

Abstract
To gain a broader insight into the role of Bcl-2 proteins in apoptosis induced after mitotic arrest, we investigated the subcellular location, oligomeric structure, and protein interactions of Bax, Bcl-2, and Bcl-xL in vinblastine-treated KB-3 cells. Vinblastine induced the translocation of Bax from the cytosol to the mitochondria, which was accompanied by conformational activation and oligomerization of Bax. Bcl-2 was located in the mitochondria, underwent multisite phosphorylation after vinblastine treatment, and was strictly monomeric under all conditions. In contrast, in control cells, Bcl-xL existed in both monomeric (30 kDa) and oligomeric (150 kDa) forms. Treatment with agents that induced Bcl-xL phosphorylation (microtubule inhibitors) caused loss of the 150-kDa form, but this species was unaffected by apoptotic stimuli that did not stimulate phosphorylation. Vinblastine also promoted Bax activation and Bax oligomerization in HCT116 colon cancer cells. Both wild-type and Bax-deficient HCT116 cells expressed the 150-kDa form of Bcl-xL, which was depleted similarly in both cell lines upon vinblastine treatment. Co-immunoprecipitation studies revealed that in untreated KB-3 cells inactive cytosolic Bax interacted with Bcl-xL, whereas in vinblastine-treated cells, activated mitochondrial Bax did not interact with Bcl-xL. Interaction of Bcl-2 with Bax was not observed under any condition. Overexpression of Bcl-xL inhibited vinblastine-induced Bax activation and Bax dimerization and in parallel inhibited apoptosis. The results indicate that vinblastine-induced apoptosis requires translocation, activation, and oligomerization of Bax and is associated with specific changes in the oligomeric properties of Bcl-xL, which occur independently of Bax.
The Bcl-2 family of proteins are key regulators of apoptosis. Anti-apoptotic members include Bcl-2, Bcl-xL, Mcl-1, A1, Nr-13, and others, and the pro-apoptotic members are represented by two subgroups: the multidomain or Bax subfamily (Bax, Bak, and Bok), which contain multiple BH domains, and the BH3-only subfamily (Bad, Bid, Bim, Noxa, Hrk, and others) (1–4). Bcl-2 proteins act in a hierarchy, with the BH3-only proteins acting as essential initiators of apoptosis (4). In healthy cells they are maintained in a latent state, and in response to apoptotic stimuli they become activated through a variety of mechanisms involving posttranslational modification or transcriptional activation. Activated BH3 proteins play a key role in the activation of multidomain Bax subfamily proteins. This leads to an increase in outer mitochondrial membrane permeability and the release of cytochrome c and other apoptogenic factors (1–4). Anti-apoptotic Bcl-2 proteins antagonize the function of the pro-apoptotic Bcl-2 proteins. Recent work has indicated that BH3-only proteins do not bind promiscuously to pro-survival members but rather bind more selectively, and, in addition to a role in neutralization of pro-survival members, they also bind and may thus directly activate Bax or Bak (4).
Bax and Bak play redundant functions and are required for mitochondrial dysfunction and apoptosis in response to death signals (5). Whereas Bak is mitochondrial, Bax undergoes conformational changes that promote its translocation from the cytosol to the mitochondria. Upon outer mitochondrial membrane association, further structural changes in Bax enable homo-oligomerization and/or interaction with other proteins to facilitate pore formation and the release of apoptosis promoting factors (1–5).
Microtubule inhibitors are widely used as anti-mitotic cancer chemotherapeutic drugs (6). At physiological concentrations, these agents have in common the ability to suppress the dynamic instability of spindle microtubules, leading to mitotic arrest and cell death by apoptosis (7). The mechanisms of apoptotic cell death resulting from mitotic arrest are complex and far from resolved (8–10). Bcl-2 proteins appear to play an important regulatory role. For example, one of the main characteristics of microtubule inhibitors is their ability to promote phosphorylation of anti-apoptotic Bcl-2 and Bcl-xL (11). However, the role of phosphorylation and the kinase(s) responsible remain obscure. Bax has been implicated in apoptosis induced by vinblastine and the nontaxane microtubule inhibitor epothilone (12, 13). In contrast, several studies have indicated that paclitaxel does not promote Bax mitochondrial translocation or Bax activation (14, 15), despite the fact that Bax expression is a predictor of paclitaxel sensitivity (16). These results suggest that factors in addition to or other than Bax activation are important for apoptosis induced by microtubule inhibitors. This notion is supported by other results indicating that Bax activation may be necessary but is often insufficient for apoptosis induction (17).
In this study we have undertaken an examination of the subcellular location, oligomeric structure, and protein-protein interactions of Bcl-2, Bcl-xL, and Bax in KB-3 cells treated with vinblastine. We show that vinblastine promotes specific changes in the subcellular location, activation status, and oligomeric structure of these key proteins. In particular, we show that although Bcl-2 is monomeric, Bcl-xL also exists in high molecular mass complexes, with discrete changes in their relative abundance evident upon treatment with vinblastine and other microtubule inhibitors. In addition, in response to vinblastine treatment, Bax undergoes mitochondrial translocation accompanied by activation and dimerization, and we provide evidence that these events are necessary for vinblastine-induced apoptosis. These studies reveal key distal steps in apoptosis induction by this important group of chemotherapeutic agents.
EXPERIMENTAL PROCEDURES
Materials
Antibodies to caspase 3 (sc-7148), Bcl-2 (sc-509), and actin (sc-1616) were obtained from Santa Cruz; antibodies to Bcl-xL (catalog number 2762) and Bax (catalog number 2772) were from Cell Signaling; antibody to cytochrome c (catalog number 556433) was from Pharmingen; and antibody to glyceraldehyde-3-phosphate dehydrogenase (GAPDH)3 (catalog number 4300) was from Ambion. 6A7 active Bax antibody was from Axxora. Anti-HA-peroxidase linked antibody (catalog number 2013819) and the DNA fragmentation apoptosis kit were obtained from Roche Applied Science. Mitotracker Red and Alexa-Fluor goat anti-rabbit IgG were from Molecular Probes. Lipo-fectamine reagents were obtained from Invitrogen. Dithiobis(succinimidyl propionate) (DSP) was from Pierce. Vinblastine, paclitaxel, vincristine, doxorubicin, etoposide, and other chemicals, unless otherwise stated, were obtained from Sigma.
Cell Culture and Transfection
The KB-3 human carcinoma cell line was maintained in monolayer culture at 37 °C and 5% CO2 in Dulbecco’s modified Eagle’s medium, supplemented with 10% fetal bovine serum, 2 mM L-glutamine, 50 units/ml penicillin, and 50 μg/ml streptomycin. Cells stably overexpressing HA-Bcl-xL (human) were prepared by transfecting KB-3 cells, at ~70% confluence, with 10 μg of plasmid DNA (HA-Bcl-xL-pcDNA3) using Lipofectamine Plus reagent in serum-free Dulbecco’s modified Eagle’s medium. After 3 h, the transfection medium was replaced with Dulbecco’s modified Eagle’s medium containing 10% fetal bovine serum. After an additional 24 h, G418 was added to the medium to a final concentration of 1 mg/ml, and the cells were maintained for 2 weeks. Drug-resistant colonies were selected, expanded, and maintained in growth medium containing 0.4 mg/ml G418. Clones were screened for Bcl-xL and HA immunoreactivity by immunoblotting using specific antibodies. HCT116 parental and Bax knock-out cell lines were maintained in McCoy’s 5A medium with 10% fetal bovine serum (12).
Preparation of Cell Extracts and Subcellular Fractions
Whole cell extracts were prepared by suspending cells in 0.25 ml of lysis buffer (25 mM HEPES, pH 7.5, 0.2% SDS, 0.5% sodium deoxycholate, 5 mM EDTA, 5 mM dithiothreitol, 20 mM β-glycerophosphate, 1 mM Na3VO4, 50 mM NaF, 1% Triton X-100, 20 μg/ml aprotinin, 50 μg/ml leupeptin, 10 μM pepstatin, 1 μM okadaic acid, and 1 mM phenylmethylsulfonyl fluoride). After 15 min on ice, the extracts were sonicated (3 × 10 s), insoluble material was removed by centrifugation (15 min at 12,000 × g), and protein concentration in the supernatant was determined using the Bio-Rad protein assay.
Cytosolic and mitochondrial extracts were prepared using a fractionation kit from Active Motif (catalog number 40015) according to the manufacturer’s instructions. Briefly, 5 × 107 cells were washed with PBS twice, resuspended in 1 ml of ice-cold cytosolic buffer, incubated on ice for 15 min, homogenized with 30 strokes of a pestle homogenizer, and centrifuged at 800 × g for 20 min. The supernatant was then centrifuged at 10,000 × g for 20 min, with the supernatant retained as the cytosolic fraction and the pellet as the mitochondria-enriched fraction. The latter was resuspended in the complete mitochondrial buffer provided in the kit.
To examine oligomeric forms of Bcl-2 proteins, washed cell pellets were incubated in buffer A (10 mM HEPES, pH 7.4, 0.15 M NaCl, 1 mM EGTA, plus phosphatase and protease inhibitors, as above) containing 0.01% digitonin for 2 min at 4 °C and centrifuged at 15,000 × g. The pellet was extracted with buffer A containing 3% CHAPS for 45 min at 4 °C to release membrane- and organelle-bound proteins, which were isolated in the supernatant after centrifugation at 15,000 × g for 15 min. The samples were prepared for SDS-PAGE under either reduced or nonreduced conditions (with or without β-mercaptoethanol, respectively) with heating to 70 °C for 5 min. In some experiments, the cross-linking agent DSP was added to a final concentration of 1 mM after cell permeabilization. After 10 min at 4 °C, DSP was quenched with 0.1 volume of 2M Tris-HCl (pH 7.4). Quantitation of protein expression from immunoblots was performed with a MultiImager densitometry system (Bio-Rad) with background values subtracted in all cases.
Immunofluorescent Localization of Bax
KB-3 cells (1.5 × 105 in 1 ml of medium) were grown on 25-mm glass coverslips placed in each well of a 6-well tissue culture plate. Following treatment, mitochondria were stained for 30 min at 37 °C with Mitotracker Red CMXRos (Molecular Probes, Eugene, OR) diluted 1:1000 in growth media. The cells were then fixed and permeablized for 30 min at room temperature in fixing medium (3.7% paraformaldehyde, 0.18% Triton X-100 in PBS) and then blocked for 30 min in blocking buffer (5% bovine serum albumin, 0.5% Triton X-100 in PBS). The cells were then incubated for 1 h at room temperature with rabbit polyclonal anti-Bax primary antibody (Cell Signaling Technology, Beverly, MA), diluted 1:100 in 5% bovine serum albumin/PBS, and then washed with PBS (3 × 5 min). Alexa Fluor 488 goat anti-rabbit IgG (H+L) secondary antibody (Molecular Probes), diluted 1:50 in 5% bovine serum albumin/PBS, was added to cells for 1 h at room temperature, and the cells were washed with PBS (3 × 5 min). The coverslips were removed from the 6-well plates and mounted on glass microscope slides using 25 μl of Vectashield mounting medium (Vector Laboratories, Burlingame, CA). Immunofluorescent protein and mitochondria were visualized using 488- and 568-nm laser lines of a Zeiss LSM410 confocal microscope.
Immunoprecipitation
To evaluate Bax activation status, the cells were lysed in 0.5 ml of 10 mM HEPES (pH 7.4), 150 mM NaCl, 1% CHAPS, plus protease inhibitors (as above) for 30 min on ice. After brief sonication, the cell extract was centrifuged at 12,000 × g for 15 min, and 500 μg of cell lysate mixed with 2 μg of 6A7 monoclonal Bax antibody in 500 μl of lysis buffer overnight at 4 °C. The lysates were then incubated with 25 μl of protein G-agarose beads (Invitrogen) for 2 h at 4 °C. The beads were pelleted by centrifugation and washed three times with 0.2 ml of lysis buffer. The beads were resuspended in 20 μl of lysis buffer plus 5 μl of 5× SDS loading buffer and electrophoresed on 12% SDS-PAGE gold precast acrylamide gels (Cambrex, Walkersville, MD). Following electrophoresis, immunoblotting was performed using rabbit anti-Bax polyclonal antibody (Cell Signaling Technology, Beverly, MA) at 1:1000 dilution.
To examine Bcl-2 protein interactions by co-immunoprecipitation, the cells were lysed in 0.3 ml of 40 mM HEPES (pH 7.5), 0.12 M NaCl, 1% Triton X-100, 1 mM EDTA, containing phosphatase and protease inhibitors as above, and after 30 min on ice centrifuged at 9,200 × g for 20 min. The extract (1.5–2 mg) was precleared with anti-rabbit goat IgG agarose beads, according to the manufacturer’s directions (eBio-sciences), and to the supernatant was added 10 μg of rabbit polyclonal antibody (to either Bcl-2 or Bcl-xL). After mixing for 2 h, anti-rabbit goat IgG-agarose beads were added and mixed overnight. The immunoprecipitate was then washed five times in the following buffers: (a) TBS containing 0.05% Tween 20; (b) 50 mM HEPES (pH 7.5), 40 mM NaCl, 2 mM EDTA, 1% Triton X-100; (c) 50 mM HEPES (pH 7.5), 40 mM NaCl, 2 mM EDTA, 0.5% Triton X-100, 0.5M LiCl; (d) 50 mM HEPES (pH 7.5), 40 mM NaCl, 2 mM EDTA, 0.5M LiCl; and (e) 50 mM HEPES (pH 7.5), 150 mM NaCl. The immunoprecipitates were incubated in SDS-PAGE sample buffer for 1 h at 37 °C and resolved by 12.5% acrylamide SDS-PAGE and transferred to polyvinylidene difluoride membrane. Immunoblotting was performed with monoclonal mouse Bcl-2, Bcl-xL, or Bax antibody.
Two-dimensional Gel Electrophoresis
KB-3 cells (~109 cells, derived from two 150-mm plates) were washed in PBS and resuspended in 0.6 ml of osmotic lysis buffer (10 mM Tris-HCl, pH 7.6, 0.3% SDS). The suspension was passed through a 1-ml syringe fitted with a 25-gauge needle 10–15 times, incubated on ice for 30 min, and sonicated 3 × 10 s, and after determination of the protein concentration, the samples were diluted to 10 μg of protein/μl. The samples were diluted 1:1 with 60 mM Tris-HCl (pH 6.8), 5% SDS, 10% glycerol, 5% β-mercaptoethanol, heated to 95 °C for 5 min, frozen in liquid nitrogen, and stored at −80 °C. Two-dimensional gel electrophoresis was performed according to the method of O’Farrell (18) by Kendrick Labs, Inc. (Madison, WI). Isoelectric focusing was carried out in 2-mm glass tubes for 9600 volt-h. For the analysis of Bcl-2, 2% pH 3.5–10 ampholines were used, and for the analysis of Bcl-xL, a mixture of 1% pH 2.2–5 and 1% pH 4–6 ampholines was used. A surface pH electrode was used to determine the pH gradient of the isoelectric focusing tube gel. After equilibration in 62.5 mM Tris-HCl (pH 6.8), 10% glycerol, and 2.3% SDS, SDS-PAGE in the second dimension was performed using 10% acrylamide gels. After transfer to polyvinylidene difluoride membrane, immunoblotting was performed for Bcl-xL or Bcl-2. Molecular mass standards were catalase (60 kDa), actin (43 kDa), and carbonic anhydrase (29 kDa).
Apoptosis Assays
KB-3 cells were trypsinized following drug treatment and diluted to a concentration of 5 × 104 cells/ml for measurement of apoptosis using a cell death detection enzyme-linked immunosorbent assay kit (Roche Applied Science). This is a quantitative photometric immunoassay for the determination of cytoplasmic histone-associated oligonucleosomes generated during apoptosis. After dilution, the cells were centrifuged at 200 × g for 5 min, and the cell pellet was resuspended in 0.5 ml of incubation buffer and incubated at room temperature for 30 min. After centrifugation at 16,000 × g for 10 min, 0.4 ml of the supernatant was removed and diluted 1:10 in incubation buffer for analysis. The enzyme-linked immunosorbent assay plate was prepared according to the manufacturer’s instructions, and 0.1 ml of sample was added to appropriate wells and incubated at room temperature for 90 min. After conjugation and incubation with substrate solution, the plate was shaken on an orbital shaker at 250 rpm for ~15 min, and then the absorbance at 405 nm was determined using a Bio-Tek ELx800 microplate reader (Bio-Tek Instruments, Winooski, VT). Apoptosis was also evaluated by quantitation of sub-G1 DNA content after propidium iodide staining and flow cytometry, as described previously (19).
RESULTS
Subcellular Localization of Bcl-2 Proteins
Cytosolic and mitochondrial fractions were prepared from control and vinblastine-treated KB-3 cells to determine the subcellular location of Bax and to monitor any changes associated with drug treatment. The integrity of the fractions was demonstrated by immunoblotting for caspase 3, which was detected in the cytosolic fraction and not in the mitochondrial fraction, and for cytochrome c, which was detected in the mitochondrial fraction and not in the cytosolic fraction (Fig. 1A). Previous studies have shown in untreated KB-3 cells that Bcl-2 is present in the mitochondrial fraction and not in the cytosolic fraction and that Bcl-xL is present in both the mitochondrial and cytosolic fractions (19). The subcellular locations of Bcl-2 and Bcl-xL are unchanged after vinblastine treatment, although both proteins undergo gel mobility shifts caused by phosphorylation (19). Bax, on the other hand, was mainly present in the cytosol of control cells and underwent mitochondrial translocation in response to vinblastine treatment, first evident at 24 h and more prominent at later time points (Fig. 1B). Release of cytochrome c to the cytosol was evident with a similar time course (Fig. 1C). Actin served as a loading control (Fig. 1C). Previous studies have shown that under these conditions, apoptosis, as judged by caspase 3 activation, poly(ADP-ribose) polymerase cleavage, and DNA fragmentation, is evident beginning at 24 h of treatment and continuing through 48 h (19).
FIGURE 1. Vinblastine induces Bax mitochondrial translocation and cytochrome c release.
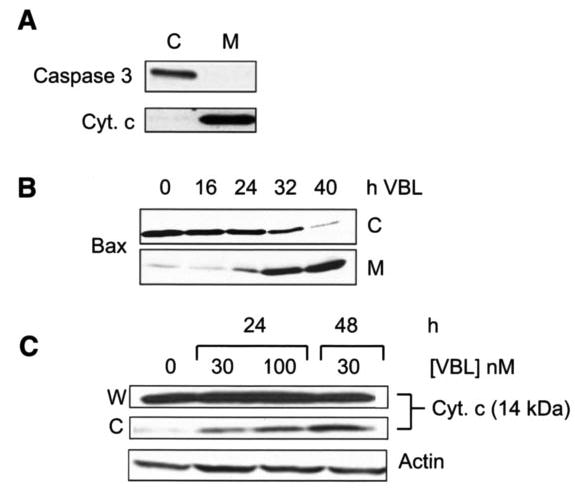
A, immunoblotting of caspase 3 and cytochrome c (Cyt. c) as markers for cytosolic (C) and mitochondrial (M) subcellular fractions from KB-3 cells. B, KB-3 cells were untreated or treated with 30 nM vinblastine for the times indicated, and cytosolic (C) and mitochondrial (M) fractions were prepared and subjected to immunoblotting for Bax. C, KB-3 cells were treated with vinblastine for the times and concentrations indicated, and whole cell extracts (W) and cytosolic fractions were subjected to immunoblotting for cytochrome c. The bottom panel shows immunoblotting for actin in whole cell extracts as a loading control.
To confirm vinblastine-induced mitochondrial translocation of Bax, control and vinblastine-treated cells were fixed and immunostained for Bax, as described under “Experimental Procedures.” The mitochondria were stained using Mitotracker Red. As shown in Fig. 2, in control untreated cells, Bax immunofluorescence, shown by the green signal, was distinct in location compared with the red mitochondrial signal. However, after vinblastine treatment, the green Bax signal coincided with the red mitochondrial signal, indicating and confirming Bax mitochondrial localization.
FIGURE 2. Immunofluorescent localization of Bax.
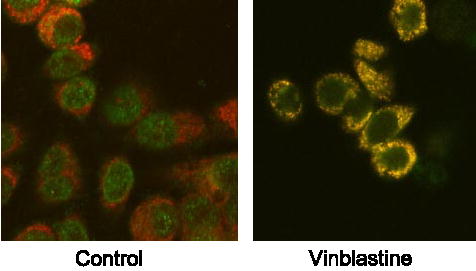
KB-3 cells were untreated (control) or treated with 30 nM vinblastine for 36 h, fixed, and permeabilized. Mitochondria (red signal) were visualized with Mitotracker Red, and Bax (green signal) was localized by immunofluorescence, as described under “Experimental Procedures.” The images were taken with a Zeiss LSM410 confocal microscope.
Bax Activation and Oligomerization
To determine whether Bax mitochondrial translocation was associated with Bax activation, active Bax was immunoprecipitated under native conditions from control and vinblastine-treated cells. For immunoprecipitation we used the anti-Bax 6A7 antibody, which recognizes the conformationally active form of Bax, and immunoprecipitates were analyzed by immunoblotting using a non-conformation-dependent Bax antibody. As shown in Fig. 3A, the active form of Bax was readily detected in cells treated with vinblastine for 36 h and also to a much lower extent in cells treated with vinblastine for 24 h, consistent with the profile of Bax mitochondrial translocation (Fig. 1), whereas active Bax was not detected in control cells. Active Bax was not observed when 6A7 antibody was omitted during immunoprecipitation, and additional controls showed equivalent Bax protein expression in whole cell extracts (Fig. 3A).
FIGURE 3. Vinblastine induces activation and dimerization of Bax.

A, KB-3 cells were untreated or treated with 30 nM vinblastine (VBL) for the times indicated and subjected to immunoprecipitation with anti-Bax 6A7 antibody (lanes 3, 6, and 9), followed by immunoblotting for Bax. Precipitates prepared in the absence of 6A7 antibody (lanes 2, 5, and 8) and whole cell extracts (lanes 1, 4, and 7) were also examined as controls. B, KB-3 cells were treated with vinblastine (VBL, 30 nM) for the times indicated, permeabilized, treated with digitonin in the absence or presence of 1 mM DSP, and CHAPS-solubilized particulate fractions subjected to SDS-PAGE. Immunoblotting for Bax was performed with the monomeric (21-kDa) and oligomeric (42-kDa) forms shown.
Studies in other systems have indicated that Bax activation is often associated with Bax oligomerization. For example, ATP depletion induced by hypoxia results in dimerization and higher oligomerization of Bax (20). To determine whether vinblastine induced Bax oligomerization, KB-3 cells were treated with or without vinblastine, permeabilized with digitonin, and then incubated with or without the cross-linking agent, DSP, as described under “Experimental Procedures.” Particulate fractions were prepared and extracted with CHAPS, and samples were subjected to immunoblotting for Bax. As shown in Fig. 3B, Bax migrated as a monomer of 21 kDa in control cells; however, after 36-h vinblastine treatment, a proportion of Bax also migrated at 42 kDa. The 42-kDa form was only observed following DSP treatment and in the absence of β-mercaptoethanol. When the cross-linker was cleaved under reducing conditions with β-mercaptoethanol, the 42-kDa species was absent (data not shown), suggesting that this species indeed represented a higher molecular mass complex of Bax, possibly a dimer.
Vinblastine Treatment Alters the Oligomeric Structure of Bcl-xL
The samples shown in Fig. 3 were also subjected to immunoblotting for Bcl-2 and Bcl-xL. No oligomeric forms of Bcl-2 were detected following either vinblastine treatment or DSP treatment (Fig. 4A, top panel). Under each condition, Bcl-2 migrated as a monomer with an apparent molecular mass of 23 kDa, with vinblastine inducing phosphorylation-dependent mobility shifts, as shown previously (19). In contrast, discrete species of differing molecular masses were found when these CHAPS-solubilized particulate fractions were probed for Bcl-xL (Fig. 4A, lower panel). In control cells, a substantial proportion of Bcl-xL migrated with an apparent molecular mass of 150 kDa compared with the 30-kDa monomeric form. In addition, several species in the range of 50–70 kDa were also observed. The 150-kDa species was eliminated by DSP treatment, suggesting that the relevant protein complex is disrupted or unstable in the presence of the chemical cross-linking reagent. Interestingly, the 150-kDa species was also eliminated after vinblastine treatment, which promotes Bcl-xL phosphorylation (19), also evident as a mobility shift of the monomeric form in Fig. 4A. Only the monomeric form of Bcl-xL was present after treatment of samples with β-mercaptoethanol to reverse the cross-links (Fig. 4A, lower panel, right two lanes).
FIGURE 4. Vinblastine and other microtubule inhibitors induce alterations in Bcl-xL oligomerization.

A, samples were prepared as described in the legend to Fig. 3B and subjected to immunoblotting for Bcl-2 (top panel) or Bcl-xL (lower panel), as indicated. The phosphorylated forms of Bcl-2 and Bcl-xL and the 150-kDa species of Bcl-xL are indicated by arrows, and molecular mass standards (in kDa) are shown on the right. The samples run in the right two lanes of each panel were treated with β-mercaptoethanol (β-ME) to reverse cross-links. VBL, vinblastine. B, KB-3 cells were untreated or treated with 30 nM vinblastine (VBL), 30 nM vincristine (VCR), 30 nM paclitaxel (TAX), 1 μM doxorubicin (DOX), or 15 μM etoposide (VP-16). The samples were prepared as for Fig. 3B and subjected to immunoblotting for Bcl-xL. Monomeric unphosphorylated and phosphorylated Bcl-xL and the 150-kDa species are indicated.
These results suggest that vinblastine treatment promotes alterations in the interaction of Bcl-xL with itself and/or with other proteins. Characteristically, microtubule inhibitors, but not other apoptotic stimuli, induce Bcl-xL phosphorylation (19). To determine whether the reduction in the 150-kDa species was a general feature of microtubule inhibition and thus possibly related to phosphorylation, we treated KB-3 cells with two other microtubule inhibitors, vincristine and paclitaxel, as well as two agents with different mechanisms, doxorubicin and etoposide. As shown in Fig. 4B, treatment with the microtubule inhibitors eliminated expression of the 150-kDa form of Bcl-xL, whereas this species remained prominent when the other agents were used.
Characterization of Bcl-xL and Bcl-2 by Two-dimensional Gel Electrophoresis
Two-dimensional PAGE was used to further characterize and compare the different oligomeric forms of Bcl-xL. One advantage is that homoligomers of a protein would be expected to migrate with a higher molecular mass but a similar pI to the monomer, whereas oligomers of mixed protein content would be expected to have a pI distinct from that of the monomeric protein. Another advantage of this analysis is that the first dimension is conducted under nondenaturing conditions. Because stability may be compromised with the denaturants used under the conditions of Fig. 4, we reasoned that two-dimensional analysis may enable a more accurate assessment of the relative abundance of the different forms of Bcl-xL. The samples were prepared for two-dimensional PAGE as described under “Experimental Procedures,” following the protocol recommended by Kendrix Laboratories. Human Bcl-xL (Swiss-Prot code Q07817) has a theoretical pI of 4.86; therefore, an acidic range of ampholines was employed in the first dimension, as described under “Experimental Procedures.” Following two-dimensional PAGE, the proteins were transferred to polyvinylidene difluoride membrane and immunoblotted for Bcl-xL. In extracts from control cells, an immunoreactive spot representing monomeric Bcl-xL was observed with an apparent molecular mass of 28 kDa and an apparent isoelectric point of 5.4, close to that expected for monomeric Bcl-xL (Fig. 5A). (It should be noted that because of the acidic conditions required to resolve Bcl-xL, the pH gradient shown, although representing a close approximation, was not linear over the whole range, and endogenous Bcl-xL may harbor modifications that alter its charge compared with the theoretical value). In addition, in control cells, substantial Bcl-xL immunoreactivity was observed with molecular masses of ~70 and 150 kDa. Several species were reproducibly observed, with pI vales in the range of 5.4–5.7. In extracts from vinblastine-treated cells, monomeric Bcl-xL migrated to a more acidic pI (estimated to be about 4.9) and a slightly higher apparent molecular mass of 30 kDa, a diagonal shift consistent with phosphorylation (19). Importantly, high molecular mass species were much less abundant after vinblastine treatment, consistent with findings from one-dimensional PAGE (Fig. 4A). These results confirm that oligomeric forms of Bcl-xL exist and that their relative abundance and interrelationship is altered after vinblastine-induced phosphorylation.
FIGURE 5. Analysis of Bcl-xL and Bcl-2 by two-dimensional gel electrophoresis.
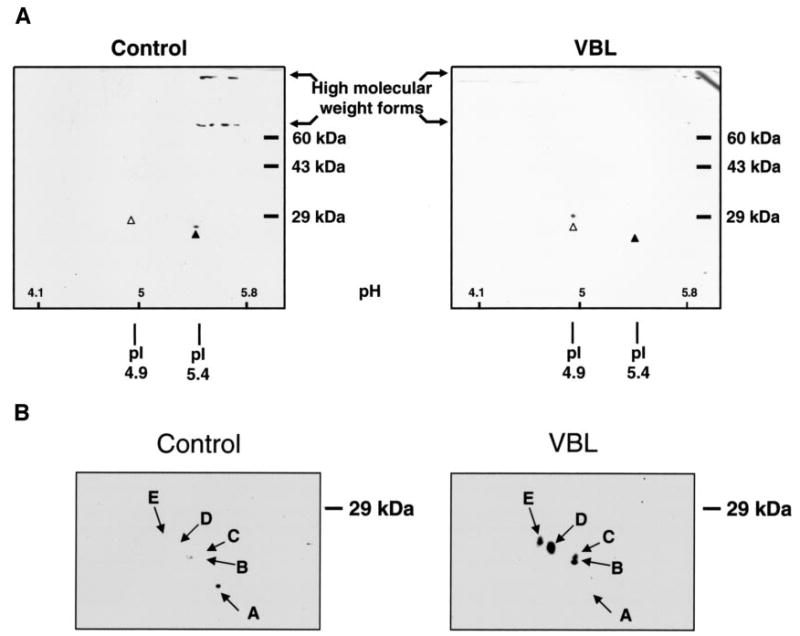
KB-3 cells were either untreated (control) or treated with 30 nM vinblastine for 36 h (VBL), and cell extracts were subjected to two-dimensional PAGE, as described under “Experimental Procedures.” A, immunoblot analysis of Bcl-xL. An acidic range of ampholines was employed, with approximate pH values shown inset at the bottom. The monomeric form of Bcl-xL, observed in control cells (pI of 5.4), is indicated with the black arrowhead, and the phosphorylated form, observed in vinblastine-treated cells (pI of 4.9), is indicated with the white arrowhead. High molecular mass species are also indicated. B, immunoblot analysis of Bcl-2. Only the relevant portion of the gel is shown, because Bcl-2 was strictly monomeric with no evidence of high molecular mass species. The pI values and apparent molecular masses of the resolved species of Bcl-2 are as follows. Spot A, pI 6.8, 23 kDa; spot B, pI 6.6, 24.5 kDa; spot C, pI 6.6, 25.1 kDa; spot D, pI 6.3, 26.9 kDa; spot E, pI 6.0, 27.5 kDa.
Bcl-2 was examined by two-dimensional PAGE in a similar fashion, except that a conventional pH gradient was utilized, as human Bcl-2 (Swiss-Prot code P10415) has a more neutral theoretical pI of 6.75. No oligomeric forms were observed, and Bcl-2 existed exclusively in monomeric form (Fig. 5B), consistent with results from one-dimensional PAGE (Fig. 4A). In extracts from control cells, a single species with an apparent molecular mass of 23 kDa and a pI of 6.8 was observed, very close to that expected. In extracts from vinblastine-treated cells, several additional spots, shifted diagonally from the parent spot, consistent with multisite phosphorylation, were observed. The apparent molecular masses and the pI values of these species are listed in the legend to Fig. 5B. Interestingly, spot B and spot C differed in apparent molecular mass but not in pI, suggesting that they may be related by a modification that does not affect charge.
Studies with HCT116 Wild-type and Bax Knock-out Cells
To confirm the main observations with another cell line, we used HCT116 colon carcinoma cells, because variants both wild-type and defective for Bax expression, via homologous disruption of the bax gene, were available for study (21). Fig. 6A shows Bax immunoblots in Bax+/− and Bax−/− HCT116 cell lines compared with KB-3 cells, with or without vinblastine treatment. Bax protein expression was comparable in KB-3 and Bax+/− cells, and absent in Bax−/− cells, as expected, with no change in overall level after vinblastine treatment. GAPDH was used as a loading control and was comparable in each lane.
FIGURE 6. Vinblastine promotes Bax activation and dimerization and alterations in Bcl-xL oligomerization in HCT116 cells.

A, extracts were made from KB-3, Bax+/−, and Bax−/− cells, treated with or without 30 nM vinblastine (VBL) for 24 h, as indicated, and subjected to immunoblotting for Bax or GAPDH. B, KB-3, Bax+/− or Bax−/− cells were untreated or treated with 30 nM vinblastine (VBL) for the times indicated and subjected to immunoprecipitation with anti-Bax 6A7 antibody (lanes 3, 6, and 9), followed by immunoblotting for Bax. Precipitates prepared in the absence of 6A7 antibody (lanes 2, 5, and 8) and whole cell extracts (lanes 1, 4, and 7) were also examined as controls. C, KB-3, Bax+/−, or Bax−/− cells were treated with vinblastine (VBL, 30 nM) for 36 h, permeabilized, and treated with digitonin in the presence or absence 1 mM DSP as indicated, and CHAPS-solubilized particulate fractions were subjected to SDS-PAGE. Immunoblotting for Bax was performed, with monomeric (21-kDa) and oligomeric (42-kDa) forms shown. GAPDH served as a loading control. D, KB-3, Bax+/− or Bax−/− cells were prepared as in the legend to Fig. 3B, and extracts were subjected to immunoblotting for Bcl-xL. The monomeric form and the 150-kDa species of Bcl-xL are indicated by arrows, and molecular mass standards (in kDa) are shown on the right.
We next determined whether vinblastine activated Bax in HCT116 cells. Immunoprecipitation was performed with anti-Bax 6A7 antibody with samples normalized to equal protein, confirmed by GAPDH immunoblotting (Fig. 6A). As shown in Fig. 6B, active Bax was detected in KB-3 cells after 36 h of vinblastine treatment, as before (Fig. 3A), and also in Bax+/− cells, although earlier at 24 h, indicating that vinblastine-induced Bax conformational activation is not restricted to KB-3 cells. As expected, active Bax was not detected in Bax−/− cells (Fig. 6B). To determine whether vinblastine-induced Bax activation in HCT116 cells was associated with Bax dimerization, the cells were permeabilized and treated with the cross-linking agent DSP and samples processed for SDS-PAGE, as described in Fig. 3. As shown in Fig. 6C, Bax migrated as a monomer of 21 kDa in KB-3 and HCT116 Bax+/− cells. After 36-h vinblastine treatment and in the presence but not the absence of DSP, a proportion of Bax also migrated at 42 kDa in both KB-3 and HCT116 Bax+/− cell lines. Thus, vinblastine induces Bax oligomerization in two independent cell lines. The absence of the 42-kDa species after vinblastine treatment of Bax−/− cells supports its identity as a Bax oligomer. Bcl-xL was next analyzed in Bax+/− and Bax−/− cell lines. Importantly, the 150-kDa form was observed in both cell lines, with gel mobility identical to that observed in KB-3 cells. In addition, the 150-kDa oligomeric form in the HCT116 variant cell lines was diminished in response to vinblastine treatment, although to a lesser extent than that observed in KB-3 cells. Quantitation of band intensity indicated that the 150-kDa species was reduced by vinblastine treatment by 60% in Bax+/− cells and by 70% in Bax−/− cells. Note that to quantitate band intensity, it was necessary to use an immunoblot where the ECL signal was linear with respect to the protein loaded. The immunoblot used and shown in Fig. 6D was more underexposed than normal, and the weaker intermediate bands (50–70 kDa) shown previously (Fig. 4B) were not observable at this intensity level, although these bands were revealed with longer ECL exposures.
Interaction of Bax with Bcl-xL but Not Bcl-2
To determine whether Bax interacts with Bcl-2 or Bcl-xL in control cells or following vinblastine-induced mitochondrial translocation, co-immunoprecipitation was employed. Bcl-2 was immunoprecipitated from control and vinblastine-treated cells and subjected to immunoblotting for both Bcl-2 and Bax. The results demonstrated that both unphosphorylated Bcl-2 from control cells and multi-phosphorylated Bcl-2 from vinblastine-treated cells were readily identified in the immunoprecipitates (Fig. 7A, upper blot, lanes 2 and 5, respectively), but not if antibody was omitted from the mixture (Fig. 7A, upper blot, lanes 3 and 6, respectively). However, after reprobing the same membrane, Bax was not detected in the immunoprecipitates, although the protein was confirmed to be present in whole cell extracts (Fig. 7A, lower blot, lanes 1 and 3). Similar experiments were performed for Bcl-xL (Fig. 7B). Bcl-xL was readily identified by immunoblotting in Bcl-xL immunoprecipitates from control and vinblastine-treated cells (Fig. 7B, upper blot). In this case, when the membrane was reprobed for Bax, Bax was detected in the immunoprecipitate from control cells, was detected to a lesser extent in the immunoprecipitate from 24-h vinblastine-treated cells, and was nearly undetectable in the immunoprecipitate from 36-h vinblastine-treated cells (Fig. 7B, lower blot). These results are of considerable significance in that in control cells both Bax (Fig. 1) and to some extent Bcl-xL (19) are present in the cytosol, and after vinblastine treatment, Bax translocates to the mitochondria (Figs. 1 and 2), where Bcl-xL is mainly resident. Because the proteins interact in control cells but not after vinblastine treatment (Fig. 7B), the results suggest that inactive cytosolic Bax, but not active mitochondrial Bax, interacts with Bcl-xL. To test this hypothesis, the 6A7 antibody was used to immunoprecipitate conformationally active Bax. As shown in Fig. 7C (upper blot), active Bax was evident in samples from vinblastine-treated cells, with a significant increase from 24 to 36 h. Reprobing of these immunoprecipitates for Bcl-xL, however, showed an absence of Bcl-xL in all cases, although Bcl-xL was readily detected in the cell extracts (Fig. 7C, lower blot). These results confirm that Bcl-xL interacts with inactive Bax but not the conformationally altered form of Bax.
FIGURE 7. Differential interactions of Bax with Bcl-xL but not Bcl-2.

A, KB-3 cells were untreated (lanes 1–3) or treated with vinblastine (VBL, 30 nM, 36 h, lanes 4 – 6) and Bcl-2 was immunoprecipitated (IP), as described under “Experimental Procedures.” Immunoprecipitations were performed with (+Ab, lanes 2 and 5) or without (−Ab, lanes 3 and 6) the relevant primary antibody as indicated. Whole cell extracts were also analyzed (lanes 1 and 4). The samples were analyzed by immunoblotting for Bcl-2 (top panel) or Bax (lower panel). B, KB-3 cells were treated with 30 nM vinblastine (VBL) for the times indicated and subjected to immunoprecipitation (IP) with anti-Bcl-xL antibody (lanes 3, 6, and 9), followed by immunoblotting for Bcl-xL (top panel) or Bax (lower panel) as indicated. Precipitates prepared in the absence of primary antibody (lanes 2, 5, and 8) and whole cell extracts (lanes 1, 4, and 7) were also examined as controls. C, KB-3 cells were treated with 30 nM vinblastine (VBL) for the times indicated and subjected to immunoprecipitation (IP) with anti-Bax 6A7 antibody (lanes 2, 5, and 8), followed by immunoblotting for Bax (top panel) or Bcl-xL (lower panel) as indicated. Precipitates prepared in the absence of primary antibody (lanes 3, 6, and 9) and whole cell extracts (lanes 1, 4, and 7) were also examined as controls.
Inhibition of Bax Activation and Bax Dimerization Inhibits Apoptosis Induction
The results presented above show that vinblastine-induced apoptosis is associated with Bax activation and Bax dimerization. To determine whether these events are required for apoptosis, we sought to inhibit them by overexpression of anti-apoptotic Bcl-2 proteins. Toward this goal, we generated cells overexpressing Bcl-xL. KB-3 cells were first transiently transfected with a plasmid encoding HA-tagged Bcl-xL. A protein of 30 kDa, immunoreactive with Bcl-xL or HA antibody, was readily detected after immunoprecipitation with either HA or Bcl-xL antibody, but not in mock-transfected cells, nor when antibody was omitted from the precipitation step (Fig. 8A). Stable transfectants, termed KB3-HA-Bcl-xL cells, were generated as described under “Experimental Procedures.” Two representative clones were used that significantly overexpressed HA-Bcl-xL relative to control cells (Fig. 7B). KB3-HA-Bcl-xL cells were untreated or treated with vinblastine, cell extracts were prepared, and Bax activation was examined by immunoprecipitation with 6A7 antibody. Compared with control cells, which showed significant Bax activation after 36 h of treatment as shown above in Fig. 3A, active Bax was nearly undetectable in KB3-HA-Bcl-xL cells. Representative results for one of the cell lines are shown in Fig. 8C, and essentially identical results were obtained with the other KB3-HA-Bcl-xL overexpressing cell line. Furthermore, Bax dimerization was also undetectable after vinblastine treatment in both Bcl-xL overexpressing cell lines relative to control cells (Fig. 7D). These results show that overexpression of anti-apoptotic Bcl-xL prevents vinblastine-induced Bax activation and Bax dimerization. To determine the effect on apoptosis, a quantitative assay for apoptosis was conducted, as described under “Experimental Procedures.” As expected, vinblastine treatment of control cells induced apoptosis, but KB3-HA-Bcl-xL cells were clearly resistant, with a much lower level of apoptosis, corresponding to 80% inhibition (clone 2) and 50% inhibition (clone 1) under the same conditions (Fig. 7E). In confirmation of these findings, sub-G1 DNA content, indicative of apoptotic DNA, was significantly reduced in the two Bcl-xL-overexpressing clones (18 and 12% sub-G1 DNA, respectively) versus wild-type cells (45% sub-G1 DNA) under similar conditions of vinblastine treatment. These results suggest that Bax activation and Bax dimerization are required for vinblastine-induced apoptosis.
FIGURE 8. Overexpression of Bcl-xL inhibits vinblastine-induced Bax activation, Bax dimerization, and apoptosis.

A, transient expression of HA-Bcl-xL. KB-3 cells were untransfected, transfected with vector only (Vec), or with HA-Bcl-xL plasmid. Whole cell extracts were subjected to immunoprecipitation with anti-HA antibody (upper two panels) or anti-Bcl-xL antibody (bottom panel). Ab (+ or −) indicates whether antibody was present or omitted in the immunoprecipitation step. Immunoprecipitates were separated by SDS-PAGE and blotted with the antibody indicated on the right. B, stable expression of HA-Bcl-xL. KB-3 cells stably expressing HA-Bcl-xL were isolated, as described under “Experimental Procedures.” Extracts from untransfected (−) or two clones of HA-Bcl-xL transfected cells (+) were subjected to immunoblotting with antibody to either Bcl-xL or the HA tag as indicated. GAPDH provided a loading control. C, KB-3 (top panel) or KB3-HA-Bcl-xL (lower panel) cells were untreated or treated with 30 nM vinblastine (VBL) for the times indicated and subjected to immunoprecipitation with anti-Bax 6A7 antibody (lanes 3, 6, and 9), followed by immunoblotting for Bax. Precipitates prepared in the absence of 6A7 antibody (lanes 2, 5, and 8) and whole cell extracts (lanes 1, 4, and 7) were also examined as controls. The data shown for KB-3 cells are the same as that presented in Fig. 3A. D, KB-3 or two independent clones of KB3-HA-Bcl-xL cells were untreated or treated with vinblastine as indicated, permeabilized, and treated with digitonin in the presence of 1 mM DSP, and CHAPS-solubilized particulate fractions were subjected to SDS-PAGE in the absence of β-mercaptoethanol. Immunoblotting for Bax was performed, with the monomeric (21-kDa) and oligomeric (42-kDa) forms shown. E, KB-3 or two clones of KB3-HA-Bcl-xL cells were left untreated (−) or treated with (+) vinblastine (30 nM, 48 h), and the relative extent of apoptosis quantitatively was assessed, as described under “Experimental Procedures.” The results are shown as the means ± S.D. (n = 6) and are representative of two independent experiments.
DISCUSSION
Microtubule inhibitors such as vinca alkaloids and paclitaxel and derivatives play an important role in cancer therapy. Recent evidence suggests that, despite binding to different sites on tubulin, physiological concentrations of these agents have in common the ability to suppress the dynamic instability of spindle microtubules, leading to mitotic arrest and apoptosis (7). However, the mechanisms of apoptosis induction after mitotic arrest are complex and far from resolved, and several different signal transduction pathways, including those involving p53, mitogen-activated protein kinases, and NFκB, have been implicated (8–10). Evidence has accumulated to suggest a role for the Bcl-2 family of apoptotic regulators, but their precise involvement and the underlying mechanisms have remained largely unexplored. One of the most prominent effects of microtubule inhibitors is the phosphorylation of Bcl-2 and Bcl-xL (11). Recent studies from our laboratory have indicated that vinblastine promotes a coordinated cycle of phosphorylation and dephosphorylation of Bcl-2 and Bcl-xL (19).
In this report we have made several novel observations that advance and add to our knowledge of the role of Bcl-2 proteins in apoptosis induction by microtubule inhibiting drugs. Our data show that vinblastine induces Bax mitochondrial translocation associated with conformational changes and dimerization of Bax. Furthermore, we show that, whereas Bcl-2 is strictly monomeric under the conditions employed, Bcl-xL exists also in high molecular mass complexes and that vinblastine treatment promotes specific changes in the oligomeric pattern of Bcl-xL. These results were paralleled in HCT116 cells, indicating that these observations are not cell type-specific and may be of more general significance. We also show in KB-3 cells that inactive Bax but not active Bax interacts with Bcl-xL and that Bax and Bcl-2 do not interact under any of the conditions employed here. Finally, we demonstrate that when Bax activation and dimerization are prevented, vinblastine-induced apoptosis is blocked.
In healthy cells, Bax is cytosolic, and in response to apoptotic stimuli the protein undergoes conformational changes and mitochondrial translocation, leading to the formation of oligomers and perhaps higher order structures that promote the release of cytochrome c and other apoptogenic factors (1–5). Although Bax activation has been widely reported as a mediator of diverse apoptotic stimuli, evidence that Bax activation is involved in apoptosis induced after mitotic arrest has only just begun to emerge. Thus, Bax activation has been demonstrated in HCT-116 cells after treatment with vinblastine or epothilone B (12), and in breast cancer cells after treatment with an epothilone B analog (13). Our biochemical data confirm a key role for Bax in vinblastine-induced apoptosis.
Studies in ATP depleted cells have shown that apoptosis induction is associated with homo-oligomerization of Bax (20, 22). Forms of Bax corresponding to dimer, trimer, and higher oligomers were observed, and these required cross-linking agents for stabilization (20, 22). We observed a 42-kDa form of Bax after vinblastine treatment in both KB-3 cells and in HCT116 Bax+/− cells that required stabilization by DSP. The 42-kDa form of Bax we observed may represent a dimer, but further work will be required to confirm this conclusion, and at higher exposure levels a species possibly corresponding to a trimer could be observed (Fig. 6C). To our knowledge, this is the first report of Bax oligomerization in response to a microtubule inhibitor. The fact that Bcl-xL overexpression prevents Bax oligomerization and in concert blocks vinblastine-induced apoptosis strongly suggests a link between these processes. Taken together with other work in the field, these results underscore the question of how such diverse stimuli as ATP depletion, DNA damage, and microtubule disruption converge on a common pathway leading to well defined changes in pro-apoptotic Bax. Because Bax and Bak can compensate for each others functions and operate in a redundant manner (5), it will be important also in future studies to examine Bak in this context.
Examination of Bcl-xL revealed the novel finding that Bcl-xL exists in several oligomeric forms. When analyzed by one-dimensional PAGE, Bcl-xL migrated with apparent molecular masses of about 30 kDa, representing the monomer, as well as an oligomer of 150 kDa and several species in the range of 50–70 kDa. The higher molecular mass forms were readily evident even in the presence of SDS and in the absence of cross-linking agent, suggesting that they are quite stable. However, we do not know whether the relative amounts observed by one-dimensional PAGE reflect the normal abundance in cells. Indeed, analysis by two-dimensional PAGE, where native conditions are used in the first dimension, suggested that the high molecular mass species of Bcl-xL are quite abundant relative to the monomer (Fig. 5A). Other reports have indicated the existence of Bcl-xL homodimers (of 52 kDa) in the cytosol of HeLa cells (23). However, our observation of a 150-kDa species is the first report to our knowledge of a distinct high molecular mass form of Bcl-xL. Analysis by two-dimensional PAGE indicated that the high molecular mass Bcl-xL immunoreactive complexes spanned a range of isoelectric points, both similar to and more basic compared with that of Bcl-xL (Fig. 5A). Thus, the data from the two-dimensional gels are somewhat equivocal and do not allow us to firmly conclude whether the high molecular mass forms represent homo- or hetero-oligomers of Bcl-xL, or both types. Of further significance is that the 150-kDa oligomer was also observed in HCT116 cells, indicating that the presence of this species may be of more general significance. Importantly, the 150-kDa form was present in both wild-type and Bax-deficient cells and thus is not dependent on the presence of Bax for its formation. In addition, the 150-kDa species was depleted after vinblastine treatment, and other microtubule inhibitors, but not DNA damaging agents, also caused the loss of this form. Microtubule inhibitors characteristically stimulate Bcl-xL phosphorylation, whereas other apoptotic stimuli do not (19). Thus, it is tempting to speculate that this change in oligomeric structure is related to phosphorylation. Further work will be required to rigorously answer this question.
In control KB-3 cells, as in other cell lines (23), Bax is cytosolic, Bcl-2 is membrane-bound, and Bcl-xL is found in both fractions (Fig. 1) (19). A prominent feature of Bcl-2 proteins is their ability to interact with each other to modulate the apoptotic threshold (1–4). It was of obvious interest to examine possible interactions of these proteins in untreated KB-3 cells and to determine whether any changes were associated with vinblastine treatment. Co-immunoprecipitation studies indicated that Bax was present in Bcl-xL immunoprecipitates from untreated KB-3 cells but not after vinblastine treatment (Fig. 7B), and consistent with this, Bcl-xL was absent from immunoprecipitates of active Bax (Fig. 7C). These results suggest that inactive cytosolic Bax interacts with cytosolic Bcl-xL, whereas activated mitochondrial Bax does not associate with Bcl-xL despite the presence of Bcl-xL in the mitochondria (19). Several factors may contribute to this loss of interaction including changes in the conformation of Bax or phosphorylation of Bcl-xL, both documented events following vinblastine treatment or perhaps other uncharacterized mechanisms. It is well established that Bax and Bcl-xL can form heterodimers, and this interaction appears to be evolutionarily conserved (24). This sequestration of Bax in its inactive form by cytosolic Bcl-xL appears to show significant similarity to the sequestration of Bak by anti-apoptotic Mcl-1 and Bcl-xL in unstressed cells where Bak is freed upon activation of BH3-only proteins (25).
In contrast to other reports that have indicated interactions between Bcl-2 and Bax (26), we did not find evidence of such an interaction in KB-3 cells. Also in contrast to Bcl-xL but consistent with another report (27), Bcl-2 existed strictly in monomeric form in KB-3 cells under all conditions employed (Figs. 4A and 5B). A single band with an apparent molecular mass of 23 kDa was observed on one-dimensional gels, and a single spot with an apparent molecular mass of 23 kDa and pI 6.8, consistent with unphosphorylated Bcl-2, was observed on two-dimensional gels. After vinblastine treatment, several bands decremented in gel mobility were observed on one-dimensional gels, and these bands have been established via phosphatase treatment to represent multi-phosphorylated forms of Bcl-2 (19). The consistent and reproducible observation of this level of phosphorylation complexity was absolutely dependent on the use of a mixture of phosphatase inhibitors in the extraction buffers. When analyzed by two-dimensional PAGE, four spots were observed that were shifted diagonally from the parent spot, and thus were consistent with being phosphorylated. However, two of the spots, B and C, differed in apparent molecular mass but not in pI. Thus, they appeared to be related to each other not by phosphorylation but by some modification that did not affect overall charge. Although much caution was taken to prevent proteolysis, it is possible that one might be a degradation product of the other. Alternately, in response to microtubule inhibition, Bcl-2 might undergo modification(s) distinct from phosphorylation. In this regard, it is interesting to note that Bcl-xL has been reported to undergo deamination, at specific asparagine residues (28). Such a modification would not change charge but might change apparent molecular mass, akin perhaps to phosphorylation, which manifests as a change in apparent mass much greater than the actual addition of a phosphate group. However, to our knowledge, there have been no reports of deamination of Bcl-2. Current efforts are underway to purify the various modified forms of Bcl-2 to locate and verify the nature of the modifications introduced in response to vinblastine treatment.
In summary, our results show that vinblastine treatment of KB-3 cells leads to selective changes in the subcellular location, activation status, oligomeric structure, and protein-protein interactions of members of the Bcl-2 family of apoptotic regulators. Alterations in Bax included mitochondrial translocation, conformational changes, and oligomerization, and these changes are similar to those observed in response to other diverse apoptotic stimuli. However, based on current evidence, several other changes, such as phosphorylation of Bcl-2 and Bcl-xL, and the changes in oligomeric structure of Bcl-xL reported here appear to be distinctive of microtubule inhibitors. Thus, the Bcl-2 proteins appear to act as a point of convergence for both stimulus-specific and core cell death pathways. Understanding how these pathways are integrated to control apoptosis in response to damage caused by cytotoxic drugs and other forms of damage remains a challenge for future study.
Acknowledgments
We are very grateful to Dr. Bert Vogelstein for the HCT116 cell lines, to Dr. Yoshinori Takahashi for advice on use of the 6A7 Bax antibody, to Dr. Dos Sarbassov for advice on immunoprecipitation, and to Dr. Elena Galitovskaya for assistance with preparation of HA-Bcl-xL cells.
Footnotes
This work was supported by National Institutes of Health Grants CA10982 and CA75577 from the National Cancer Institute.
The abbreviations used are: GAPDH, glyceraldehyde-3-phosphate dehydrogenase; DSP, dithiobis(succinimidyl propionate); PBS, phosphate-buffered saline; HA, hemagglutinin; pI, isoelectric point; CHAPS, 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonic acid.
References
- 1.Kroemer G, Reed JC. Nat Med. 2000;6:513–519. doi: 10.1038/74994. [DOI] [PubMed] [Google Scholar]
- 2.Gross A, McDonnell JM, Korsmeyer SJ. Genes Dev. 1999;13:1899–1911. doi: 10.1101/gad.13.15.1899. [DOI] [PubMed] [Google Scholar]
- 3.Cory S, Huang DCS, Adams JM. Oncogene. 2003;22:8590–8607. doi: 10.1038/sj.onc.1207102. [DOI] [PubMed] [Google Scholar]
- 4.van Delft MF, Huang DCS. Cell Res. 2006;16:203–213. doi: 10.1038/sj.cr.7310028. [DOI] [PubMed] [Google Scholar]
- 5.Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB, Korsmeyer SJ. Science. 2001;292:727–730. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rowinsky EK, Donehower RC. The Chemotherapy Source Book. 2nd Ed. Williams and Wilkins; Baltimore, MD: 1998. pp. 387–423. [Google Scholar]
- 7.Jordan MA, Wilson L. Nat Rev Cancer. 2004;4:253–265. doi: 10.1038/nrc1317. [DOI] [PubMed] [Google Scholar]
- 8.Blagosklonny MV, Fojo T. Int J Cancer. 1999;83:151–156. doi: 10.1002/(sici)1097-0215(19991008)83:2<151::aid-ijc1>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 9.Wang LG, Liu XM, Kreis W, Budman DR. Cancer Chemother Pharmacol. 1999;44:355–361. doi: 10.1007/s002800050989. [DOI] [PubMed] [Google Scholar]
- 10.Bhalla KN. Oncogene. 2003;22:9075–9086. doi: 10.1038/sj.onc.1207233. [DOI] [PubMed] [Google Scholar]
- 11.Ruvolo PP, Deng X, May WS. Leukemia. 2001;15:515–522. doi: 10.1038/sj.leu.2402090. [DOI] [PubMed] [Google Scholar]
- 12.Yamaguchi H, Chen J, Bhalla K, Wang HG. J Biol Chem. 2004;279:39431–39437. doi: 10.1074/jbc.M401530200. [DOI] [PubMed] [Google Scholar]
- 13.Yamaguchi H, Paranawithana SR, Lee MW, Huang Z, Bhalla KN, Wang HG. Cancer Res. 2002;62:466–471. [PubMed] [Google Scholar]
- 14.Longuet M, Serduc R, Riva C. Int J Oncol. 2004;25:309–317. [PubMed] [Google Scholar]
- 15.Salah-Eldin AE, Inoue S, Tsukamoto S, Aoi H, Tsuda M. Int J Cancer. 2003;103:53–60. doi: 10.1002/ijc.10782. [DOI] [PubMed] [Google Scholar]
- 16.Taguchi T, Kato Y, Baba Y, Nishimura G, Tanigaki Y, Horiuchi C, Mochimatsu I, Tsukuda M. Oncol Rep. 2004;11:421–426. [PubMed] [Google Scholar]
- 17.Makin GW, Corfe BM, Griffiths GJ, Thistlethwaite A, Hickman JA, Dive C. EMBO J. 2001;20:6306–6315. doi: 10.1093/emboj/20.22.6306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.O’Farrell PH. J Biol Chem. 1975;250:4007–4021. [PMC free article] [PubMed] [Google Scholar]
- 19.Du L, Lyle CS, Chambers TC. Oncogene. 2005;24:107–117. doi: 10.1038/sj.onc.1208189. [DOI] [PubMed] [Google Scholar]
- 20.Mikhailov V, Mikhailova M, Pulkrabek DJ, Dong Z, Venkatachalam MA, Saikumar P. J Biol Chem. 2001;276:18361–18374. doi: 10.1074/jbc.M100655200. [DOI] [PubMed] [Google Scholar]
- 21.Zhang L, Yu J, Park BH, Kinzler KW, Vogelstein B. Science. 2000;290:989–992. doi: 10.1126/science.290.5493.989. [DOI] [PubMed] [Google Scholar]
- 22.Mikhailov V, Mikhailova M, Degenhardt K, Venkatachalam MA, White E, Saikumar P. J Biol Chem. 2003;278:5367–5376. doi: 10.1074/jbc.M203392200. [DOI] [PubMed] [Google Scholar]
- 23.Jeong SY, Gaume B, Lee YJ, Hsu YT, Ryu SW, Yoon SH, Youle RJ. EMBO J. 2004;23:2146–2155. doi: 10.1038/sj.emboj.7600225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Takeda N, Yamaguchi H, Shida K, Terajima D, Satou Y, Kasuya A, Satoh N, Satake M, Wang HG. Apoptosis. 2005;10:1211–1220. doi: 10.1007/s10495-005-1391-4. [DOI] [PubMed] [Google Scholar]
- 25.Willis SN, Chen L, Dewson G, Wei A, Naik E, Fletcher JI, Adams JM, Huang DCS. Genes Dev. 2005;19:1294–1305. doi: 10.1101/gad.1304105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Diaz JL, Oltersdorf T, Horne W, McConnell M, Wilson G, Weeks S, Garcia T, Fritz LC. J Biol Chem. 1997;272:11350–11355. doi: 10.1074/jbc.272.17.11350. [DOI] [PubMed] [Google Scholar]
- 27.Conus S, Kaufmann T, Fellay I, Otter I, Rosse T, Borner C. EMBO J. 2000;19:1534–1544. doi: 10.1093/emboj/19.7.1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Takehara T, Takahashi H. Cancer Res. 2003;63:3054–3057. [PubMed] [Google Scholar]