

Abstract
Histone-modifying enzymes play essential roles in physiological and aberrant gene regulation. Since histone deacetylases (HDACs) are promising targets of cancer therapy, it is important to understand the mechanisms of HDAC regulation. Selective modulators of HDAC isoenzymes could serve as efficient and well-tolerated drugs. We show that HDAC2 undergoes basal turnover by the ubiquitin–proteasome pathway. Valproic acid (VPA), in addition to selectively inhibiting the catalytic activity of class I HDACs, induces proteasomal degradation of HDAC2, in contrast to other inhibitors such as trichostatin A (TSA). Basal and VPA-induced HDAC2 turnover critically depend on the E2 ubiquitin conjugase Ubc8 and the E3 ubiquitin ligase RLIM. Ubc8 gene expression is induced by both VPA and TSA, whereas only TSA simultaneously reduces RLIM protein levels and therefore fails to induce HDAC2 degradation. Thus, poly-ubiquitination and proteasomal degradation provide an isoenzyme-selective mechanism for downregulation of HDAC2.
Keywords: HDAC2/HDAC inhibitor/histone deacetylase/proteasomal degradation/valproic acid
Introduction
The recruitment of histone acetyltransferases (HATs) and histone deacetylases (HDACs) is considered as a key element in the dynamic regulation of many genes playing important roles in cellular proliferation and differentiation (Glass and Rosenfeld, 2000; Kouzarides, 2000). Hyperacetylation of the N-terminal tails of histones H3 and H4 correlates with gene activation, whereas deacetylation mediates transcriptional repression (Strahl and Allis, 2000). Consequently, many diseases have been linked to changes in gene expression caused by mutations affecting transcription factors. Aberrant repression by leukemia fusion proteins such as PML-RAR, PLZF-RAR, AML-ETO and Stat5-RAR serves as prototypical examples in this regard. In all of these cases, chromosomal translocations result in fusion proteins which convert transcriptional activators into repressors. These constitutively repress genes important for hematopoietic differentiation via recruitment of HDACs (Gelmetti et al., 1998; Grignani et al., 1998; Guidez et al., 1998; He et al., 1998; Lin et al., 1998; Lutterbach et al., 1998; Wang et al., 1998; Hildebrand et al., 2001; Maurer et al., 2002). Similar events could also contribute to pathogenesis in other types of cancer.
Mammalian histone deacetylases can be divided into three subclasses (Gray and Ekström, 2001). HDACs 1, 2, 3 and 8, which are homologs of the yeast RPD3 protein, constitute class I. HDACs 4, 5, 6, 7, 9 and 10 are related to the yeast Hda 1 protein and form class II. Recently, mammalian homologs of the yeast Sir2 protein have been identified forming a third class of deacetylases. All of these HDACs apparently exist in the cell as subunits of multiprotein complexes. In particular, class I and II HDACs have been shown to interact with the transcriptional corepressors mSin3, N-CoR and SMRT, which recruit HDACs to transcription factors (Alland et al., 1997; Heinzel et al., 1997; Laherty et al., 1997; Nagy et al., 1997; Huang et al., 2000; Kao et al., 2000).
Currently, only limited information is available about the isoenzyme-specific or redundant biological functions of these HDACs. Class II HDACs have been shown to translocate from the cytoplasm to the nucleus in response to external stimuli (McKinsey et al., 2000). In particular, HDAC9 acts as a signal-responsive suppressor of the transcriptional program governing cardiac hypertrophy and heart failure (Zhang et al., 2002). Class I HDACs are constitutively nuclear and play important roles in dynamic gene regulation. The essential biological function of class I HDACs is emphasized by the finding that targeted deletion of the HDAC1 gene leads to early embryonic lethality in mice, possibly due to a proliferation defect upon unrestricted expression of the cell cycle inhibitors p21 and p27 (Lagger et al., 2002).
Since recruitment of HDACs leads to transcriptional repression, inhibitors of this enzymatic activity can reverse aberrant repression and lead to re-expression of genes inducing differentiation. Therefore, HDAC inhibitors are considered as candidate drugs in cancer therapy (Krämer et al., 2001; Marks et al., 2001; Melnick and Licht, 2002). The benefit of these enzyme inhibitors has been established by many in vitro experiments, experimental therapy and ongoing clinical trials (Warrell et al., 1998; Melnick and Licht, 2002). However, many HDAC inhibitors such as trichostatin A (TSA) do not exhibit isoenzyme selectivity and are of limited therapeutic value due to poor bioavailability in vivo as well as toxic side-effects at high doses.
Recently, we discovered that the well-tolerated antiepileptic drug valproic acid (VPA) is a class I selective HDAC inhibitor (Göttlicher et al., 2001). This activity can be distinguished from its therapeutically exploited antiepileptic activity (Göttlicher et al., 2001; Phiel et al., 2001). Since VPA has been used clinically for over two decades, the pharmacology and side-effects of this drug have been studied in detail. As expected for HDAC inhibitory compounds, VPA induces differentiation of carcinoma cells, transformed hematopoietic progenitor cells and leukemic blasts from acute myeloid leukemia (AML) patients. Moreover, tumor growth and metastasis formation are significantly reduced in animal experiments (Göttlicher et al., 2001). Interestingly, VPA was also reported to have beneficial effects for patients suffering from neuroblastomas and glioblastomas even before its HDAC inhibitory properties were established (Driever et al., 1999).
During our analysis of VPA effects on HDACs, we discovered that VPA but not TSA triggers proteasome-mediated degradation of HDAC2. Thus, VPA appears to act as an isoenzyme-selective downmodulator of HDAC2 at therapeutically useful concentrations by both inhibiting HDAC catalytic activity and inducing specific degradation of HDAC2.
Results
Reduction of HDAC2 protein levels by VPA treatment
We investigated whether HDAC inhibitors would not only inhibit activity but also affect expression of HDACs. We found a significant downregulation of HDAC2 protein levels in cells treated with the carboxylic acids VPA or butyrate, whereas other HDAC inhibitors such as TSA and MS-27-275 failed to induce this effect (Figure 1A). Reduction of protein levels to ∼30% of untreated cells is found 24 h after VPA exposure and persists for at least 48 h (Figure 1B). The delayed response suggests the involvement of intermediary steps such as induction of protein expression. In murine F9 teratocarcinoma and human embryonic kidney HEK293T cells (Figure 1B and C), as well as in 14 additional cell lines (data not shown), a time- and dose-dependent reduction in HDAC2 protein levels upon VPA treatment is apparent. Protein levels of HDAC1 and HDAC3 were not reduced but even showed a transient induction in some experiments (Figure 1B). Furthermore, VPA treatment does not cause a reduction in protein levels of HDACs 4, 5 and 8 (data not shown). VPA doses required for reduction of HDAC2 protein levels are similar to those required for inhibition of HDAC enzymatic activity and clear effects are detected at 0.5–1 mM (Figure 1C). In HEK 293T cells which tolerate higher doses of VPA a more rapid and slightly more pronounced reduction is found if VPA concentrations are increased to 5 mM. Treatment with TSA for up to 48 h does not reduce HDAC2 levels in either F9 or HEK293T cells (Figure 1D). A significant reduction of HDAC2 protein levels is also found in vivo after treatment of mice with VPA (Figure 1E).

Fig. 1. VPA but not TSA leads to reduction of HDAC2 protein levels. (A) K562 human erythroleukemia cells were treated for 24 h as indicated with the HDAC inhibitors VPA (1.5 mM), TSA (100 nM), butyrate (1.5 mM) or MS-27–275 (5 µM). Amounts of HDAC2 protein were determined by western blot analysis of whole-cell extracts. Actin protein levels were determined to verify equal loading of samples. (B) F9 mouse teratocarcinoma or HEK293T human embryonic kidney carcinoma cells were exposed to 1 mM VPA for the indicated periods of time. Protein levels of HDAC2 as well as HDAC1, HDAC3 and actin were determined by western blot analysis. (C) The dose-dependent reduction of HDAC2 protein levels was determined in F9 or HEK293T cells after exposure to VPA for 30 or 24 h, respectively. (D) Time course analyses in F9 and HEK293T cells confirmed that TSA (100 nM) does not affect the amount of HDAC2 protein. (E) Reduction of HDAC2 protein levels after treatment of mice with VPA was tested by western blot analysis of tissue extracts and immunohistochemistry. Similar results were obtained in at least two sets of independent experiments.
Induction of proteasomal degradation of HDAC2
Under conditions leading to a reduction in HDAC2 protein levels, no reduction of HDAC2 mRNA levels was found in F9 and HEK293T cells (data not shown). This finding suggests that VPA affects the rate of protein synthesis or degradation. HDAC2 protein synthesis rates with and without VPA pretreatment for 24 h were compared by pulse labeling with [35S]methionine in F9 or HEK293T cells. No substantial difference in HDAC2 synthesis rate between control or VPA-treated cells was found (Figure 2A).
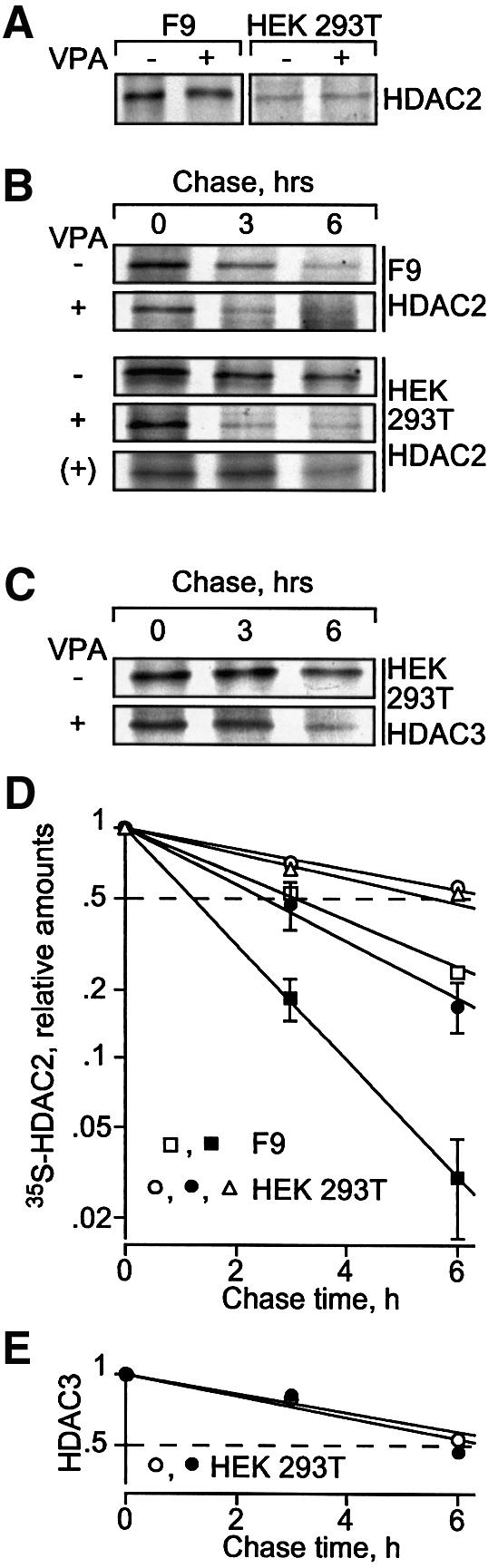
Fig. 2. VPA induces degradation of HDAC2 protein. Synthesis and degradation rates of HDAC2 were determined by [35S]methionine labeling followed by chase analyses. [35S]HDAC2 was detected by HDAC2-specific immunoprecipitation, SDS–PAGE and autoradiography. (A) Pulse labeling for 1 h was performed in F9 and HEK293T cells, which were precultured for 24 h in the absence or presence of 1 mM VPA (F9) or 1.5 mM VPA (HEK293T). (B) For pulse–chase analysis, cells were cultured for 24 h either in the absence or presence of 1 mM VPA (F9) or 1.5 mM VPA (HEK293T) and labeled with [35S]methionine for an additional hour without or with VPA. After removal of [35S]methionine and addition of non-labeled methionine, the elimination of radiolabeled HDAC2 was followed over a period of 6 h. A pulse–chase analysis was also performed in non-pretreated HEK293T cells to which VPA was added only at the time when the chase was started [row marked (+)]; VPA at time of chase, triangles. Efficiency of immunoprecipitations was confirmed by proving depletion of HDAC2 from the precipitation supernatants. Comparable loading between lanes was controlled by Coomassie staining of gels for the amounts of antibodies. Similar results were obtained in at least a second independent experiment. (C) HDAC3 was also precipitated from HEK293T cell extracts. (D and E) Phosphoimager analyses of the experiments in (B) and (C) were quantitatively evaluated by subtraction of local background. The graphs show averages of two or three experiments (control, open symbols; VPA, filled symbols; VPA at time of chase, triangles) and were used for half-life determination. Standard deviations are shown if bars exceed symbol sizes.
Protein half-life of HDAC2 was determined by pulse– chase analysis and was substantially decreased by pretreatment of cells with VPA (Figure 2B). In F9 cells, a decrease from 3 to 1.2 h was found and in HEK293T cells the change was from 6.8 to 2.4 h (Figure 2D). VPA had no effect on the HDAC2 protein degradation rate when added only at the time of chase (Figure 2B and D). This experiment provided additional evidence that the response of HDAC2 protein levels to VPA is indirect rather than being a direct response, for example, to a conformational change of HDAC2 upon interaction with VPA. Degradation of HDAC3 was not affected by VPA-pretreatment of HEK293T cells (Figure 2C and E), consistent with a lack of reduction in steady-state HDAC3 protein levels upon VPA treatment.
To investigate whether HDAC2 degradation is due to either protease-dependent or proteasomal degradation, several inhibitors of proteases or the proteasome were applied in combination with VPA (Figure 3A). None of the protease inhibitors pepstatin A, leupeptin or ALLM had an effect on HDAC2 levels in the presence or absence of VPA (Figure 3A). Treatment of HEK293T cells with the proteasome inhibitors ALLN or MG-132 either abolished or significantly reduced VPA-induced degradation of HDAC2 (Figure 3A). Thus, increased proteasomal degradation by a mechanism involving synthesis of intermediary factors appears to be the most likely cause of VPA-induced degradation of HDAC2.

Fig. 3. VPA induces polyubiquitination and proteasome-dependent degradation of HDAC2. (A) HEK293T cells were treated for 24 h with VPA or left untreated. The protease inhibitors pepstatin A (20 µM), leupeptin (10 µM), or ALLM (20 µM), or the proteasome inhibitors ALLN (5 µM), or MG 132 (10 µM), were added at the same time as VPA. HDAC2 protein levels were determined by western blot analysis. (B) Precipitation of poly-ubiquitinated proteins with anti-HDAC2 antibody as well as high molecular weight anti-HDAC2-reactive proteins with anti-ubiquitin antibody was tested by immunoprecipitation analysis from extracts of HEK293T cells that had been left untreated or were treated with 1.5 mM VPA for 36 h. The proteasome inhibitors ALLN (25 µM) or MG 132 (20 µM) were added 4 h before cell harvest. The presence of ubiquitinated proteins in anti-HDAC2 immunoprecipitates is shown in the middle panel. The left panel shows the corresponding control experiment with preimmune serum instead of the anti-HDAC2 antibody. The right panel shows precipitation with an anti-ubiquitin or non-immune (pre) serum and detection of poly-ubiquitinated HDAC2 protein by western blot analysis. (C) Ubiquitinated HDAC2 was also precipitated from F9 cells that had been treated with 1 mM VPA in experiments comparable to those in (B). One representative example of two independent experiments is shown.
The most common mechanism of targeting proteins for degradation by the proteasome depends on poly-ubiquitination (Hicke, 2001). Therefore, the presence and VPA-dependent induction of HDAC2 ubiquitination were tested. Immunoprecipitates generated with an anti-HDAC2 antibody were analyzed by western blot against ubiquitin for the presence of high molecular weight ubiquitinated proteins. Untreated cells contain only a small amount of ubiquitinated proteins that precipitate with anti-HDAC2 antibody (Figure 3B, middle panel). Pretreatment of cells with VPA alone reduced this signal, possibly due to reduced levels of HDAC2 at the time of analysis. The proteasome inhibitor ALLN alone had no significant effect. Only cotreatment with VPA and proteasome inhibitors dramatically increased the amount of precipitated high molecular weight ubiquitinated proteins. The signal is specific for immunoprecipitates formed with an antibody against HDAC2 since preimmune serum did not precipitate detectable amounts of anti-ubiquitin reactive proteins (Figure 3B, left panel). TSA has no effect on the amount of immunoprecipitated anti-ubiquitin-reactive material (data not shown). These data suggest that VPA treatment induces ubiquitination of one or several proteins precipitated with an antibody against HDAC2. However, this assay does not distinguish whether HDAC2 itself or proteins associated with HDAC2 are poly-ubiquitinated.
To obtain evidence for direct ubiquitination of HDAC2, an anti-ubiquitin antibody was used for immunoprecipitation and western blots were probed with an anti-HDAC2 antibody. A high molecular weight band migrating slower than unmodified HDAC2 was detected, which is consistent with the presence of poly-ubiquitinated HDAC2 (Figure 3B, right panel). Similar to the result of the anti-HDAC2 precipitation, the intensity of the HDAC2-reactive signal was increased in the anti-ubiquitin precipitation only when cells had been treated with both VPA and a proteasome inhibitor. However, mono- and oligo-ubiquitinated forms of HDAC2 could not be detected, possibly due to the small amount of material that can be precipitated and/or insufficient sensitivity of the assay. Similar results were obtained from experiments in F9 cells (Figure 3C). In addition, an increase in high molecular weight material is already seen only after treatment with proteasome inhibitor, suggesting that HDAC2 is also to some extent ubiquitinated in the absence of VPA. In addition to those bands seen in HEK293T cell extracts, F9 cells show prominent bands consistent with mono- and oligo-ubiquitinated HDAC2 in both immunoprecipitations. Similar results were obtained when HEK293T cells were transfected with a His6-tagged form of ubiquitin and Ni2+-NTA–agarose was used for precipitation (data not shown). Under these conditions, bands that are consistent with the mobility of mono- or oligo-ubiquitinated HDAC2 are also detectable in HEK293T cells.
Identification of the ubiquitination machinery for HDAC2
Ubiquitin-conjugating and -ligating enzymes involved in the modification of HDAC2 were identified by two approaches. A systematic search for VPA-inducible genes in F9 cells by suppression subtractive hybridization (SSH) had revealed that expression of the E2 ubiquitin-conjugating enzyme Ubc8 (the ortholog of yeast Ubc4/Ubc5) is induced by VPA (M.Golebiewski, unpublished data). This was confirmed by real-time RT–PCR analysis (Figure 4A) and northern blot analysis of murine F9 and human HEK293T cells using species-specific probes for murine Ubce8 and its human homolog UbcH8, respectively (Figure 4B). Subsequently, the term Ubc8 will refer to either the human or the murine form, depending on which cell type is used. Although the induction of the E2 ligase Ubc8 could account for the increased degradation of HDAC2, it could not explain the lack of an effect in TSA-treated cells since Ubc8 is also induced by TSA at least as efficiently as by VPA (Figure 4A). Ubc8 is also induced at the protein level and, again, there is no substantial difference between VPA and TSA (Figure 4C).

Fig. 4. The HDAC inhibitors VPA and TSA induce expression of the E2 ubiquitin-conjugating enzyme Ubc8, whereas the E3 ligase RLIM is differentially regulated by TSA and VPA. (A) F9 cells were treated for 17 h with 1 mM VPA (V) or 100 nM TSA (T) and expression levels of Ubce8 mRNA were determined by real-time RT–PCR assuming a 1.5-fold amplification per cycle. The amplicon was part of the coding region of the Ubce8 mRNA. Results were normalized for GAPDH expression. Average values ± range of two independent experiments, each with triplicate determinations. (B) Northern blot analysis of 5 µg of poly(A)+ mRNA from F9 or HEK293T cells treated for the indicated times with 1 mM VPA (F9 cells) or 1.5 mM VPA (HEK293T cells) was performed to confirm RT–PCR results. Probes were derived from the 3′-UTR of murine Ubce8 or the 5′ coding region of human UbcH8. Phosphoimager analysis was performed for quantitative evaluation and relative values for Ubc8 mRNA abundance normalized for GAPDH are presented below the panels. One of two experiments with similar results is shown. (C) Ubce8 protein levels were determined by western blot analysis in total extracts of F9 cells that had been treated for 24 h as indicated. (D) HEK293T cells were treated for 24 h with VPA, TSA or ALLN (2.5 µM) as indicated. Abundance of the E3 ubiquitin ligase RLIM was determined by western blot analysis. Actin protein levels were determined to verify equal loading.
The second approach to finding potential ubiquitin ligases for HDAC2 relied on the previous identification of RLIM as a negative regulator of LIM homeodomain transcription factors (LIM-HDs). RLIM represses LIM-HD-dependent gene expression by several apparently complementary mechanisms, which include recruitment of the mSin3 corepressor (Bach et al., 1999) and E3 ubiquitin ligase activity towards the CLIM coactivators of LIM-HDs (Ostendorff et al., 2002). Since RLIM has been shown to interact with a corepressor in vivo, we speculated that it might also act towards other components of corepressor complexes, such as HDACs. The analysis of RLIM protein expression in cells treated with the HDAC inhibitors VPA or TSA revealed a differential response. VPA did not alter RLIM expression, whereas TSA reduced RLIM protein levels (Figure 4D, left panel). Steady-state mRNA levels of RLIM were not changed by any of the HDAC inhibitors (data not shown). Therefore, TSA is likely to affect either synthesis or stability of RLIM protein. The fact that a proteasome inhibitor prevents TSA-dependent downregulation of RLIM protein levels suggests that TSA, in contrast to VPA, induces proteasomal degradation of RLIM by an as yet unidentified mechanism (Figure 4D, right panel). Interestingly, Ubc8 serves as an E2-conjugating enzyme for RLIM so that both proteins could act in concert (Ostendorff et al., 2002).
In order to demonstrate that the regulation of both Ubc8 and RLIM by HDAC inhibitors provides a plausible explanation for the selective downregulation of HDAC2, it is essential to show that they can act on HDAC2 as a substrate. The first indication for that came from the finding that RLIM can be coprecipitated with antibodies against HDAC2 (Figure 5A). RLIM does not coprecipitate with HDACs 1 and 3 (Figure 5A), which is likely to contribute to the selective degradation of HDAC2. We subsequently tested whether HDAC2 is a substrate for Ubc8- and RLIM-dependent ubiquitination by an in vitro ubiquitination assay using bacterially expressed proteins and in vitro translated HDAC2 as a substrate (Figure 5B). HDAC2 was ubiquitinated in vitro, as indicated by both the reduction in non-modified HDAC2 levels and the appearance of slower migrating proteins of the expected mobility of mono-, oligo- and poly-ubiquitinated HDAC2. Efficient ubiquitination was only found when both Ubc8 and RLIM were included in the reactions, but not if only ubiquitin and E1 enzyme were present. This assay appears to be specific, since the GATA-4 transcription factor and its cofactor FOG-2 were not ubiquitinated in a control experiment (data not shown).

Fig. 5. RLIM interacts with HDAC2 and induces poly-ubiquitination in vitro. (A) Interaction of HDACs 1, 2 and 3 with RLIM was tested by co-immunoprecipitation from whole-cell extracts of HEK293T cells that had been pretreated with the proteasome inhibitor ALLN (25 µM) for 4 h. Immunoprecipitations efficiently depleted HDACs from the cell extracts (not shown) and coprecipitated RLIM was detected by western blot analysis. Control immunoprecipitations were performed with non-immune serum. The left lane shows 5% of the extract used for immunoprecipitation. (B) In vitro translated [35S]methionine-labeled HDAC2 was incubated for 2 h in buffer with E1 ubiquitin ligase and ubiquitin only, or in reactions containing the recombinantly expressed E2 ubiquitin ligase Ubce8 without or with the E3 ligase RLIM. For control (left lane), the reaction was stopped immediately after addition of ubiquitin and E1 ligase. Loss of input HDAC2 and appearance of high molecular weight radiolabeled proteins were determined by SDS–PAGE followed by autoradiography.
To test whether HDAC2 is also a substrate of Ubc8 and RLIM in the cell and whether the abundance of these enzymes is rate limiting for HDAC2 degradation, both enzymes were overexpressed by transient transfection. A rate-limiting role of Ubc8 for HDAC2 degradation could be confirmed since increasing amounts of transfected Ubc8 expression vector gradually decrease the abundance of HDAC2 in murine F9 or human HEK293T cells in the absence of VPA (Figure 6A). Interestingly, murine Ubce8 and human UbcH8 exhibit relevant differences since they are fully functional towards HDAC2 only if expressed in cells from their species of origin. Unchanged levels of β-actin and HDACs 1 and 3 (data not shown) indicate that the effect on HDAC2 is specific and not due to general protein degradation.
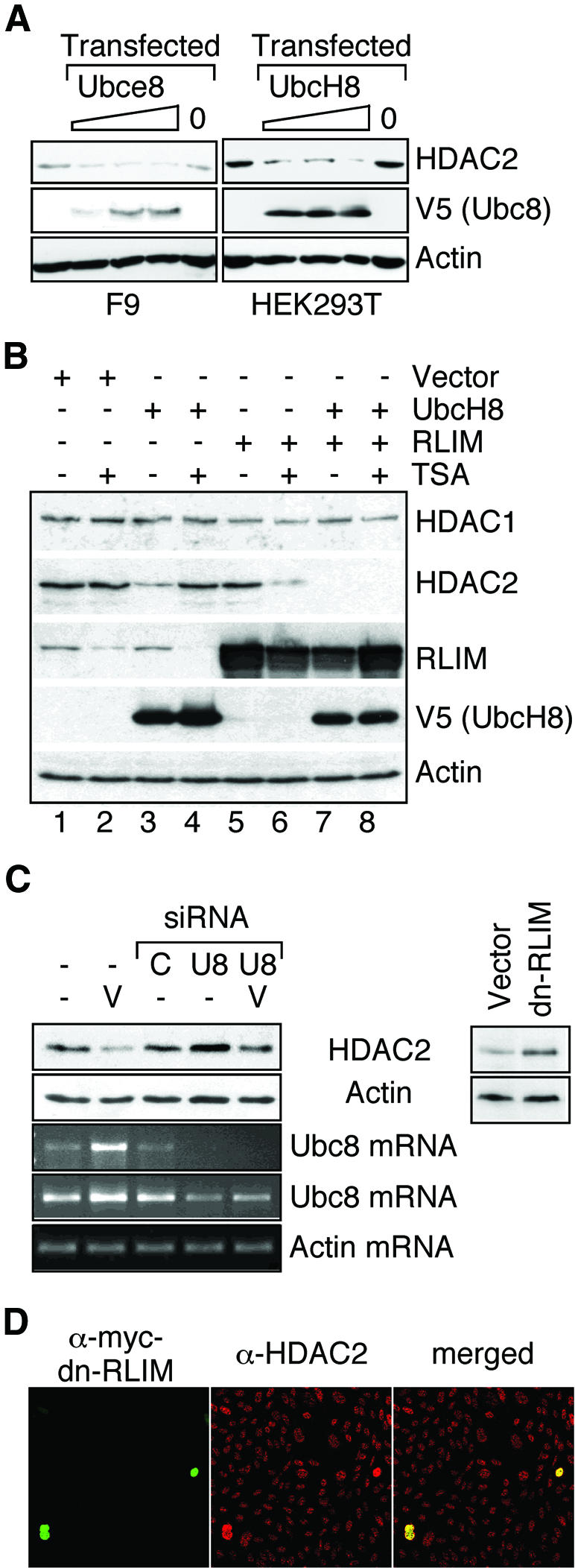
Fig. 6. Identification of HDAC2-ubiquitinating E2 and E3 enzymes as limiting factors for HDAC2 degradation. (A) F9 or HEK293T cells were either left untransfected (left lanes) or transfected with 0.5, 1 or 1.5 µg of Ubce8, UbcH8 or empty expression vector (0). Twenty-four hours after transfection, HDAC2 levels were determined in whole-cell lysates by western blot analysis. The amount of transfected murine Ubce8 or human UbcH8 was analyzed by western blotting against the V5 epitope tag. Similar results were obtained in at least three independent experiments. (B) HEK293T cells were transfected as indicated with expression vectors for UbcH8 or RLIM and empty expression vector. If indicated, cells were treated with 300 nM TSA 24 h after transfection and whole-cell extracts for western blot analysis were prepared an additional 24 h later. One representative of two similar experiments is shown. (C) Endogenous Ubc8 levels were lowered in HEK293T cells (left panel) by transfection of two siRNAs (AACCTCCCTACC ACCTGAAAG and AACTGGAAGCCTTGCACCAAG) directed against the Ubc8 mRNA (U8) or a non-related mRNA (C). HDAC2 levels were determined by western blot analysis in cells without further treatment or after treatment with 1.5 mM VPA (V). Semi-quantitative analysis of Ubc8 mRNA was performed by 30 (upper) or 35 (lower) cycle RT–PCR reactions in comparison to actin (25 cyles). Right panel: HEK293T cells were transfected at high efficiency with an expression vector for a myc-tagged dominant-negative mutant form of RLIM or the empty expression vector. HDAC2 levels were determined 48 h later by western blot analysis. The panels show one representative of at least three independent experiments. Quantitative values for HDAC2 levels were normalized for actin signals. (D) CHO cells were cultured on glass slides and transfected at low efficiency with the myc-tagged dominant-negative mutant form of RLIM. Expression of the transgene (left panel) and endogenous HDAC2 (middle panel) was detected by immunostaining. Representative frames and a merged picture (right panel) are shown.
The functional significance of Ubc8 and RLIM expression levels for HDAC2 degradation was further substantiated by transfection experiments in HEK293T cells. In mock-transfected cells in the absence of HDAC inhibitors, Ubc8 levels are limiting for the degradation of HDAC2 since expression of recombinant Ubc8 causes a decrease in HDAC2 protein concentration (Figure 6B, lanes 1 and 3). Thus VPA-induced overexpression of Ubc8 (Figure 4C) is most likely a determining factor for the selective degradation of HDAC2. Treatment with TSA also leads to an increase in Ubc8 expression (Figure 4C) and simultaneously to a significantly enhanced degradation of RLIM (Figure 6B, lane 2). As a consequence, RLIM apparently becomes a limiting factor and HDAC2 protein levels are virtually unaffected despite induced Ubc8 levels (compare lanes 1 and 2). Furthermore, TSA-mediated degradation of the E3 ligase RLIM (lane 4) renders HDAC2 levels resistant towards overexpression of Ubc8 (compare lanes 3 and 4). Thus, Ubc8 appears to be unable to target HDAC2 for degradation in the absence of RLIM. Apparently, RLIM is not limiting in untreated HEK293T cells since overexpression has no substantial impact on HDAC2 levels (Figure 6B, compare lanes 1 and 5). Only under conditions of elevated Ubc8 expression, as a consequence of either TSA treatment or transfection of an expression vector, does RLIM become limiting and overexpression further reduces HDAC2 levels (Figure 6B, lanes 6, 7 and 8). In this case, the massive overexpression of RLIM seems to exceed the capacity of the TSA-induced degradation mechanism (lanes 6 and 8). Similar results on the role of RLIM were also obtained after overexpression in F9 cells, with the minor difference that both Ubc8 and RLIM appear to be limiting in untreated cells (data not shown). The observation that RLIM is limiting the degradation of HDAC2 only in untreated F9 but not in untreated HEK293T cells is most likely due to lower levels of Ubc8 expression in untreated HEK293T cells. Neither HDAC1 (Figure 6B) nor HDAC3 (data not shown) protein levels were affected under these conditions.
Overexpression and induction studies indicate that ubiquitination by the E2 conjugase Ubc8 and the E3 ligase RLIM are required for proteasomal degradation of HDAC2. Proteasomal degradation apparently also affects HDAC2 levels in cells not treated with VPA since breakdown of HDAC2 upon inhibition of de novo synthesis by cycloheximide is prevented by the proteasome inhibitor ALLN (data not shown). To test whether Ubc8 and RLIM are limiting for HDAC2 turnover in untreated cells, Ubc8 was knocked down by siRNA and RLIM function was impaired by the expression of a dominant-negative mutant of RLIM in which a deletion within the RING finger abolishes E3 ligase activity (Ostendorff et al., 2002). Transfection of siRNA directed against Ubc8 but not a control siRNA induced HDAC2 levels reproducibly by 40% in HEK293T cells (Figure 6C, left panel). In VPA-treated HEK293T cells, the effect of the siRNA was more pronounced (2.3-fold induction of HDAC2). This is consistent with a significant reduction but not complete depletion of Ubc8 mRNA upon siRNA transfection (Figure 6C). Expression of mutant RLIM in HEK293T cells induces a >2-fold increase of endogenous HDAC2 levels, demonstrating that HDAC2 is an in vivo target for RLIM-mediated ubiquitination and subsequent degradation (Figure 6C, right panel). This observation was confirmed at single-cell level by immunocytochemistry (Figure 6D). In contrast to the biochemical analysis, this experiment was performed under low-efficiency transfection conditions. Transfection of the empty expression vector coding for the myc tag and the nuclear localization signal had no effect on HDAC2 levels (data not shown).
The transfection experiments indicate that Ubc8 and RLIM act on HDAC2 in cells. Ubc8 is limiting for HDAC2 degradation. Hence it is plausible that induced expression either by transfection or by VPA treatment leads to a reduction of HDAC2 levels. RLIM is not necessarily limiting in untreated cells, but becomes limiting for HDAC2 turnover after TSA treatment, which induces significant overexpression of Ubc8 and reduces protein levels of RLIM. Therefore, the abundance of both E2-conjugating enzyme and E3 ligase is likely to determine final HDAC2 turnover rates. We conclude that the induction of Ubc8 by VPA is a likely cause for reduction of HDAC2 levels, whereas the combination of TSA-dependent induction of Ubc8 together with reduction of RLIM protein levels leaves HDAC2 turnover virtually unchanged (Figure 7).
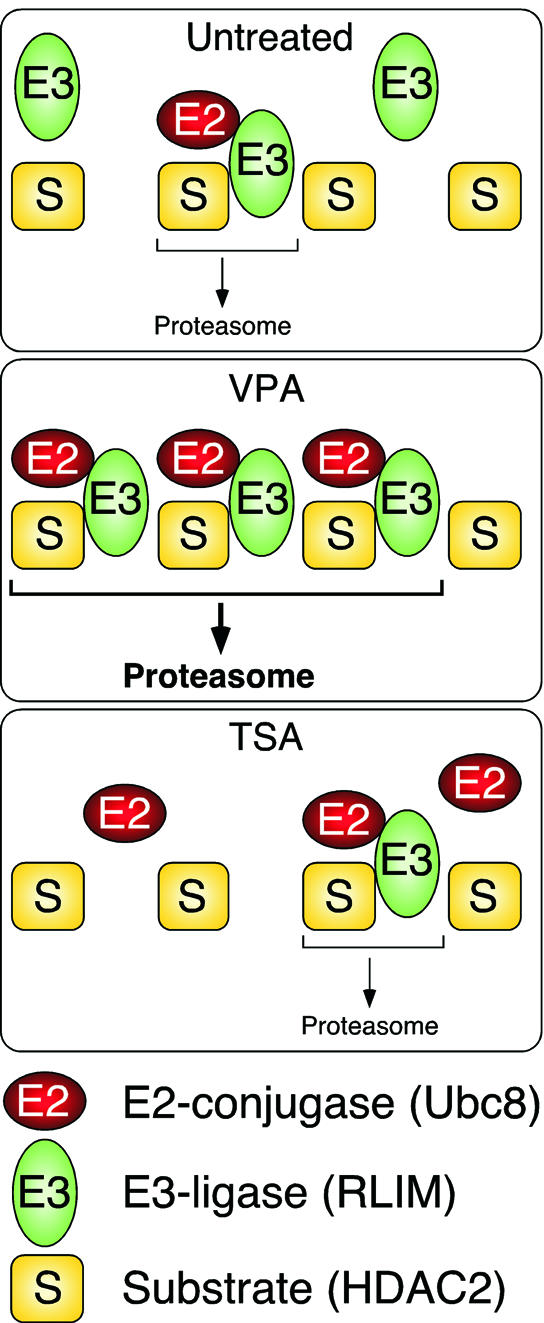
Fig. 7. Schematic representation of the mechanism of HDAC2 degradation. Proteasomal degradation of HDAC2 involves the E3 ubiquitin ligase RLIM and in untreated cells is limited by the abundance of an E2 ubiquitin conjugase, presumably Ubc8. We cannot exclude the involvement of additional E2 and E3 enzymes. In VPA-treated cells, Ubc8 is induced and the degradation of HDAC2 is enhanced. In TSA-treated cells, the amount of RLIM is reduced in addition to induction of Ubc8. Under these conditions, the abundance of the ubiquitin ligase RLIM limits the rate of HDAC2 degradation to a level similar to that in untreated cells.
Discussion
Our study shows the tagging of HDAC2 for proteasomal degradation by the Ubc8 E2 ubiquitin conjugase and the RLIM E3 ubiquitin ligase. This degradation contributes to basal turnover of HDAC2 and is differentially regulated by compounds that also inhibit the catalytic activity of HDACs (Figure 7). The amount of Ubc8 appears to be limiting for the degradation rate of HDAC2 under untreated conditions. Therefore, induction of Ubc8 results in an increased turnover of HDAC2, when VPA is used as an HDAC inhibitor. Under conditions of induced E2 enzyme levels, the E3 ligase RLIM, which is associated with Ubc8, becomes limiting for HDAC2 degradation. Since some HDAC inhibitors such as TSA reduce the amounts of RLIM, the induction of the E2 enzyme is compensated for and no significant effect on HDAC2 protein levels is observed. However, RLIM is not downregulated if the E2 enzyme is induced by VPA and therefore a net increase in HDAC2 degradation occurs.
Ubiquitination and SUMO modification have been found in HDACs 1 and 4 (David et al., 2002; Kirsh et al., 2002). Induced proteasomal degradation of HDAC1 upon treatment with quinidine has also been reported (Zhou et al., 2000). The proteins responsible for ubiquitination of HDAC1 have not been identified and are obviously different from those that are limiting for basal and induced HDAC2 turnover. Conditions inducing HDAC2 degradation do not affect levels of the other HDACs that were analyzed (HDACs 1, 3, 4, 5 and 8). Specificity with respect to the HDAC isoenzyme targeted for degradation is most likely established by the selective interaction of RLIM with HDAC2 but not with the other HDACs.
Since all HDAC inhibitors tested induce Ubc8 mRNA expression, this upregulation is likely to be due to inhibition of HDAC-dependent repression. The lack of induction when protein synthesis is inhibited by cycloheximide (data not shown) and a lag period in mRNA induction suggest that HDAC inhibition could induce an intermediary gene product rather than Ubc8 gene expression itself.
Our results indicate that the proteasome-dependent downregulation of RLIM protein levels determines the differential effects of HDAC inhibitors on HDAC2 degradation. TSA, MS-27-275 and probably also other HDAC inhibitors (see Figure 1A) lead to reduced RLIM levels, whereas VPA and butyrate apparently do not. Thus, specificity of HDAC2 degradation with respect to the inducing compound is established by the selective downregulation of RLIM by TSA and other HDAC inhibitors, but not by VPA. Proteasome-dependent degradation of RLIM is consistent with the finding that RLIM can be poly-ubiquitinated (Ostendorff et al., 2002). However, it is still unknown how RLIM degradation is induced by TSA and whether this is associated with the HDAC inhibitory activity of TSA. Since downregulation of RLIM appears to occur with a delay of several hours after treatment of cells with TSA, it is likely that induction of intermediary gene products is required. A plausible explanation would be the induction of an as yet unidentified ubiquitin conjugase or ligase that is relevant for the turnover of RLIM. VPA, in contrast to most other HDAC inhibitors, preferentially inhibits class I HDACs (Göttlicher et al., 2001). A likely explanation for the failure of VPA to downregulate RLIM could be that induction of this putative ubiquitinating enzyme depends preferentially on inhibition of class II HDACs. Another possible explanation could be that stabilization of RLIM is an HDAC class II-dependent process. However, this is unlikely, since protein acetylation has been described to protect for example p53 from degradation. We are not aware of the opposite case, for example, that increased acetylation as a consequence of HDAC inhibition would promote protein degradation. Finally, a complicated model might involve the degradation of RLIM in a complex together with its HDAC2 substrate. The type of HDAC inhibitor bound to HDAC2 might cause a conformational change that alters the susceptibility of RLIM to degradation.
Irrespective of the mechanistic details of RLIM downregulation by TSA, the ubiquitination and turnover rates of HDAC2 upon HDAC inhibition are regulated by the abundance of relevant E2 or E3 enzymes. There are many examples where post-translational modification of the substrate protein (e.g. the cell cycle-regulating proteins NF-κB, p53 or β-catenin) or the ubiquitinating enzymes is essential for the control of turnover rates. However, there are only few examples where abundance of a ubiquitin ligase or conjugase appears to play a critical role; for example, overexpression of UbcH10 leads to a decrease in levels of geminin H and cyclins A and B (reviewed in Glickman and Ciechanover, 2002). Furthermore, Skp2, a component of an SCF E3–ligase complex that is rate limiting for degradation of the p27Kip1 cyklin/CDK inhibitor, is expressed in a cell cycle-dependent fashion (Carrano et al., 1999; reviewed in Glickman and Ciechanover, 2002). Regulation of HDAC2 turnover by induction of the E2 ubiquitin conjugase is expected to be relevant for mediating the activity of drugs such as VPA in cells, mice and presumably also VPA-treated patients. Aberrant ubiquitination is thought to play a role in many diseases such as cancer or neurodegenerative diseases. Interestingly, Ubc8 was found to interact with the ring-finger ubiquitin ligase Parkin that has been linked to the pathogenesis of Parkinson’s disease (reviewed in Glickman and Ciechanover, 2002). Faulty ubiquitination is mostly caused by defects in the ligating enzymes or the substrates. The notion that expression of ubiquitin conjugases or ligases is subject to conditional regulation suggests an additional mechanism for disease development.
The physiological and pathophysiological consequences of aberrant expression of individual HDACs are difficult to judge at present. Different sets of HDAC isoenzymes are probably relevant for the regulation of different target genes or proteins. This notion is supported by pilot experiments indicating that the isoenzyme-selective inhibitor VPA alters the expression of only a small subset of those target genes that are affected by the non-specific inhibitor TSA (M.Golebiewski, O.H.Krämer and T.Heinzel, unpublished data). Despite significant similarities in a large family of HDAC enzymes, individual HDACs (e.g. HDAC1 and 9) appear to be limiting for distinct physiological processes during embryonic development (Lagger et al., 2002; Zhang et al., 2002). Selective modulators of individual HDACs would be useful tools for the dissection of those aspects of genetic programs that critically depend on individual HDACs during adult life in addition to those that are disrupted by knockout strategies during embryonic development.
HDAC inhibitors are being considered as anticancer drugs because of their potential to induce differentiation or apoptosis preferentially in cancer cells and not in non-transformed cells (Krämer et al., 2001; Marks et al., 2001; Melnick and Licht, 2002). The selective modulation of only a critical subset of HDACs is likely to determine therapeutic efficiency and side-effects. By one mechanism of action, VPA inhibits the catalytic activity of HDACs, preferentially of class I, presumably by binding to the catalytic center of the enzyme (Göttlicher et al., 2001). Here we show as an additional mechanism downregulation of HDAC2 by proteasomal degradation induced by VPA. The combined effects apparently suffice to induce differentiation or apoptosis in transformed cells of hematopoietic or non-hematopoietic origin (Regan, 1985; Driever et al., 1999; Göttlicher et al., 2001). The specific downmodulation of HDAC2 by degradation and inhibition of the residual enzyme may be particularly relevant since only HDAC2, but not HDACs 1 and 3, was found to be overexpressed in tumor cell lines as compared with normal cells originating from corresponding tissues (Yang et al., 1997). Furthermore, HDAC2 appears to be expressed at increased levels in many human colonic tumor samples, where the adenomatosis polyposis coli (APC) β-catenin pathway appears to contribute to regulation of HDAC2 expression (P.Zhu, J.Mengwasser, E.Martin and M.Göttlicher, unpublished data). Finally, in the case of the polycomb group protein EZH2, which appears to be involved in the progression of prostate cancer, the repression of a series of target genes has been associated with the recruitment of HDAC2 into EZH2 protein complexes (van der Vlag and Otte, 1999; Varambally et al., 2002). Some of those genes repressed by ectopic expression of EZH2 may be expected to be de-repressed in VPA-treated cells. The human genes repressed upon EZH2 expression (Varambally et al., 2002) were compared with those induced by VPA in murine F9 or a human melanoma cell line. Despite the use of different cell and array types, 38 and 50 genes, respectively, were detected in our analysis and of those 7 (18%) or 18 (36%), respectively, were indeed induced by VPA (O.H.Krämer and M.Golebiewski, unpublished data).
In summary, the present study shows that HDAC2 activity can not only be modulated by the binding of inhibitors to the catalytic center, but also by selective regulation of its protein levels. Apparently, fine-tuned degradation through the ubiquitin proteasome pathway with limiting amounts of the E2 ubiquitin conjugase Ubc8 and the E3 ubiquitin ligase RLIM maintains a balanced steady-state protein level of HDAC2 that is susceptible to modulation by low molecular weight compounds such as VPA. Ultimately, we expect that our findings could aid in the definition of HDAC isoenzyme-specific target genes and contribute to the development of novel strategies for the treatment of malignant disease linked to the overexpression of HDAC2.
Materials and methods
Cell culture and standard extract preparation have been described (Göttlicher et al., 2001). For transient transfections, cells were seeded in 12-well dishes 24 h before transfection. At 60–70% confluency, cells were transfected with a total of 2 µg of plasmid DNA per well using 5 µl of Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. For the experiment shown in Figure 6B, HEK293T cells were seeded in 10 cm dishes and transfected using polyethyleneimine with expression vectors for UbcH8, RLIM or empty expression vector with a total of 15 µg of DNA. Cells were lysed 24 h later in SDS–PAGE loading buffer or NET-N buffer to prepare whole-cell extracts for western blot analysis or immunoprecipitation.
Western blot, immunoprecipitations and in vivo ubiquitination analysis
Antibodies for western blot analysis were obtained from Santa Cruz Biotechnology if not stated otherwise: HDAC1, sc6298; HDAC2, sc7899, sc9959; HDAC3, sc8138; HDAC4, sc5245; HDAC5, Upstate 07-045; HDAC8, sc11405; Ubce8, sc16200; ubiquitin, Sigma U5379, Chemicon MAB1510; v5-tag, Invitrogen 46-0705; actin, sc1616 and Sigma A2066/A5060. The immune serum against RLIM has been described (Ostendorff et al., 2002). All western blots were probed for actin to ensure equal loading of samples. Immunoprecipitations were performed as described using 1 µg of antibody (Heinzel et al., 1997). For co-immunoprecipitations of RLIM and HDACs, 293T cells (10 cm dish, 60% confluency) were crosslinked with 1% formaldehyde in phosphate-buffered saline for 10 min at room temperature. The reaction was stopped by adding 1 M glycine for an additional 5 min. Cells were harvested and lysed in NETN buffer. Following co-immunoprecipitation, crosslinks were reversed prior to loading by boiling the samples in Laemmli buffer.
Immunohistochemistry
Female BALB/c-mice were treated with 800 mg/kg VPA subcutaneously for 10 days. Organs were fixed in formaldehyde and embedded in paraffin. Deparaffinized sections were incubated with a primary anti-HDAC2 antibody (Zymed, 51-5100) at a dilution of 1:30. A peroxidase-labeled secondary antibody (Dianova 111-065-045) was used for detection.
Metabolic labeling and pulse–chase analysis
Appropriately pretreated cells were starved in methinonine/cysteine-deficient Dulbecco’s modified Eagle’s medium (Sigma) for 30 min. Metabolic labeling was performed for 1 h (pulse-labeling experiments) or 3 h (pulse–chase analysis) in the presence of 3.7 MBq/ml Pro-mix [35S] (Amersham Pharmacia). For pulse–chase analysis, cells were washed and incubated in normal methionine- and cysteine-containing medium for 3 or 6 h. Radiolabeled HDACs were detected by specific immunoprecipitation from cell lysates in RIPA buffer followed by SDS–PAGE and autoradiography.
Cloning of Ubc8 open reading frames
First-strand cDNA from murine F9 cells or a human fetal brain cDNA library were used as templates in 30 cycle PCR reactions with annealing temperatures of 60°C (mouse) or 58°C (human). Primers for murine Ubce8 covered the start codon (CACGCTCCAACCCGAGATG) and the last 18 bases before the TAA stop codon (AGAGGGCCGGTCC ACTCC). PCR of human UbcH8 included the same 3′ primer and the 5′ primer CACGGGTGCCACACACTCGGT. The amplicons of 475 bp (Ubce8) and 486 bp (UbcH8) were cloned into pcDNA3.1/V5-His-Topo-TA (Invitrogen) and verified by sequencing.
Quantitative real-time PCR
First-strand cDNA (1–50 ng) from treated F9 cells was used as the template together with 250 nM of each primer in a quantitative PCR with Sybr Green to monitor progress of DNA synthesis. A hot-start two-step cycle program with 15 s at 95°C and 60 s at 60°C was performed on a Perkin–Elmer ABI PRISM 7700 light cycler. Each reaction product was checked on agarose gels stained with ethidium bromide for specificity of amplification. Delta-Ct values of each sample were normalized by subtraction of the delta-Ct values obtained from the amplification of a GAPDH fragment from the same first-strand cDNA synthesis. Ubce8 primers flanking the coding region were used (GGAGTTCAGA GAGCGGTGTCC and GACGATCCAGGCTTCAGAACA). GAPDH primers for a 387 bp amplicon were GCCGATGCCCCCATGTTTGT and CTTGGCAGGTTTCTCCAGGCG.
Acknowledgments
Acknowledgements
The authors thank Annemarie Schimpf and Margarethe Litfin for excellent technical assistance, Irene Huber for help with the cloning of UbcH8 and Claudia Bürger for technical advice. Elke Martin and Alexander Maurer, G2M Cancerdrugs AG, were invaluable discussion partners throughout the entire project. Matthias Truss generously provided siRNAs and Dirk Bohman provided the expression vector for His-tagged ubiquitin. Support by the Forschungszentrum Karlsruhe through PhD fellowships to P.Z. and M.Gol. is gratefully acknowledged. This work was supported by grants from the NGFN (German National Genome Research Network) and the EC to T.H.
References
- Alland L., Muhle,R., Hou,H.,Jr, Potes,J., Chin,L., Schreiber-Agus,N. and DePinho,R.A. (1997) Role for N-CoR and histone deacetylase in Sin3-mediated transcriptional repression. Nature, 387, 49–55. [DOI] [PubMed] [Google Scholar]
- Bach I. et al. (1999) RLIM inhibits functional activity of LIM homeodomain transcription factors via recruitment of the histone deacetylase complex. Nat. Genet., 22, 394–399. [DOI] [PubMed] [Google Scholar]
- Carrano A.C., Eytan,E., Hershko,A. and Pagano,M. (1999) SKP2 is required for ubiquitin-mediated degradation of the CDK inhibitor p27. Nat. Cell Biol., 1, 193–199. [DOI] [PubMed] [Google Scholar]
- David G., Neptune,M.A. and DePinho,R.A. (2002) SUMO-1 modification of histone deacetylase 1 (HDAC1) modulates its biological activities. J. Biol. Chem., 277, 23658–23663. [DOI] [PubMed] [Google Scholar]
- Driever P.H., Knüpfer,M.M., Cinatl,J. and Wolff,J.E. (1999) Valproic acid for the treatment of pediatric malignant glioma. Klin. Pädiatr., 211, 323–328. [DOI] [PubMed] [Google Scholar]
- Gelmetti V., Zhang,J., Fanelli,M., Minucci,S., Pelicci,P.G. and Lazar,M.A. (1998) Aberrant recruitment of the nuclear receptor corepressor-histone deacetylase complex by the acute myeloid leukemia fusion partner ETO. Mol. Cell Biol., 18, 7185–7191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass C.K. and Rosenfeld,M.G. (2000) The coregulator exchange in transcriptional functions of nuclear receptors. Genes Dev., 14, 121–141. [PubMed] [Google Scholar]
- Glickman M.H. and Ciechanover,A. (2002) The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol. Rev., 82, 373–428. [DOI] [PubMed] [Google Scholar]
- Göttlicher M. et al. (2001) Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J., 20, 6969–6978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray S.G. and Ekström,T.J. (2001) The human histone deacetylase family. Exp. Cell Res., 262, 75–83. [DOI] [PubMed] [Google Scholar]
- Grignani F. et al. (1998) Fusion proteins of the retinoic acid receptor-α recruit histone deacetylase in promyelocytic leukaemia. Nature, 391, 815–818. [DOI] [PubMed] [Google Scholar]
- Guidez F., Ivins,S., Zhu,J., Söderström,M., Waxman,S. and Zelent,A. (1998) Reduced retinoic acid-sensitivities of nuclear receptor corepressor binding to PML- and PLZF-RARα underlie molecular pathogenesis and treatment of acute promyelocytic leukemia. Blood, 91, 2634–2642. [PubMed] [Google Scholar]
- He L.Z., Guidez,F., Tribioli,C., Peruzzi,D., Ruthardt,M., Zelent,A. and Pandolfi,P.P. (1998) Distinct interactions of PML-RARα and PLZF-RARα with co-repressors determine differential responses to RA in APL. Nat. Genet., 18, 126–135. [DOI] [PubMed] [Google Scholar]
- Heinzel T. et al. (1997) A complex containing N-CoR, mSin3 and histone deacetylase mediates transcriptional repression. Nature, 387, 43–48. [DOI] [PubMed] [Google Scholar]
- Hicke L. (2001) Protein regulation by monoubiquitin. Nat. Rev. Mol. Cell Biol., 2, 195–201. [DOI] [PubMed] [Google Scholar]
- Hildebrand D., Tiefenbach,J., Heinzel,T., Grez,M. and Maurer,A.B. (2001) Multiple regions of ETO cooperate in transcriptional repression. J. Biol. Chem., 276, 9889–9895. [DOI] [PubMed] [Google Scholar]
- Huang E.Y., Zhang,J., Miska,E.A., Guenther,M.G., Kouzarides,T. and Lazar,M.A. (2000) Nuclear receptor corepressors partner with class II histone deacetylases in a Sin3-independent repression pathway. Genes Dev., 14, 45–54. [PMC free article] [PubMed] [Google Scholar]
- Kao H.Y., Downes,M., Ordentlich,P. and Evans,R.M. (2000) Isolation of a novel histone deacetylase reveals that class I and class II deacetylases promote SMRT-mediated repression. Genes Dev., 14, 55–66. [PMC free article] [PubMed] [Google Scholar]
- Kirsh O. et al. (2002) The SUMO E3 ligase RanBP2 promotes modification of the HDAC4 deacetylase. EMBO J., 21, 2682–2691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzarides T. (2000) Acetylation: a regulatory modification to rival phosphorylation? EMBO J., 19, 1176–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krämer O.H., Göttlicher,M. and Heinzel,T. (2001) Histone deacetylase as a therapeutic target. Trends Endocrinol. Metab., 12, 294–300. [DOI] [PubMed] [Google Scholar]
- Lagger G. et al. (2002) Essential function of histone deacetylase 1 in proliferation control and CDK inhibitor repression. EMBO J., 21, 2672–2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laherty C.D., Yang,W.M., Sun,J.M., Davie,J.R., Seto,E. and Eisenman,R.N. (1997) Histone deacetylases associated with the mSin3 corepressor mediate mad transcriptional repression. Cell, 89, 349–356. [DOI] [PubMed] [Google Scholar]
- Lin R.J., Nagy,L., Inoue,S., Shao,W., Miller,W.H.,Jr. and Evans,R.M. (1998) Role of the histone deacetylase complex in acute promyelocytic leukaemia. Nature, 391, 811–814. [DOI] [PubMed] [Google Scholar]
- Lutterbach B. et al. (1998) ETO, a target of t(8;21) in acute leukemia, interacts with the N-CoR and mSin3 corepressors. Mol. Cell. Biol., 18, 7176–7184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks P.A., Richon,V.M., Breslow,R. and Rifkind,R.A. (2001) Histone deacetylase inhibitors as new cancer drugs. Curr. Opin. Oncol., 13, 477–483. [DOI] [PubMed] [Google Scholar]
- Maurer A.B., Wichmann,C., Gross,A., Kunkel,H., Heinzel,T., Ruthardt,M., Groner,B. and Grez,M. (2002) The Stat5-RARα fusion protein represses transcription and differentiation through interaction with a corepressor complex. Blood, 99, 2647–2652. [DOI] [PubMed] [Google Scholar]
- McKinsey T.A., Zhang,C.L., Lu,J. and Olson,E.N. (2000) Signal-dependent nuclear export of a histone deacetylase regulates muscle differentiation. Nature, 408, 106–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melnick A. and Licht,J.D. (2002) Histone deacetylases as therapeutic targets in hematologic malignancies. Curr. Opin. Hematol., 9, 322–332. [DOI] [PubMed] [Google Scholar]
- Nagy L., Kao,H.Y., Chakravarti,D., Lin,R.J., Hassig,C.A., Ayer,D.E., Schreiber,S.L. and Evans,R.M. (1997) Nuclear receptor repression mediated by a complex containing SMRT, mSin3A and histone deacetylase. Cell, 89, 373–380. [DOI] [PubMed] [Google Scholar]
- Ostendorff H.P., Peirano,R.I., Peters,M.A., Schlüter,A., Bossenz,M., Scheffner,M. and Bach,I. (2002) Ubiquitination-dependent cofactor exchange on LIM homeodomain transcription factors. Nature, 416, 99–103. [DOI] [PubMed] [Google Scholar]
- Phiel C.J., Zhang,F., Huang,E.Y., Guenther,M.G., Lazar,M.A. and Klein,P.S. (2001) Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer and teratogen. J. Biol. Chem., 276, 36734–36741. [DOI] [PubMed] [Google Scholar]
- Regan C.M. (1985) Therapeutic levels of sodium valproate inhibit mitotic indices in cells of neural origin. Brain Res., 347, 394–398. [DOI] [PubMed] [Google Scholar]
- Strahl B.D. and Allis,C.D. (2000) The language of covalent histone modifications. Nature, 403, 41–45. [DOI] [PubMed] [Google Scholar]
- van der Vlag J. and Otte,A.P. (1999) Transcriptional repression mediated by the human polycomb-group protein EED involves histone deacetylation. Nat. Genet., 23, 474–478. [DOI] [PubMed] [Google Scholar]
- Varambally S. et al. (2002) The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature, 419, 624–629. [DOI] [PubMed] [Google Scholar]
- Wang J., Hoshino,T., Redner,R.L., Kajigaya,S. and Liu,J.M. (1998) ETO, fusion partner in t(8;21) acute myeloid leukemia, represses transcription by interaction with the human N-CoR/mSin3/HDAC1 complex. Proc. Natl Acad. Sci. USA, 95, 10860–10865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warrell R.P., He,L.Z., Richon,V., Calleja,E. and Pandolfi,P.P. (1998) Therapeutic targeting of transcription in acute promyelocytic leukemia by use of an inhibitor of histone deacetylase. J. Natl Cancer Inst., 90, 1621–1625. [DOI] [PubMed] [Google Scholar]
- Yang W.M., Yao,Y.L., Sun,J.M., Davie,J.R. and Seto,E. (1997) Isolation and characterization of cDNAs corresponding to an additional member of the human histone deacetylase gene family. J. Biol. Chem., 272, 28001–28007. [DOI] [PubMed] [Google Scholar]
- Zhang C.L., McKinsey,T.A., Chang,S., Antos,C.L., Hill,J.A. and Olson,E.N. (2002) Class II histone deacetylases act as signal-responsive repressors of cardiac hypertrophy. Cell, 110, 479–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q., Melkoumian,Z.K., Lucktong,A., Moniwa,M., Davie,J.R. and Strobl,J.S. (2000) Rapid induction of histone hyperacetylation and cellular differentiation in human breast tumor cell lines following degradation of histone deacetylase-1. J. Biol. Chem., 275, 35256–35263. [DOI] [PubMed] [Google Scholar]