activities were determined as in (A). The % activation compared with that mediated by GAL–Jun + c-Jun, which was set to 100%, is shown (means ± SD of three independent experiments).
Once c-Jun is de-repressed by sequestration of the repressor or limiting components of a larger repressor complex, GAL–Jun1–256 activated transcription in wild-type and JNK-deficient cells, further supporting the interpretation that JNK binding and/or phosphorylation is not essentially required for the productive interaction of positively acting co-factors with c-Jun. Thus, the main function of the kinase appears to be the signal-dependent removal of repressor activity. To confirm this model, we investigated whether or not the interaction between c-Jun and repressor can still be destabilized by JNK signaling in the knock-out cells. As shown above (Figure 3B, lower part), the ability of over-expressed c-Jun to sequester the repressor can be suppressed by stimulation of the JNK pathway in wild-type 3T3 cells. This inhibition of repressor titration was indeed mediated via JNK, since it is almost completely abolished in jnk1–/–, jnk2–/– cells (Figure 4B).
Although JNK is not an essential component of the repressor, it might mediate the stabilizing effect of the δ-domain on the interaction of repressor with c-Jun. To test this idea, we performed competition experiments in either wild-type or jnk1–/–, jnk2–/– cells. As observed in 293 cells (Figure 3C), the δ-domain stabilized the interaction with the repressor in 3T3 wild-type and also in jnk1–/–, jnk2–/– cells (Figure 4C). This finding suggests that the δ-domain facilitates repressor binding independently of JNK.
HDAC3 can bind to c-Jun and functionally interacts with the ε-domain to suppress the transcriptional activity of c-Jun
What could the molecular nature of the c-Jun repressor be? Transcriptional repressor complexes often associate with HDACs. In mammalian cells three different classes of HDACs have been characterized. Class I includes HDACs 1, 2, 3 and 8; class II HDACs 4–7, 9 and 10. NAD-dependent HDACs are grouped in class III (Khochbin et al., 2001). Because class I HDACs are ubiquitously expressed, whereas class II are mostly tissue specific, we concentrated on the class I enzymes HDAC1 and HDAC3 and examined if they might repress transcription activation by c-Jun. As shown above, co-expression of GAL– Jun1–256, and full-length c-Jun as a competitor efficiently activated reporter gene expression (Figure 5A, left panel). Under conditions where the activity of Gal–Jun1–256 is de-repressed by the presence of a c-Jun competitor, ectopic expression of HDAC3, but not of the closely related HDAC1, can restore the repressed state. The observation that HDAC3 expression can reverse the effect of repressor titration suggests that this enzyme is a part of the repressor.

Fig. 5. HDAC3 binds to c-Jun and represses transcription activation. (A) HDAC3 but not HDAC1 suppresses c-Jun activity dependent on the ε- domain. As described in Figure 2B, cells were transfected with the indicated expression constructs. UAS reporter activation mediated by GAL–Jun + c-Jun was measured in the absence and presence of co-expressed HDAC1 or 3. The activation compared with that mediated by GAL–Jun + c-Jun, which was set to 100%, is shown (means ± SD of 3–4 independent experiments). Lysates of transfected cells were analyzed by immunoblotting using either antibodies specific for the Flag or HA epitope (lower panel). (B) HDAC3 binds to the N-terminal region (aa 1–101) of c-Jun. 293 cells were transfected with expression constructs either coding for full-length c-Jun, HDAC3 or empty expression vector (left panel) or coding for HDAC3 and various c-Jun deletions (right panel). Lysates were subjected to immunoprecipitation (IP) and subsequent immunoblot analysis (IB) using the indicated antibodies. (C) HDAC3 suppresses c-Jun dependent on its ε-domain. UAS-reporter activation mediated by GAL–Junε– + c-Jun (left panel) was quantitated in the presence or absence of co-expressed HDAC3 as described in (A). (D) Loss-of-HDAC3-function releases c-Jun from repression. Expression constructs either coding for GAL–Jun or GAL–Junε– were co-transfected with empty or HDAC3 siRNA expression vectors. Reporter activation was measured three days after transfection. The fold activation compared with that mediated by GAL–Jun which was set to 1, is shown (average ± average deviation of two independent experiments). (E) Expression of HDAC3 siRNA efficiently depletes endogenous HDAC3 protein levels in 293 cells. Lysates of transfected cells were analysed three days after transfection by immunoblotting using an anti-HDAC3 antibody.
To test whether c-Jun might physically associate with HDAC3, we performed co-immunoprecipitation experiments on extracts from transiently transfected 293 cells. Hemagglutinin (HA)-tagged c-Jun was specifically co-immunoprecipitated with Flag-tagged HDAC3 (Figure 5B, left panel). To identify the HDAC3 interaction domain of c-Jun, a series of N- and C-terminal c-Jun truncation mutants was expressed and assayed for association with HDAC3 (Figure 5B, right panel). Interestingly, a c-Jun mutant which lacks the dimerization and DBD (HA- c-Jun1–256) could still interact with HDAC3. This result demonstrates that HDAC3 interacts specifically with c-Jun and does not require the interaction with other AP-1 family members. Furthermore, the N-terminal fragments HA-c-Jun1–145 as well as HA-c-Jun1–101 associated with HDAC3, suggesting that the ε-domain is not essential for HDAC3–c-Jun complex formation. Consistent with this, HDAC3 can also bind to a c-Jun mutant in which the ε-domain was specifically deleted (HA-c-Jun1–256ε–). In conclusion, the first 101 amino acids of c-Jun are required and sufficient to mediate interaction with HDAC3 (see also Figure 5B, schematic diagram).
Based on the functional assays and interaction studies shown above, we surmise that HDAC3 is a component of the c-Jun repressor. However, it does not require the ε-domain for binding and thus cannot by itself represent the titratable repressor moiety. This suggests that HDAC3 might need to interact with other components of the repressor complex bound to the ε-domain in order to be active. Consistently, HDAC3 is known to operate in large protein complexes and requires interaction with co-factors, such as NCOR and SMRT, for full HDAC activity (Guenther et al., 2001; Zhang et al., 2002). To test whether such a scenario might apply in the case of c-Jun, we examined whether HDAC3 requires the ε-domain to inhibit transcriptional activity. Increasing amounts of HDAC3 were co-expressed with a Gal–Jun mutant lacking the ε-domain (Figure 5C). Monitoring Gal4 reporter activation indicated that in contrast to the repression of Gal–Jun1–256 observed upon HDAC3 co-expression (Figure 5A, right panel), there is no suppression of the mutant lacking the ε-domain. Therefore, while HDAC3 binds to c-Jun independently of the ε-domain, it requires this domain to repress transcription activation by c-Jun. In addition to the ε-domain the δ-domain functionally interacts with the repressor complex by stabilizing its interaction with c-Jun (Figure 3C). Therefore, we asked whether HDAC3 is involved in this process. In the absence of the δ-domain c-Jun still interacts with and is suppressed by HDAC3 (data not shown). Thus, in contrast to the ε-domain, the supportive function of the δ-domain in c-Jun repression is independent of HDAC3 and presumably relies on its interplay with additional repressor components.
To directly examine the contribution of endogenous HDAC3 to c-Jun repression, we performed loss-of-function studies. In order to decrease endogenous HDAC3 protein levels, we constructed a vector directing the expression of a small double stranded inhibitory RNA (siRNA) designed to target the cellular HDAC3-encoding mRNA. Transient transfection of this vector lead to an efficient depletion of HDAC3 levels in 293 cells (Figure 5E). Concomitantly, expression of HDAC3 siRNA almost completely abrogated transcriptional repression of co-expressed Gal–Jun1–256 resulting in a transcriptional activity comparable to the activity of the ε-domain deletion mutant (Figure 5D, left panel). Furthermore, HDAC3 siRNA expression did not increase transcription activation by Gal–Jun1–256 lacking the ε-domain (Figure 5D, right panel). This result along with the gain-of-function experiments described above confirms that HDAC3 is a crucial functional component of the c-Jun repressor complex. HDAC3 presumably acts in conjunction with one or more ε-domain-bound factors to suppress the transcriptional activity of c-Jun.
Discussion
The events leading from c-Jun phosphorylation to enhanced target gene transcription are poorly understood (Davis, 2000). Here we propose a model in which the primary control of the transactivation potential of c-Jun is mediated by a repressor complex that physically interacts with the N-terminal region of c-Jun and is dissociated by JNK-mediated phosphorylation.
Transcription activation by de-repression, as described here for JNK–Jun, is not uncommon in eukaryotic gene regulation. Similar principles are well described in the cases of NF-κB, nuclear receptors and numerous other transcription factors. Repression as a ground state might help to avoid inadvertent or spurious activation of target genes, which, in the case of c-Jun, could cause inappropriate apoptosis or contribute to cellular transformation. Activation by de-repression might also apply to other transcription factors of the AP-1 family. Sequence alignments of the various Jun protein members reveal a high similarity of c-Jun and Jun D, but less pronounced in the case of Jun B, within the ε-domain region. Even among different species such as Drosophila, Xenopus, rat, mouse and human the ε-domain of c-Jun homologues is highly conserved.
Interaction assays as well as gain- and loss-of-function studies identified HDAC3 as a repressor of c-Jun. The function of HDAC3 as a negative transcriptional co-factor is best described in the context of nuclear receptors such as the thyroid hormone or retinoic acid receptors (Glass and Rosenfeld, 2000). In the absence of ligand these nuclear receptors recruit a multisubunit repressor complex consisting of HDAC3 together with accessory proteins such as TBL, GSP2 and the co-repressors NCOR or SMRT (Zhang et al., 2002). HDAC3 is active only in the context of this multisubunit complex and does not display discernable enzymatic activity in isolation (Guenther et al., 2001). Repressor association results in target gene repression that can be relieved by ligand binding of the receptors and the resulting exchange of repressor with co-activator complexes.
Based on the data presented here, we propose a novel model of c-Jun activation by JNK that might provide a general concept of transcription factor activation by phosphorylation. Similar to NR activation through ligand binding, signal-dependent phosphorylation of c-Jun by JNK causes the dissociation of a suppressor complex that assembles on c-Jun and requires the ε-domain for this interaction. An essential component of this repressor complex is HDAC3. Even though HDAC3 can bind to c-Jun independently of the ε-domain, it requires the presence of the ε-domain to suppress the activity of c-Jun. An attractive model which combines these data would suggest that phosphorylation antagonizes the binding of a co-factor to the ε-domain, possibly related to NCOR or SMRT, which is essential to activate HDAC3 repressor function.
c-Jun can be relieved of repression either by signal-dependent, JNK-mediated phosphorylation or, signal independently, by titrating out one or more limiting components of the repressor complex through increased c-Jun protein levels. Both mechanisms are not mutually exclusive and might in principle operate in conjunction to increase transcription activation by c-Jun. Consistently, JNK signaling does not only increase phosphorylation but also the protein levels of c-Jun thus counteracting repression in two ways. Gene activation upon dissociation of the inhibitor, which presumably involves interactions with positively acting components of the transcription machinery, can occur in the absence of functional JNK as illustrated by experiments in knock-out cell lines. Such protein–protein interactions can evidently occur in a phosphorylation-independent manner. Consistent with this model, the interaction of c-Jun with the co-activator CBP and with RHII/Gu-RNA-helicase, which we recently have identified as a novel co-factor of c-Jun, does not require c-Jun phosphorylation (Bannister et al., 1995; Westermarck et al., 2002).
Interestingly, c-Jun-deficient mice, which die early in development due to hepatic failure, are rescued by expression of a non-phosphorylatable mutant of c-Jun (Behrens et al., 1999). Previous studies on c-Jun suppression suggested a cell type specificity of repressor activity, which for example was not found in the human liver cell line HepG2 (Baichwal and Tjian, 1990; Baichwal et al., 1992). Thus, lack of repressor activity in mouse liver might even allow transcription activation by a non-phosphorylatable mutant of c-Jun. Therefore, the puzzling observation that c-Jun phosphorylation seems to be non-essential for liver development might be explained by our findings that also non-phosphorylated c-Jun can activate transcription once repressor binding is abrogated.
The model of Jun activation by repressor displacement also offers a satisfying model to explain the increased transforming potential of v-Jun compared with c-Jun. The decisive mutation that turns v-jun into an oncogene during the course of retroviral transduction is the deletion of the δ-domain, the JNK docking site (Bos et al., 1990). This was counter-intuitive, as in consequence v-Jun is not efficiently phosphorylated by JNK, a step that has until now been thought to be required for target gene activation. Our data show that the δ-domain deletion, resembling v-Jun, destabilizes the interaction with the repressor complex and thus could explain why v-Jun may act as an activated, signal-independent allele of c-Jun.
Materials and methods
Plasmids and construction of expression vectors
Expression vectors for HA-tagged c-Junwt, different HA-tagged c-Jun phosphorylation mutants, His6-tagged c-Jun-δ– and constitutively active ΔMEKK1 have been described (Westermarck et al., 2002). Mammalian expression constructs for different GAL–c-Jun mutants were constructed by fusing the appropriate PCR-generated fragments of c-Jun to the Gal4 DBD and an HA epitope. These fusions were cloned into an expression vector driven by a human ubiquitin C promoter (Schorpp et al., 1996) which we found, in contrast to many other promoters tested, not to be influenced by additional co-expression of ΔMEKK1 or c-Jun. Deletion constructs encoding different truncations of c-Jun lacking the bZIP region and HA-c-Jun1–256ε– were subcloned into pcDNA3.1 (Invitrogen) and fused with an HA-tag and a SV40 NLS. Expression and phosphorylation of the different c-Jun mutants were analyzed by western blotting using antibodies specific for the GAL-DBD (Santa Cruz), the HA epitope (Santa Cruz) or phosphorylated c-Jun (New England Biolabs). To generate pBS/U6/SiHDAC3, first, one 21-nucleotide double strand complementary to the nucleotides 1194–1214 of human HDAC3 was inserted into the blunted ApaI and HindIII sites of pBS/U6 (Sui et al., 2002). The second oligo which has the inverted sequence of the first oligo was cloned into the HindIII and EcoRI site.
Reporter gene assays and transient transfection
293 cells were transfected with 5 µg of reporter plasmid pf2Luc (Stratagene) together with 0.5 µg pSVneo. After selection of G418 resistant clones, individual lines were transiently transfected with 10 ng GAL–Jun along with 25 ng CMVΔMEKK1 or empty expression vector for functional characterization. One of three similarly responding clones, 293 clone 6, was used in further experiments. 293 clone 6 cells were transiently transfected with 10 ng expression vector encoding different GAL–Jun mutants either together with 25 ng CMVΔMEKK1 or with empty expression vector. For competition assays, additionally 250 ng CMV HA c-Junwt, CMV HA c-JunAla, CMV HIS c-Jun–δ or empty expression vector were co-transfected. For HDAC repression assays 0.5, 1.0 and 10-fold excess of HDAC1 or HDAC3 expression plasmids (Yang et al., 2002) were co-transfected with CMV HA c-Jun. For HDAC3 RNAi experiments 10 µg pBS/U6/SiHDAC3 were incubated with 106 cells in 300 µl Dulbecco’s modified Eagle’s medium/10% fetal calf serum and electroporated at 200 V. Cell lysates were prepared three days post electroporation and analyzed by western blotting using anti-HDAC3 (Wen et al., 2000) and anti-actin antibodies (Sigma). For reporter gene assays in 293 cells 10 ng GAL–Jun were co-transfected with 1 µg pBS/U6/SiHDAC3 and luciferase activity was measured 3 days after transfection.
Activation of the human collagenase I promoter in 293 cells was analyzed by transfecting 1 µg of the collagenase reporter construct Coll-Luc (region –517 to +73, kindly provided by P.Angel) together with 100 ng CMV HA-c-Jun or empty expression vector.
For competition assays, 100 ng of GAL–Jun were co-transfected in addition. 3T3 cells were transiently transfected with the same amount of expression plasmids and 1 µg of pf2Luc using the FuGene transfection reagent (Roche). Transfection efficiency was controlled by the inclusion of 10 ng (293 cells) or 100 ng (3T3 cells) ubiquitin promoter-driven Renilla luciferase reporter construct as an internal standard (Westermarck et al., 2002).
Co-immunoprecipitation assays
293 cells were transfected with 5 µg expression plasmids coding for Flag-tagged HDAC3 and/or HA-tagged c-Jun and were lysed after 24 h in 1 ml of lysis buffer [50 mM HEPES–KOH pH 7.4, 50 mM NaCl, 1% Tween 20, 2.5 mM EGTA, 1 mM EDTA, 1 mM NaF, 10 mM β-glycerolphosphate, 0.1 mM Na3VO4, 1 mM phenylmethylsulfonyl fluoride (PMSF), protease inhibitors (Roche), 1 mM dithiothreitol (DTT)]. After sonification, lysates were cleared by centrifugation (100 000 g, 20 min, 4°C) and the supernatants were incubated overnight at 4°C either with 15 µl of HA-epitope-specific (M20, Santa Cruz) or Flag-tag-specific antibody (Sigma) conjugated to an agarose column. Sedimented beads were washed five times with 1 ml IP-washing buffer [50 mM HEPES–KOH pH 7.4, 450 mM NaCl, 1% Tween 20, 2.5 mM EGTA, 1 mM EDTA, 1 mM NaF, 10 mM β-glycerolphosphate, 0.1 mM Na3VO4, 1 mM PMSF, protease inhibitors (Roche), 1 mM DTT], resuspended in 100 µl sample buffer (2×) and eluted proteins were analyzed by western blotting using anti-HDAC3 (Santa Cruz), anti-c-Jun (H-79, Santa Cruz) and anti-HA (Roche) antibodies.
Acknowledgments
Acknowledgements
The authors thank Catherine Ovitt, Jiyong Zhao, Jean Pierre David, Jukka Westermarck, Vijay R.Baichwal and Hartmut Land for critical reading of the manuscript. This work was supported by an EMBO long-term fellowship (C.W.).
References
- Baichwal V.R. and Tjian,R. (1990) Control of c-Jun activity by interaction of a cell-specific inhibitor with regulatory domain delta: differences between v- and c-Jun. Cell, 63, 815–825. [DOI] [PubMed] [Google Scholar]
- Baichwal V.R., Park,A. and Tjian,R. (1992) The cell-type-specific activator region of c-Jun juxtaposes constitutive and negatively regulated domains. Genes Dev., 6, 1493–1502. [DOI] [PubMed] [Google Scholar]
- Bannister A.J., Oehler,T., Wilhelm,D., Angel,P. and Kouzarides,T. (1995) Stimulation of c-Jun activity by CBP: c-Jun residues Ser63/73 are required for CBP induced stimulation in vivo and CBP binding in vitro. Oncogene, 11, 2509–2514. [PubMed] [Google Scholar]
- Behrens A., Sibilia,M. and Wagner,E.F. (1999) Amino-terminal phosphorylation of c-Jun regulates stress-induced apoptosis and cellular proliferation. Nat. Genet., 21, 326–329. [DOI] [PubMed] [Google Scholar]
- Binetruy B., Smeal,T. and Karin,M. (1991) Ha-Ras augments c-Jun activity and stimulates phosphorylation of its activation domain. Nature, 351, 122–127. [DOI] [PubMed] [Google Scholar]
- Bos T.J., Monteclaro,F.S., Mitsunobu,F., Ball,A.R.,Jr, Chang,C.H., Nishimura,T. and Vogt,P.K. (1990) Efficient transformation of chicken embryo fibroblasts by c-Jun requires structural modification in coding and noncoding sequences. Genes Dev., 4, 1677–1687. [DOI] [PubMed] [Google Scholar]
- Chang L. and Karin,M. (2001) Mammalian MAP kinase signalling cascades. Nature, 410, 37–40. [DOI] [PubMed] [Google Scholar]
- Dai T., Rubie,E., Franklin,C.C., Kraft,A., Gillespie,D.A., Avruch,J., Kyriakis,J.M. and Woodgett,J.R. (1995) Stress-activated protein kinases bind directly to the delta domain of c-Jun in resting cells: implications for repression of c-Jun function. Oncogene, 10, 849–855. [PubMed] [Google Scholar]
- Davis R.J. (2000) Signal transduction by the JNK group of MAP kinases. Cell, 103, 239–252. [DOI] [PubMed] [Google Scholar]
- Derijard B., Hibi,M., Wu,I.H., Barrett,T., Su,B., Deng,T., Karin,M. and Davis,R.J. (1994) JNK1: a protein kinase stimulated by UV light and Ha-Ras that binds and phosphorylates the c-Jun activation domain. Cell, 76, 1025–1037. [DOI] [PubMed] [Google Scholar]
- Eferl R., Ricci,R., Kenner,L., Zenz,R., David,J.P., Rath,M. and Wagner,E.F. (2003) Liver tumor development. c-Jun antagonizes the proapoptotic activity of p53. Cell, 112, 181–192. [DOI] [PubMed] [Google Scholar]
- Glass C.K. and Rosenfeld,M.G. (2000) The coregulator exchange in transcriptional functions of nuclear receptors. Genes Dev., 14, 121–141. [PubMed] [Google Scholar]
- Guenther M.G., Barak,O. and Lazar,M.A. (2001) The SMRT and N-CoR corepressors are activating cofactors for histone deacetylase 3. Mol. Cell. Biol., 21, 6091–6101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibi M., Lin,A., Smeal,T., Minden,A. and Karin,M. (1993) Identification of an oncoprotein- and UV-responsive protein kinase that binds and potentiates the c-Jun activation domain. Genes Dev., 7, 2135–2148. [DOI] [PubMed] [Google Scholar]
- Janknecht R., Wells,N.J. and Hunter,T. (1998) TGF-beta-stimulated cooperation of smad proteins with the coactivators CBP/p300. Genes Dev., 12, 2114–2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jochum W., Passegue,E. and Wagner,E.F. (2001) AP-1 in mouse development and tumorigenesis. Oncogene, 20, 2401–2412. [DOI] [PubMed] [Google Scholar]
- Kallunki T., Deng,T., Hibi,M. and Karin,M. (1996) c-Jun can recruit JNK to phosphorylate dimerization partners via specific docking interactions. Cell, 87, 929–939. [DOI] [PubMed] [Google Scholar]
- Khochbin S., Verdel,A., Lemercier,C. and Seigneurin-Berny,D. (2001) Functional significance of histone deacetylase diversity. Curr. Opin. Genet. Dev., 11, 162–166. [DOI] [PubMed] [Google Scholar]
- Leppä S. and Bohmann,D. (1999) Diverse functions of JNK signaling and c-Jun in stress response and apoptosis. Oncogene, 18, 6158–6162. [DOI] [PubMed] [Google Scholar]
- Madhani H.D., Styles,C.A. and Fink,G.R. (1997) MAP kinases with distinct inhibitory functions impart signaling specificity during yeast differentiation. Cell, 91, 673–684. [DOI] [PubMed] [Google Scholar]
- Mayr B. and Montminy,M. (2001) Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat. Rev. Mol. Cell. Biol., 2, 599–609. [DOI] [PubMed] [Google Scholar]
- Ouwens D.M. et al. (2002) Growth factors can activate ATF2 via a two-step mechanism: phosphorylation of Thr71 through the Ras–MEK–ERK pathway and of Thr69 through RalGDS–Src-p38. EMBO J., 21, 3782–3793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papavassiliou A.G., Treier,M. and Bohmann,D. (1995) Intramolecular signal transduction in c-Jun. EMBO J., 14, 2014–2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schorpp M., Jager,R., Schellander,K., Schenkel,J., Wagner,E.F., Weiher,H. and Angel,P. (1996) The human ubiquitin C promoter directs high ubiquitous expression of transgenes in mice. Nucleic Acids Res., 24, 1787–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smeal T., Binetruy,B., Mercola,D.A., Birrer,M. and Karin,M. (1991) Oncogenic and transcriptional cooperation with Ha-Ras requires phosphorylation of c-Jun on serines 63 and 73. Nature, 354, 494–496. [DOI] [PubMed] [Google Scholar]
- Sui G., Soohoo,C., Affar el,B., Gay,F., Shi,Y. and Forrester,W.C. (2002) A DNA vector-based RNAi technology to suppress gene expression in mammalian cells. Proc. Natl Acad. Sci. USA, 99, 5515–5520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tournier C. et al. (2000) Requirement of JNK for stress-induced activation of the cytochrome c-mediated death pathway. Science, 288, 870–874. [DOI] [PubMed] [Google Scholar]
- Treisman R. (1996) Regulation of transcription by MAP kinase cascades. Curr. Opin. Cell Biol., 8, 205–215. [DOI] [PubMed] [Google Scholar]
- Vogt P.K. (2001) Jun, the oncoprotein. Oncogene, 20, 2365–2377. [DOI] [PubMed] [Google Scholar]
- Wen Y.D., Perissi,V., Staszewski,L.M., Yang,W.M., Krones,A., Glass,C.K., Rosenfeld,M.G. and Seto,E. (2000) The histone deacetylase-3 complex contains nuclear receptor corepressors. Proc. Natl Acad. Sci. USA, 97, 7202–7207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westermarck J. et al. (2002) The DEXD/H-box RNA helicase RHII/Gu is a co-factor for c-Jun-activated transcription. EMBO J., 21, 451–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W.M., Tsai,S.C., Wen,Y.D., Fejer,G. and Seto,E. (2002) Functional domains of histone deacetylase-3. J. Biol. Chem., 277, 9447–9454. [DOI] [PubMed] [Google Scholar]
- Zhang J., Kalkum,M., Chait,B.T. and Roeder,R.G. (2002) The N-CoR–HDAC3 nuclear receptor corepressor complex inhibits the JNK pathway through the integral subunit GPS2. Mol. Cell, 9, 611–623. [DOI] [PubMed] [Google Scholar]