

Abstract
Herein, we define how MEKK1, a MAPK kinase kinase, regulates cell migration. MEKK1 is associated with actin fibers and focal adhesions, localizing MEKK1 to sites critical in the control of cell adhesion and migration. EGF-induced ERK1/2 activation and chemotaxis are inhibited in MEKK1–/– fibroblasts. MEKK1 deficiency causes loss of vinculin in focal adhesions of migrating cells, increased cell adhesion and impeded rear-end detachment. MEKK1 is required for activation of the cysteine protease calpain and cleavage of spectrin and talin, proteins linking focal adhesions to the cytoskeleton. Inhibition of ERK1/2 or calpain, but not of JNK, mimics MEKK1 deficiency. Therefore, MEKK1 regulates calpain-mediated substratum release of migrating fibroblasts.
Keywords: calpain/MEKK1/rear-end detachment
Introduction
Cell migration involves at least four basic components: extension of the leading edge, adherence to the substratum, release of adherence at the trailing edge and retraction of the trailing uropod. While adherence is necessary to exert force on a surface and produce forward movement, release of adherence at the uropod must occur for cell migration to continue. Control of adherence, and release thereof, is thus a critical regulatory function for migrating cells. Whereas extension and adherence have been studied extensively, few proteins have been characterized as regulators of the adhesion-release process during cell migration. Recently, the cysteine proteinase calpain has emerged as an important regulator of cell adhesion (Huttenlocher et al., 1997; Bialkowska et al., 2000). Originally referred to as Ca2+-dependent neutral protease (Beckerle et al., 1987), the calpain family includes two ubiquitously expressed isoforms, µ- and m-calpain (murine calpains 1 and 2, respectively) (Huang and Wang, 2001). Calpains have been suspected to regulate cell adhesion due to their ability to cleave several focal adhesion and cytoskeletal proteins (Pfaff et al., 1999; Wang and Yuen, 1999). Further, calpain has been shown to be associated with cell adhesion complexes (Beckerle et al., 1987), and calpain-deficient fibroblasts display inhibited migration (Arthur et al., 2000; Dourdin et al., 2001). Overexpression of the calpain inhibitor calpastatin also impairs cell detachment and migration (Potter et al., 1998). Calpain has been proposed to regulate cell detachment through proteolytic cleavage of adhesion complex proteins in the trailing edge of migrating cells (Palecek et al., 1998). An understanding of calpain regulation during migration has remained elusive. Recently, the MEK/ERK1/2 signaling pathway has been linked to calpain activation as the pharmacological MEK inhibitor PD98059 was shown to inhibit EGF-induced calpain activation (Glading et al., 2000). Subsequent studies revealed that membrane-proximal ERK1/2 activity is required for EGF-induced m-calpain activation and cell migration (Glading et al., 2001).
MEKK1 is a MAPK kinase kinase that is activated in response to changes in cell shape and the microtubule cytoskeleton (Yujiri et al., 1999). Dual regulation of JNK and ERK1/2 pathways by low concentrations of growth factors like EGF and by microtubule-disrupting agents like nocodazole is mediated by MEKK1 (Yujiri et al., 1998). Targeted gene disruption of MEKK1 demonstrated that it functionally regulates cell migration both in vivo and in vitro (Yujiri et al., 2000). Our results reveal an association of MEKK1 with focal adhesions and actin fibers entering the focal adhesions. MEKK1 regulates the limited proteolysis of the focal adhesion proteins talin and spectrin. Characterization of MEKK1-null mouse embryo fibroblasts demonstrates that MEKK1 regulates the ERK1/2 pathway for control of calpain-catalyzed rear-end detachment.
Results
MEKK1 regulates cell adhesion
Comparison of wild-type and MEKK1–/– fibroblasts readily defines a function for MEKK1 in regulating cell adhesion and migration. We found that MEKK1–/– fibroblasts have a marked increase in adherence to the culture substratum relative to wild-type fibroblasts (Figure 1A). Using wild-type and MEKK1–/– cells that had been serum starved and then challenged with or without serum, the culture plates were inverted and centrifuged. The remaining attached cells were counted as a measure of adherence, with greater number of cells after centrifugation being indicative of increased adherence (Lotz et al., 1989). This simple assay clearly demonstrates the increased adherence of MEKK1–/– cells relative to wild-type fibroblasts. Even though MEKK1–/– cells show an increased adherence in the centrifugation assay, their rate of attachment on fibronectin or tissue culture plastic alone is similar to the rate of attachment of wild-type cells (Figure 1B). The increased adherence in the centrifugation assay, but similar attachment rate of MEKK1–/– compared with wild-type fibroblasts, suggested MEKK1 regulates release of cell adhesion. We used live cell microscopy to show that MEKK1–/– fibroblasts are, indeed, inhibited in random migration and rear-end detachment (see Supplementary data available at The EMBO Journal Online). Both wild-type and MEKK1–/– fibroblasts are capable of extending lamellipodia necessary for forward movement, and both cell types develop tails or uropods at the trailing edge as the cell moves in the direction of the leading edge. However, as wild-type fibroblasts have the ability to detach and retract their trailing edge, MEKK1–/– cell forward progress is impeded by an inability of the uropod to detach from the substrate, thereby giving the appearance that MEKK1–/– cells are tethered at their trailing end. Thus, the increased adherence of MEKK1–/– cells is, at least in part, a result of defective detachment from the substratum.
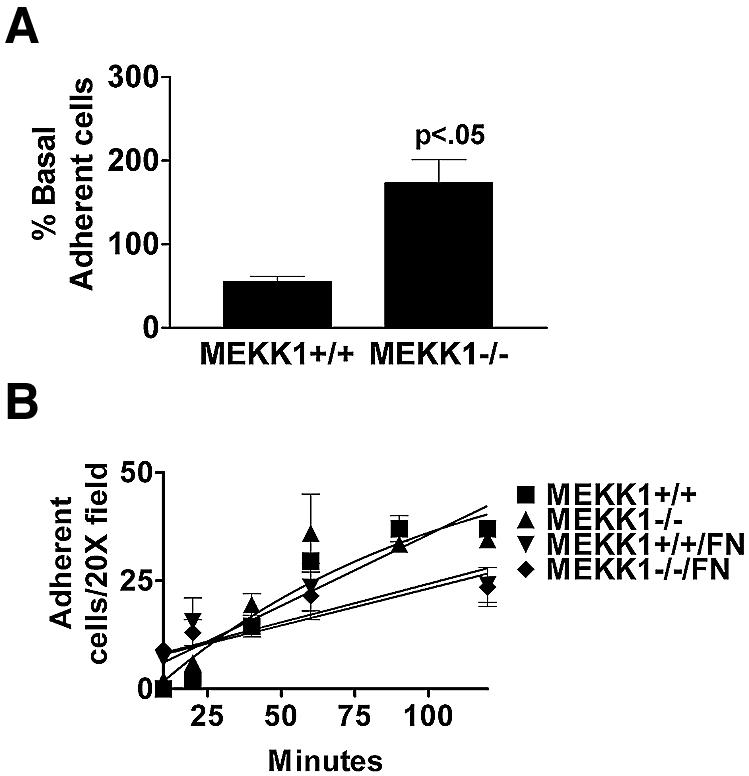
Fig. 1. MEKK1-deficient fibroblasts show increased adherence characteristic of a defect in rear-end detachment. (A) Wild-type or MEKK1–/– MEFs were serum starved for 8 h, then treated with media with or without 10% fetal bovine serum (FBS). The plates were then inverted and centrifuged at 2300 g for 5 min. Adherent cells remaining attached to the well surface were stained with Wright’s stain and quantitated. Cell adherence is represented as the percent of the total serum-treated cells compared with the non-treated cells; 100% was taken as the number of wild-type cells in the dish before serum challenge, inversion and centrifugation. MEKK1–/– cells with serum challenge is >100% because more cells are retained after centrifugation than for the non-serum-stimulated wild-type cells, indicative of the increased adherence of MEKK1–/– cells. Results shown are the mean ± SEM of at least three independent experiments, and the statistical significance was determined by Student’s t-test. (B) Fibroblasts were resuspended in complete media and allowed to attach to either untreated or fibronectin-coated tissue culture plates. Cells were monitored for 2 h and the number of attached cells determined by phase microscopy. A digital movie of migrating MEKK1+/+ and MEKK1–/– fibroblasts is available as Supplementary data.
MEKK1 regulates directed cell migration
Inhibited movement of MEKK1–/– fibroblasts was readily demonstrated using both transwell migration and in vitro wound response assays. Chemotaxis toward serum is inhibited in MEKK1–/– compared with wild-type fibroblasts (Figure 2A), consistent with a defect in cell movement that would be observed with a loss of rear-end detachment. Further, when soluble fibronectin is used as a chemotactic agent, migration is reduced by 50% in MEKK1–/– versus wild-type cells (Figure 2B). Although EGF by itself is a weak chemotactic agent, EGF combined with fibronectin produces a synergistic effect relative to fibronectin alone to induce chemotaxis (Maheshwari et al., 1999) (Figure 2B). This synergistic effect is completely absent in MEKK1-deficient cells, thus demonstrating that MEKK1 is required for the EGF/fibronectin-induced fibroblast migration.

Fig. 2. MEKK1 expression is necessary for fibroblast migration. (A) Fibroblasts were seeded into the upper chamber of a Transwell migration plate with 5% FBS in the lower chamber. Cells traversed after 5 h to the lower surface of the membrane were quantitated. The results shown are the mean ± SEM of at least three independent experiments. (B) Fibroblasts were treated as in (A) except that the bottom well of the Transwell contained either 1 nM EGF, 100 µg/ml fibronectin or the combination of EGF and fibronectin. (C and E) Wild-type or MEKK1–/– fibroblasts were seeded onto coverslips and allowed to grow overnight. In addition, MEKK1–/– fibroblasts stably transfected with full-length MEKK1 (Add-back) were analyzed. Each confluent culture was ‘wounded’ with a razor and observed over the course of 5 h for migration into the wound space (in vitro wound healing assay). (C) is a DIC image of migrating cells. (D) The time required for confluent fibroblasts in a tissue culture plate to close a standardized wound (200 µm) is represented by the graph. Results shown are the mean ± SEM of at least three independent experiments, and the statistical significance was determined by Student’s t-test (*P < 0.05). (E) Cells were first stained with the fluorescent vital dyes PKH26 (red; MEKK1+/+) or PKH67 (green; MEKK1–/–) and then mixed in equal numbers before seeding onto coverslips, and treated as described above. The fluorescence image depicts the migration of MEKK1+/+ (wild type) and MEKK1–/– cells in co-culture. The data are representative samples from at least three independent experiments.
When a confluent contact-inhibited fibroblast culture is ‘wounded’ using a razor swipe, the cells along the edge of the wound are contact inhibited on all sides save one, and will migrate into the wound opening. Wild-type fibroblasts will migrate into the wound space within 5 h after initiation of the wound (Figure 2C). In contrast, MEKK1–/– fibroblast migration into the open area of the wound is markedly inhibited. Indeed, the time required by MEKK1–/– fibroblasts to completely close a standardized wound (24.5 h) is significantly (P < 0.05) prolonged compared with the MEKK1+/+ cells (11 h) (Figure 2D). By 12 h post-wounding, some MEKK1–/– fibroblasts have migrated into the wound site but do not reach numbers or distances achieved by MEKK1+/+ fibroblasts. This indicates that MEKK1–/– fibroblasts are markedly defective, but not completely inhibited in directed migration. The loss of migration is a direct consequence of MEKK1 deficiency because MEKK1 add-back by transfection of MEKK1–/– cells restores migration into the wound (Figure 2C and D). It is possible that the migration defect of MEKK1–/– cells was due to the loss of expression of a secreted factor such as a cytokine, protease or extracellular matrix protein. To determine whether the migration defect in MEKK1–/– cells was due to loss of a secreted factor, MEKK1–/– and wild-type fibroblasts were co-cultured (Figure 2E) and their respective migration analyzed by the in vitro wound healing assay. MEKK1–/– and wild-type fibroblasts were stained with different vital fluorescent dyes so they could be readily distinguished when co-cultured. Strikingly, after 5 h post-wounding, wild-type but not MEKK1–/– fibroblasts have extensively moved into the wound space. Virtually no MEKK1–/– fibroblasts in co-culture with MEKK1+/+ fibroblasts have migrated into the wound space, demonstrating that co-culture with wild-type cells can not restore migration to MEKK1–/– fibroblasts. Thus, the migration defect of MEKK1–/– fibroblasts is not rescued by secreted factors from wild-type cells. This result strongly suggests that the defective migration of MEKK1–/– fibroblasts is not due to an inability to secrete a required protein.
Vinculin content in focal adhesions is diminished in migrating MEKK1–/– fibroblasts
The MEKK1–/– phenotype is characterized by increased adherence, defective rear-end detachment and inhibited migration, suggesting a defect in the regulation of focal adhesions. To address the consequence of MEKK1 deficiency in the regulation of focal adhesions of migrating cells, we used quantitative immunofluorescence analysis to measure vinculin content in focal adhesions of migrating wild-type (Figure 3A) and MEKK1–/– (Figure 3C) fibroblasts. The experiment required a 12 h incubation after inflicting the razor swipe to allow migration of MEKK1–/– cells into the wound site, and a larger wound than that of Figure 2D. Analysis of 58 wild-type and 97 MEKK1–/– fibroblasts migrating into the scrape wound of confluent monolayers indicated 2.8-fold (P < 0.0001) less vinculin in focal adhesions of MEKK1–/– versus wild-type fibroblasts (Figure 3D). This result is consistent with the inability of migrating MEKK1–/– fibroblasts to properly organize the complex of proteins in focal adhesions. While vinculin may also be organized at cell-to-cell contacts, we have found that vinculin consistently co-localizes with the focal adhesion protein talin in mouse embryo fibroblasts (data not shown) and as such serves as a surrogate assay for focal adhesion formation. Further, as organized vinculin staining is indicative of focal adhesions, loss of vinculin staining indicates that the increased adherence of MEKK1-deficient cells is not due to an increased number or size of focal adhesions as has been observed in FAK–/– fibroblasts. This indicates that the change in adherence and focal adhesion composition resulting from MEKK1 deficiency is different from that observed with loss of FAK expression. Importantly, the add-back of MEKK1 expression restores vinculin content to focal adhesions in migrating fibroblasts (Figure 3B and D). Stationary MEKK1–/– and wild-type fibroblasts in confluent monolayers have similar vinculin staining (data not shown), consistent with MEKK1 signaling being important for regulation of the turnover of focal adhesions during migration and not in the ability to form focal adhesions. The partial restoration of vinculin to focal adhesions is likely due to the level of MEKK1 expressed in the add-back clone, which is only 20% of MEKK1 protein in wild-type fibroblasts. Stable expression of MEKK1 by add-back to MEKK1–/– cells is extremely difficult. This is predictably due to the regulation of MEKK1 expression during the cell cycle that is not mimicked by using other promoters or viral LTRs to express MEKK1, and the toxicity of MEKK1 overexpression (Yujiri et al., 1999). Despite intensive effort, we have not been able to achieve stable add-back clones that express >20% of wild-type MEKK1 protein. Interestingly, our preliminary findings suggest that the level of talin and FAK, unlike vinculin, may be similar in MEKK1–/– and wild-type fibroblasts (data not shown). To determine whether MEKK1 does indeed differentially regulate protein composition in focal adhesions, it will be necessary to perform quantitative immunofluorescence analysis of several focal adhesion proteins in stationary and migrating wild-type, MEKK1–/– and add-back fibroblasts.

Fig. 3. Vinculin content in focal adhesions is diminished in migrating MEKK1–/– fibroblasts. A 0.4 µm deconvolved image section of the cell having the brightest focal adhesion staining was used for the measurement of integrated intensity of vinculin content for MEKK1+/+ (A), add-back (B) and MEKK1–/– (C) fibroblasts. The intensity of vinculin staining was measured per cell area of the section. The add-back clone stably expresses full-length MEKK1 and was derived from the MEKK1–/– fibroblasts. The bar graph in (D) shows the analysis from three experiments where 58 wild-type, 97 MEKK1–/– and 96 add-back cells were analyzed for integrated vinculin staining intensity per cell area. Vinculin content in the MEKK1–/– clone is diminished at a statistically significant level from wild-type MEKK1+/+ cells (*P < 0.0001) and add-back cells (**P < 0.001). Bar = 50 µm.
EGF stimulates the formation of MEKK1–FAK complexes
Figure 4A shows that when serum-starved fibroblasts are stimulated with EGF, MEKK1 is co-immunoprecipitated with FAK. Co-expression of MEKK1 and FAK in HEK293 cells by transient transfection demonstrated that they could be reciprocally co-immunoprecipitated (Yujiri et al., 2003). These findings demonstrate the interaction of MEKK1 with FAK-associated protein complexes and that endogenous MEKK1 recruitment into FAK complexes is regulated by a growth factor that stimulates both FAK and MEKK1 kinase activities (Fanger et al., 1997). Thus, EGF induces the stable association of MEKK1 with FAK, altering the protein composition of focal adhesions.

Fig. 4. MEKK1 and FAK form a complex in EGF-stimulated fibroblasts. (A) Wild-type mouse embryo fibroblasts were treated with 100 ng/ml EGF for the indicated times. After cell lysis, endogenous FAK was immunoprecipitated with anti-FAK antibody, and associated MEKK1 detected by MEKK1 immunoblotting. Total immunoprecipitated FAK was measured by anti-FAK immunoblotting using the same blots as that for MEKK1 analysis. (B) Wild-type (MEKK1+/+) and MEKK1–/– fibroblasts stably expressing papilloma virus E6/E7 proteins were immunoblotted for FAK protein expression using an antibody recognizing the N- or C-terminal domain of FAK. FAK is a 125 kDa protein with N- and C-terminal cleavage fragments of 68 and 57 kDa, respectively. (C) Serum-starved MEKK1+/+ or MEKK1–/– MEFs were allowed to adhere to fibronectin-coated bacterial plates for 30 min, and then lysed. Endogenous FAK was immunoprecipitated with anti-FAK antibodies, and the level of tyrosine-phosphorylated FAK determined by phosphotyrosine immunoblotting. The membrane was then stripped and total FAK was assessed by FAK immunoblotting. (D) MEKK1+/+ and MEKK1–/– fibroblasts ± E6/E7 protein expression were analyzed for migration in Transwell assays using 5% FBS as described for Figure 2A. *Inhibition of MEKK1–/– E6/E7 cell migration is statistically significant relative to FAK–/– cells (P < 0.05) and MEKK1–/– cells (P < 0.001).
Focal adhesion turnover, and therefore cell adhesion, is proposed to be modulated not only by signaling pathways involving protein phosphorylation, but by proteolysis of focal adhesion components as well. Interestingly, FAK and MEKK1 are regulated both by phosphorylation and proteolytic cleavage (Cooray et al., 1996; Schlesinger et al., 1998; Cary et al., 2002). The discovery that MEKK1 becomes stably associated with FAK suggested that MEKK1 functions in focal adhesion signaling. The increased adherence and inhibition of migration resulting from MEKK1 deficiency also demonstrates that MEKK1 has an important role in the function of focal adhesions.
MEKK1 localizes with focal adhesions
Expression of EGFP–MEKK1 in MEKK1–/– MEFs demonstrates its association with focal adhesions and actin fibers entering the focal adhesions (Figure 5). The localization of EGFP–MEKK1 with focal adhesions is consistent with its co-immunoprecipitation with FAK (Figure 4). We did not observe a measurable increase of EGFP–MEKK1 in focal adhesions of EGF-stimulated cells despite an increased stable association of MEKK1 in FAK immunoprecipitates. This suggests that the EGF stimulation of MEKK1–FAK co-immunoprecipitation may be related to increased stabilization of the complex, possibly due to phosphorylation-related responses, rather than a re-localization of MEKK1 to focal adhesions. It should also be noted that caspase-mediated cleavage of MEKK1 compelled us to use a mutant MEKK1 protein for these studies. The caspase-cleavage site residues had been mutated to alanines (Widmann et al., 1998). The caspase-cleaved fragments do not appear to localize to focal adhesions. Cumulatively, the co-immunoprecipitation and immunofluorescence studies demonstrate that MEKK1 localizes, in part, to focal adhesions and actin filaments entering focal adhesions for the control of cell adherence.

Fig. 5. MEKK1 localizes to focal adhesions. MEKK1–/– fibroblasts were transfected with EGFP–MEKK1, incubated in serum-free media for 12 h, then processed as described in Materials and methods and subjected to immunofluorescence analysis. (A) An MEKK1–/– MEF transfected with EGFP–MEKK1 and treated with anti-FAK antibodies (FAK displayed as red fluorescence). Bar = 10 µm. (B) Displayed are two representative examples of co-localization of EGFP–MEKK1 with endogenous FAK (purple) and actin (red). Bar = 1 µm.
E6/E7 papilloma virus proteins induce FAK proteolysis in MEKK1–/– fibroblasts
E6/E7 expression immortalizes primary fibroblasts by inducing the degradation of p53. We had used E6/E7 for immortalization of both wild-type and MEKK1–/– mouse embryo fibroblasts. We quickly realized that there was a significant difference in the phenotype of MEKK1–/– fibroblasts but not wild-type fibroblasts that had been immortalized with papillomavirus E6/E7 proteins versus immortalization by serial passage. In addition to stimulating p53 degradation, the E6 oncoprotein binds to the focal adhesion protein paxillin (Turner, 2000). One outcome of E6 expression is the disruption of paxillin association with vinculin and FAK (Tong et al., 1997; Turner, 2000). Relative to primary MEKK1–/– fibroblasts, or MEKK1–/– fibroblasts immortalized by serial passage, MEKK1–/– fibroblasts expressing E6/E7 were extremely flat and highly adherent to the substratum, reminiscent of FAK–/– cells (Ilic et al., 1995). In contrast, E6/E7-expressing wild-type fibroblasts were more rounded and less adherent to substratum. Figure 4B shows that E6/E7 expression in MEKK1–/– fibroblasts causes a near quantitative cleavage of FAK. This contrasts with E6/E7-expressing MEKK1+/+ fibroblasts where FAK remains intact. This result is consistent with MEKK1 being associated with FAK–paxillin–vinculin complexes and playing a protective role against E6/E7-induced FAK degradation. This suggests that EGF-induced MEKK1 association with focal adhesions alters the organization and interaction of focal adhesion proteins.
Functionally, MEKK1 protects FAK from E6/E7-mediated degradation, but MEKK1 expression is not required for fibronectin binding-induced stimulation of FAK tyrosine phosphorylation (Figure 4C). With MEKK1–/– fibroblasts immortalized by serial passage, fibronectin stimulation of FAK tyrosine phosphorylation is similar to that observed with MEKK1+/+ fibroblasts. This result indicates that FAK tyrosine phosphorylation is largely independent of the role MEKK1 plays in organization, signaling and proteolytic susceptibility of focal adhesion proteins.
E6/E7-induced FAK cleavage in MEKK1–/– fibroblasts causes a migration defect more severe than that observed with FAK–/– or MEKK1–/– fibroblasts not expressing the E6/E7 oncoproteins (Figure 4D). FAK–/– fibroblasts, like MEKK1–/– fibroblasts, exhibit a marked reduction in migration in transwell assays (Figure 4D). Unlike E6/E7 wild-type fibroblasts that readily migrate toward serum, E6/E7 MEKK1–/– fibroblasts display a migration defect more severe than either MEKK1–/– or FAK–/– fibroblasts that do not express the E6/E7 proteins (Figure 4D). Thus, MEKK1 is important in maintaining the integrity and protein composition of focal adhesion complexes and is functionally required for fibroblast migration. The combined loss of MEKK1 and FAK exacerbates the migration defect resulting from the loss of either kinase alone.
MEKK1 regulates calpain activation
Previous studies have suggested that the intracellular cysteine protease calpain is involved in the regulation of rear-end detachment of migrating cells (Huttenlocher et al., 1997; Frame et al., 2002; Glading et al., 2002), the migration defect of MEKK1–/– fibroblasts. Using a cell-permeable fluorogenic calpain substrate, we discovered that MEKK1–/– cells have significantly less calpain proteinase activity than wild-type fibroblasts (Figure 6A). Similarly, in culture conditions identical to the in vitro wound assay, MEKK1–/– fibroblasts have significantly less calpain activity than wild-type fibroblasts. Cleavage of the fluorogenic substrate is blocked using a calpain-specific inhibitor, PD150606, which prevents Ca2+ binding to calpain and does not significantly inhibit either cathepsins or caspases (Wang et al., 1996), confirming that the assay is specifically measuring calpain activity (Figure 6A). Just as add-back of MEKK1 protein by transfection of MEKK1–/– fibroblasts rescued adherence and migration, add-back of MEKK1 also restored calpain regulation (Figure 6A). Even though the add-back clone expresses MEKK1 protein at only 20% of the MEKK1 expression of wild-type fibroblasts, it is sufficient to at least partially restore calpain activation, and rescue both migration and adherence. As we had demonstrated that MEKK1 is associated with focal adhesions (Figures 4 and 5), we asked whether FAK was necessary for calpain activation. Since FAK-deficient fibroblasts have been found to have severe migration defects (Ilic et al., 1995), we wanted to determine whether loss of FAK from the signaling complex affected calpain activity. Strikingly, calpain activity in serum-treated FAK-deficient fibroblasts was dramatically reduced compared with that of FAK-expressing cells (Figure 6B). Taken together, our data support the existence of a complex containing FAK and MEKK1 that is required for calpain control of cell adherence and migration.

Fig. 6. MEKK1-deficient fibroblasts show reduced calpain activity, and calpain inhibition mimics MEKK1 deficiency. In vivo calpain activity was assessed in fibroblasts using the cell-permeable, fluorescent calpain substrate SLLVY-AMC (A and B) and by anti-spectrin or anti-talin immunoblotting (C). (A) MEKK1+/+ cells in the presence and absence of the calpain inhibitor PD150606 (50 µM), MEKK1–/– and MEKK1 add-back cells were used for measurement of calpain activity. (B) FAK+/+ and FAK–/– cells were used to measure calpain activity as in (A). (C) The anti-spectrin and anti-talin immunoblots were stripped and reprobed with anti-m-calpain antibodies to verify protein levels. The immunoblots are representative of at least three independent experiments. (D) Wild-type fibroblasts grown to confluency on coverslips were pre-treated for 1 h with 50 µM PD150606 (left panel) or 2 µM GM6001, a matrix metalloproteinase inhibitor (right panel), and then analyzed for migration using the in vitro wound healing assay following a razor swipe in the continuous presence of inhibitor. Results are representative of at least three independent experiments for each set of experiments in (A–D).
α2β2 spectrin (fodrin) is a structural protein linking the cytoskeleton to membranes and is a defined calpain substrate (Wang and Yuen, 1999; Wang, 2000). Calpain cleavage of spectrin produces a specific pattern of proteolytic peptide fragments of 145–150 kDa (Wang and Yuen, 1999; Wang, 2000) that are unique to calpain and are different in size from those generated by caspases (Wang and Yuen, 1999; Wang, 2000). We found that calpain-specific spectrin fragments generated in wild-type fibroblasts were nearly undetectable in MEKK1–/– fibroblasts (Figure 6C). This finding demonstrates the loss of cleavage of an endogenous calpain substrate in MEKK1–/– fibroblasts. Cleavage of another calpain substrate, talin, was similarly reduced in the MEKK1-deficient cells (Figure 6C). While calpain activity was diminished in MEKK1–/– fibroblasts, immunoblotting indicated that total calpain protein expression was similar in wild-type and MEKK1–/– fibroblasts (Figure 6C). This result is consistent with the existence of a MEKK1-dependent signaling complex that regulates calpain activity but not calpain expression. Together, the loss of both spectrin and talin cleavage, coupled with reduced hydrolysis of the fluorogenic calpain-specific substrate, clearly demonstrates a loss of calpain activation in MEKK1–/– cells. Importantly, PD150606, the calpain-specific inhibitor, blocks the migration of wild-type fibroblasts in the in vitro wound assay (Figure 6D). However, GM6001, an inhibitor of metalloproteinases that function to remodel the extracellular matrix, did not inhibit fibroblast migration into the wound space, thereby demonstrating the specific requirement of calpain inhibition for disrupted migration in this assay. The findings demonstrate that the MEKK1-dependent regulation of calpain activity is required for cell migration.
MEKK1 regulates ERK1/2 activity
The MEK inhibitor PD98059 blocks ERK1/2 activation, inhibits calpain activation and cell motility (Glading et al., 2000). However, as PD98059 may influence Ca2+ influx independently of MEK activity (Pereira et al., 2002), and thereby possibly alter calpain activity, we tested the effect of UO126, an unrelated MEK inhibitor that does not non-specifically alter Ca2+ levels (Pereira et al., 2002) (Figure 7). U0126 inhibits calpain activation (Figure 7B) and dramatically reduces fibroblast chemotaxis towards serum (Figure 7A). In contrast, SP600125, a JNK inhibitor, does not diminish fibroblast migration or calpain activity (Figure 7B and C). Thus, while MEKK1 has been shown to regulate both the ERK1/2 and JNK signaling pathways, calpain activation and consequent cell migration may be attributed specifically to MEKK1 regulation of ERK1/2 signaling.

Fig. 7. Fibroblast migration and calpain activity are dependent on MEK, but not JNK activity. (A) Wild-type fibroblasts were loaded into Transwell migration chambers (105 cells/well) and allowed to migrate for 5 h, using 5% serum as a chemotactic agent. Calpain inhibitor PD150606 (50 µM), MEK inhibitor UO126 (10 µM) or matrix metalloproteinase inhibitor GM6001 (2 µM) were added to both the upper and lower chambers of the designated wells. (B) Wild-type fibroblasts were pre-incubated for 1 h with JNK inhibitor SP600125 (10 µM) or MEK inhibitor UO126 (10 µM), and calpain activity was assessed by SLLVY-AMC cleavage, and compared with that of non-treated cells. The results of both (A) and (B) are the mean ± SEM of at least three independent experiments. (C) Wild-type fibroblasts were pre-incubated with 10 µM SP600125 or 10 µM UO126 for 1 h and then analyzed for migration using the in vitro wound healing assay in the continuous presence of inhibitor. (D) Serum-starved wild-type, MEKK1–/– or MEKK1 add-back fibroblasts were treated with EGF or FGF-2 for 10 min and then lysed. ERK1/2 activation was then assessed by phospho-ERK immunoblotting. The membrane was then stripped and the total ERK2 level determined by ERK2 immunoblotting. The data are representative of at least three independent experiments. NS, no stimulus.
Given that numerous studies have identified Raf as the MAPK kinase kinase responsible for growth factor-mediated ERK1/2 activation, what role might MEKK1 play in EGF-induced ERK1/2 activation? We asked whether a portion of EGF-induced ERK1/2 activation might actually be attributable to MEKK1. By treating the cells with low (0.1–5 ng/ml) concentrations of EGF or FGF-2, we found that induced ERK1/2 phosphorylation and consequent activation by low levels of growth factors in mouse embryo fibroblasts is significantly dependent on MEKK1 expression (Figure 7D), while at higher levels of growth factor (100 ng/ml or higher), ERK1/2 phosphorylation levels of MEKK1+/+ and MEKK1–/– MEFs were similar (data not shown). Thus, MEKK1 control of ERK1/2 activation is readily apparent at low growth factor concentrations and verified by comparison of wild-type and MEKK1–/– fibroblasts. Further, densitometric analysis of Figure 7D reveals that MEKK1+/+ cells stimulated with the level of EGF used in our migration experiments (5 ng/ml) (Figure 2B) display a 2.5-fold greater level of pERK than the MEKK1–/– MEFs. Our data show that the loss of MEKK1 regulation of ERK1/2 signaling in MEKK1–/– fibroblasts contributes significantly to inhibition of calpain activation required for regulation of adherence and rear-end detachment of migrating cells. Importantly, the add-back of MEKK1 completely restores ERK1/2 activation by low doses of growth factor.
Discussion
Cell adhesion to the extracellular matrix is mediated by integrins, which, in turn, are linked to the cytoskeleton through proteins in focal adhesion complexes (Yamada and Miyamoto, 1995). The strength of cell adherence to the extracellular matrix is modulated by signal transduction pathways involving proteins within focal adhesions (Yamada and Miyamoto, 1995). One such pathway of particular importance to adhesion-mediated signaling is the ERK1/2 pathway. v-Src, a potent activator of ERK1/2 signaling, has been shown to drive MEK-dependent ERK activation and localization to focal adhesions in chicken embryo fibroblasts (Fincham et al., 2000). One explanation for the importance of membrane-localized ERK activity in cell adhesion, and by extension cell migration, is that ERK1/2 signaling is required for efficient release of cellular adhesion to the matrix substratum (Glading et al., 2000). We have previously reported that MEKK1 regulates ERK1/2 activation, and that MEKK1-deficient cells display reduced motility (Yujiri et al., 1998, 2000). In this paper, our findings demonstrate that MEKK1–/– fibroblasts have an increased adherence but display normal extension of the leading edge during migration. Thus, the loss of migration in MEKK1–/– fibroblasts is due to the increased adherence and a diminished ability to detach the trailing end of the cell, rather than decreased forward extension. We have demonstrated by co-immunoprecipitation that MEKK1 is associated with FAK-associated protein complexes, and that these complexes were localized in adhesion complexes. MEKK1 has also been proposed to co-localize with α-actinin and actin stress fibers (Christerson et al., 1999), and our immunofluorescence studies support that contention. Thus, our findings reveal that the cellular location of MEKK1 is well suited to regulate signal transduction in response to cell adhesion or cytoskeletal changes. Indeed, we have previously shown MEKK1 to regulate signaling in response to cytoskeletal alteration (Yujiri et al., 1999, 2000). Activation of the MEK–ERK1/2 pathway initiated by changes in integrin interactions with focal adhesion proteins is well characterized (Howe et al., 2002), and our findings with MEKK1 knockout fibroblasts demonstrate this response is, in part, dependent on the expression of MEKK1.
The protease calpain has recently been implicated in the control of cell migration through regulation of de-adhesion from matrix substratum. We have demonstrated that MEKK1 expression is critical for calpain activity regulation. Glading et al. (2001) have recently shown that membrane-proximal ERK1/2 activity is required for EGF-induced m-calpain activity, and we demonstrate that MEKK1 is required for regulation of calpain activation. While our studies did not rule out that MEKK1-dependent MEK activity might phosphorylate proteins other than ERK1/2 that are required for calpain activity, recent studies (Glading et al., 2001) indicate that appropriate localization of ERK activity is the key MEK-dependent factor in calpain activation. It is possible that MEKK1 might also influence calpain cellular location, but we were unable to detect any MEKK1-dependent calpain localization (data not shown). Importantly, reconstitution of the wild-type phenotype by expression of MEKK1 confirms that the defects we have measured in MEKK1–/– fibroblasts are a direct consequence of MEKK1 deficiency and not an epigenetic difference between cell lines.
Calpain has been shown to be associated with focal adhesions, and two primary substrates for calpain in fibroblasts are the structural proteins spectrin and talin (Glading et al., 2002). Through vinculin, both spectrin and talin are linked to α-actinin and the actin cytoskeleton (Petit and Thiery, 2000). Calpain-dependent cleavage of these two proteins thus alters the tethering of the cytoskeleton to focal adhesion complexes and the plasma membrane. Regulation of limited calpain cleavage of these proteins requires MEKK1 regulation of the ERK1/2 pathway. The requirement of MEKK1 expression for the control of adhesion probably explains, at least in part, the defective eyelid closure of MEKK1-deficient mice due to impaired epithelial cell migration. Furthermore, other MEKK1–/– cell types including neutrophils and macrophages appear to have compromised migration (our unpublished observations), indicating that MEKK1 contributes to the regulation of migration of mutiple cell types.
The significance of our work is that a focal adhesion-regulated pathway controlling cell adherence and migration has now been genetically defined. Targeted disruption of FAK (Ilic et al., 1995), MEKK1 (Yujiri et al., 2000), MEK1 (Giroux et al., 1999), ERK2 (Saba El-Leil, 2002) and calpain (Dourdin et al., 2001) demonstrates defects in cell migration (Figure 8). Pharmacological inhibitors of MEK1 and calpain mimic the loss of expression of component members of this pathway by blocking migration and influencing adherence. Interestingly, only the loss of MEKK1 expression yields viable animals, the other knockouts are embryonic lethal. This result is most likely related to the requirement of FAK to initiate multiple signals from focal adhesions, and the involvement of MEK1 and ERK2 in many diverse functions regulated by members of the Raf family (A-Raf, B-Raf and c-Raf1), Mos, Tpl-2 and MEKK1, all MAPK kinase kinases defined genetically to regulate the ERK1/2 pathway (Schlesinger et al., 1998). At low growth factor levels, which are most likely physiologically relevant, it is apparent that MEKK1 is critical for normal ERK1/2 activation. At higher growth factor concentrations, the activation of Raf proteins probably rescues the MEKK1 deficiency by activating the ERK1/2 pathway. Thus, pharmacological inhibition of MAPK kinase kinases such as MEKK1 would predictably have greater specificity in selectively inhibiting migration in pathological states such as cancer metastasis than inhibition of MEK1, ERK1/2 or calpain.

Fig. 8. Model depicting the FAK–MEKK1–MEK1/2–ERK2 pathway controlling calpain activation and the disruption of focal adhesion-actin cytoskeletal complexes. MEKK1–/– cells are defective in focal adhesion composition and regulation of the ERK2–calpain activation pathway (see text for details).
Materials and methods
Antibodies and reagents
Anti-phospho ERK, ERK2 and FAK (C-terminal) antibodies used for immunoblotting were from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Anti-phosphotyrosine and anti-FAK (N-terminal) antibodies were from Upstate Biotechnology (Lake Placid, NY). Fibronectin-coated tissue culture plates were from BD Biosciences (Bedford, MA). Anti-vinculin (V-4505) and FITC-conjugated donkey anti-mouse antibodies were from Sigma (St Louis, MO). HRP–sheep anti-mouse IgG was from Amersham (Piscataway, NJ). Protein A–HRP conjugate was from Zymed Laboratories (San Francisco, CA). PKH67 and PKH26 vital dyes were from Sigma. PD150606 and SP600125 were from Calbiochem (San Diego, CA). UO126 was from Promega (Madison, WI). SLLVY-AMC was from Peptides International (Louisville, KY). The MEKK1 sequence DTVD (amino acids 871–874) was substituted with alanines by a PCR strategy, and then subcloned into pEGFP-C1 (BD Biosciences Clontech, Palo Alto, CA).
Fibroblasts
The development of MEKK1–/– mice has been described previously (Yujiri et al., 2000). Mouse embryo fibroblasts were isolated from E14.5 embryos. Immortalized fibroblasts were isolated after continuous passage for 3 months or by expression of papilloma E6/E7 oncoproteins. All results comparing wild-type fibroblasts and MEKK1–/– fibroblasts were characterized with similar results in primary and immortalized fibroblasts. The MEKK1 add-back fibroblasts were made by stable transfection of immortalized MEKK1–/– fibroblasts using a full-length mouse MEKK1 cDNA in pLHCX. MEKK1 expression as determined by immunoblotting was ∼20% of the MEKK1 expression of wild-type fibroblasts. The FAK–/– fibroblasts were a generous gift from Dr Dusko Ilic, University of California at San Francisco.
Cell culture
Fibroblasts were cultured in IMDM medium (Gibco, Grand Island, NY) containing penicillin/streptomycin (1%; Gibco), l-glutamine (2 mM; Gibco), monothioglycerol (0.0012%; Sigma) and 10% (v/v) fetal calf serum (Gemini Bioproducts, Woodland, CA) at 37°C in a humidified atmosphere. Human FGF-2 was purchased from Upstate Biotechnology (Lake Placid, NY) and murine EGF from Sigma.
Immunoprecipitation
After stimulation, fibroblasts were washed twice with cold PBS and lysed in 20 mM Tris–HCl pH 7.6, 0.5% NP-40, 250 mM NaCl, 3 mM EDTA, 3 mM EGTA, 1 mM PMSF, 2 mM sodium orthovanadate, 20 µg/ml aprotinin, 1 mM DTT and 5 µg/ml leupeptin. Lysates were cleared by centrifugation at 14 000 g for 10 min at 4°C, incubated with the appropriate antibody for 16 h at 4°C, and then with protein G–Sepharose for 1 h. The beads were washed three times, then resuspended in sample buffer (125 mM Tris–HCl pH 6.8, 20% glycerol, 4.6% SDS, 0.1% bromophenol blue and 10% 2-mercaptoethanol) for SDS–PAGE.
Immunoblotting
After stimulation, cells were lysed in 0.5 ml of sample buffer and incubated at room temperature for 20 min. After centrifugation at 14 000 g for 5 min, post-nuclear detergent cell lysates were collected. Proteins were separated by SDS–PAGE and transferred to nitrocellulose (Scleicher & Schuell, Keene, NH). Membranes were blocked in 5% milk (diluted in Tris-buffered saline and 0.1% Tween-20) and incubated with the appropriate antibody at 4°C overnight. HRP–protein A or HRP–sheep anti-mouse IgG was used as secondary reagent. After extensive washing, the targeted proteins were detected by enhanced chemiluminescence (ECL). Where indicated, blots were stripped by treatment with 2% SDS and 100 mM 2-mercaptoethanol in TBS, and then reprobed with desired antibodies and detected by ECL.
In vitro wound healing assessment
Fibroblasts were grown on coverslips (106 cells) until confluent. The culture was ‘wounded’ with a razor blade by swiping the coverslip to generate an open area with no cells. The coverslips were rinsed and cells allowed to migrate into the wound site in complete media with or without inhibitors for 5 h unless noted otherwise. Fibroblast migration was assessed by live cell microscopy. For the mixed cell experiments, the fibroblasts were stained with either PKH26 or PKH67 fluorescent vital dyes (Sigma), mixed and allowed to grow together overnight before wounding and assessment by fluorescence microscopy. To measure healing time, cells were grown in tissue culture dishes until confluent, wounded with an 18g needle and then periodically examined until the wound was completely sealed. The data were analyzed by Student’s t-test.
Transwell chemotaxis assay
Fibroblasts were typsinized, washed and suspended in IMDM with 5% BSA. A total of 105 cells were then loaded into the upper chamber of a Transwell (Corning, Corning, NY) migration plate (8 µm pore) and allowed to migrate toward IMDM ± 5% fetal bovine serum for 5 h at 37°C. After migration, the upper surface of the membrane was thoroughly cleaned with a cotton swab, stained with Wright stain (Sigma) and the cells that migrated to the lower surface counted (five random 20× fields/well).
Calpain fluorescence assay
Fibroblasts were seeded into 96-well plates (104 cells/well) and allowed to attach overnight. The cells were then serum starved for 8 h before treatment with 10% FBS in IMDM for 30 min. The medium was removed and 200 µl of 62.5 mM SLLVY-AMC, a cell-permeable calpain substrate, in reaction buffer (115 mM NaCl, 1 mM KH2PO4, 5 mM KCl, 2 mM CaCl2 1.2 mM MgSO4 and 25 mM HEPES pH 7.25) were added and fluorescence assessed using a Perkin Elmer 7000 BioAssay Reader (360/465 nm). When inhibitors were used, the cells were incubated with the inhibitor for 1 h prior to addition of the substrate, and the assay conducted in the presence of the inhibitor.
Centrifugation adherence assay
Cells were seeded into a 96-well plate (2000 cells/well) and allowed to attach overnight. Cells were serum starved for 8 h before treatment with or without 10% FBS in IMDM for 30 min. The plate was then sealed, inverted and centrifuged at 2300 g for 5 min at 37°C. After rinsing, the remaining attached cells were stained with Wright stain (Sigma) and counted. Cell adherence is represented as the percent of the total remaining serum-treated cells compared with the non-treated cells.
Fibroblast attachment rate
MEKK1+/+ (wild-type) or MEKK1-deficient fibroblasts were resuspended in complete media and allowed to attach in either untreated or fibronectin-coated tissue culture plates (Becton Dickinson, Bedford, MA). The number of attached cells was determined over the course of 2 h by phase microscopy of a defined plate area.
Immunofluorescence
Cells were transfected with lipofectamine and grown on glass coverslips. Two days after transfection, the cells were treated with 50 ng/ml EGF or serum-free medium alone for 30 min, then the medium was removed and the cells fixed in 3% paraformaldehyde/3% sucrose in phosphate-buffered saline (PBS) for 10 min. Following three PBS washes, the cells were permeabilized for 10 min with 0.1%Triton X-100 in PBS. After washing, the cells were blocked in 10% donkey serum in PBS for 1 h at room temperature. The coverslips were then incubated with anti-FAK (BD Transduction Laboratories) in 3% BSA/PBS for 1 h. After three washes, the coverslips were incubated with Cy5-conjugated anti-mouse antibodies (Jackson Immunoresearch) and rhodamine–phalloidin (Molecular Probes) in 3% BSA/PBS for 1 h. Following washing, cells were mounted in 75% glycerol/25% PBS/Tris pH 7.5.
Quantitation of vinculin in focal adhesions
Cells migrating into the wound area resulting from the razor swipe of a confluent monolayer of fibroblasts were paraformaldehyde fixed at 12 h post-wounding. Cells were stained with the mouse anti-vinculin antibody followed by FITC-conjugated donkey anti-mouse antibody. Images were taken every 0.4 µm along the Z-axis and deconvolved using the nearest-neighbors algorithm Slidebook program from Intelligent Imaging, Inc. (Denver, CO). The section at the substratum interface with the best in-focus vinculin staining was selected for measurement of the integrated fluorescence intensity in focal adhesions. The cell area for the section was also determined using Slidebook. The fluorescence intensity of vinculin per cell area was calculated for MEKK1+/+, MEKK1–/– and add-back clones from three independent experiments. The data were pooled and analyzed statistically using Student’s t-test.
Supplementary data
Supplementary data are available at The EMBO Journal Online.
References
- Arthur J.S., Elce,J.S., Hegadorn,C., Williams,K. and Greer,P.A. (2000) Disruption of the murine calpain small subunit gene, Capn4: calpain is essential for embryonic development but not for cell growth and division. Mol. Cell. Biol., 20, 4474–4481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckerle M.C., Burridge,K., DeMartino,G.N. and Croall,D.E. (1987) Colocalization of calcium-dependent protease II and one of its substrates at sites of cell adhesion. Cell, 51, 569–577. [DOI] [PubMed] [Google Scholar]
- Bialkowska K., Kulkarni,S., Du,X., Goll,D.E., Saido,T.C. and Fox,J.E. (2000) Evidence that β3 integrin-induced Rac activation involves the calpain-dependent formation of integrin clusters that are distinct from the focal complexes and focal adhesions that form as Rac and RhoA become active. J. Cell Biol., 151, 685–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cary L.A., Klinghoffer,R.A., Sachsenmaier,C. and Cooper,J.A. (2002) SRC catalytic but not scaffolding function is needed for integrin-regulated tyrosine phosphorylation, cell migration and cell spreading. Mol. Cell. Biol., 22, 2427–2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christerson L.B., Vanderbilt,C.A. and Cobb,M.H. (1999) MEKK1 interacts with α-actinin and localizes to stress fibers and focal adhesions. Cell Motil. Cytoskeleton, 43, 186–198. [DOI] [PubMed] [Google Scholar]
- Cooray P., Yuan,Y., Schoenwaelder,S.M., Mitchell,C.A., Salem,H.H. and Jackson,S.P. (1996) Focal adhesion kinase (pp125FAK) cleavage and regulation by calpain. Biochem. J., 318, 41–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dourdin N., Bhatt,A.K., Dutt,P., Greer,P.A., Arthur,J.S., Elce,J.S. and Huttenlocher,A. (2001) Reduced cell migration and disruption of the actin cytoskeleton in calpain-deficient embryonic fibroblasts. J. Biol. Chem., 276, 48382–48388. [DOI] [PubMed] [Google Scholar]
- Fanger G.R., Johnson,N.L. and Johnson,G.L. (1997) MEK kinases are regulated by EGF and selectively interact with Rac/Cdc42. EMBO J., 16, 4961–4972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fincham V.J., James,M., Frame,M.C. and Winder,S.J. (2000) Active ERK/MAP kinase is targeted to newly forming cell-matrix adhesions by integrin engagement and v-Src. EMBO J., 19, 2911–2923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frame M.C., Fincham,V.J., Carragher,N.O. and Wyke,J.A. (2002) v-Src’s hold over actin and cell adhesions. Nat. Rev. Mol. Cell Biol., 3, 233–245. [DOI] [PubMed] [Google Scholar]
- Giroux S. et al. (1999) Embryonic death of Mek1-deficient mice reveals a role for this kinase in angiogenesis in the labyrinthine region of the placenta. Curr. Biol., 9, 369–372. [DOI] [PubMed] [Google Scholar]
- Glading A., Chang,P., Lauffenburger,D.A. and Wells,A. (2000) Epidermal growth factor receptor activation of calpain is required for fibroblast motility and occurs via an ERK/MAP kinase signaling pathway. J. Biol. Chem., 275, 2390–2398. [DOI] [PubMed] [Google Scholar]
- Glading A., Uberall,F., Keyse,S.M., Lauffenburger,D.A. and Wells,A. (2001) Membrane proximal ERK signaling is required for M-calpain activation downstream of epidermal growth factor receptor signaling. J. Biol. Chem., 276, 23341–23348. [DOI] [PubMed] [Google Scholar]
- Glading A., Lauffenburger,D.A. and Wells,A. (2002) Cutting to the chase: calpain proteases in cell motility. Trends Cell Biol., 12, 46–54. [DOI] [PubMed] [Google Scholar]
- Howe A.K., Aplin,A.E. and Juliano,R.L. (2002) Anchorage-dependent ERK signaling—mechanisms and consequences. Curr. Opin. Genet. Dev., 12, 30–35. [DOI] [PubMed] [Google Scholar]
- Huang Y. and Wang,K.K. (2001) The calpain family and human disease. Trends Mol. Med., 7, 355–362. [DOI] [PubMed] [Google Scholar]
- Huttenlocher A., Palecek,S.P., Lu,Q., Zhang,W., Mellgren,R.L., Lauffenburger,D.A., Ginsberg,M.H. and Horwitz,A.F. (1997) Regulation of cell migration by the calcium-dependent protease calpain. J. Biol. Chem., 272, 32719–32722. [DOI] [PubMed] [Google Scholar]
- Ilic D. et al. (1995) Reduced cell motility and enhanced focal adhesion contact formation in cells from FAK-deficient mice. Nature, 377, 539–544. [DOI] [PubMed] [Google Scholar]
- Lotz M.M., Burdsal,C.A., Erickson,H.P. and McClay,D.R. (1989) Cell adhesion to fibronectin and tenascin: quantitative measurements of initial binding and subsequent strengthening response. J. Cell Biol., 109, 1795–1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maheshwari G., Wells,A., Griffith,L.G. and Lauffenburger,D.A. (1999) Biophysical integration of effects of epidermal growth factor and fibronectin on fibroblast migration. Biophys. J., 76, 2814–2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palecek S.P., Huttenlocher,A., Horwitz,A.F. and Lauffenburger,D.A. (1998) Physical and biochemical regulation of integrin release during rear detachment of migrating cells. J. Cell Sci., 111, 929–940. [DOI] [PubMed] [Google Scholar]
- Pereira D.B., Carvalho,A.P. and Duarte,C.B. (2002) Non-specific effects of the MEK inhibitors PD098,059 and U0126 on glutamate release from hippocampal synaptosomes. Neuropharmacology, 42, 9–19. [DOI] [PubMed] [Google Scholar]
- Petit V. and Thiery,J.P. (2000) Focal adhesions: structure and dynamics. Biol. Cell, 92, 477–494. [DOI] [PubMed] [Google Scholar]
- Pfaff M., Du,X. and Ginsberg,M.H. (1999) Calpain cleavage of integrin β cytoplasmic domains. FEBS Lett., 460, 17–22. [DOI] [PubMed] [Google Scholar]
- Potter D.A., Tirnauer,J.S., Janssen,R., Croall,D.E., Hughes,C.N., Fiacco,K.A., Mier,J.W., Maki,M. and Herman,I.M. (1998) Calpain regulates actin remodeling during cell spreading. J. Cell Biol., 141, 647–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saba El-Leil M.K. (2002) Targeted inactivation of the Erk2 MAP kinase gene in the mouse. In Abstracts of the Meeting on Protein Phosphorylation and Mechanisms of Cellular Regulation, March 5–10, 2002, Taos, NM. Keystone Symposia. [Google Scholar]
- Schlesinger T.K., Fanger,G.R., Yujiri,T. and Johnson,G.L. (1998) The TAO of MEKK. Front. Biosci., 3, D1181–D1186. [DOI] [PubMed] [Google Scholar]
- Tong X., Salgia,R., Li,J.L., Griffin,J.D. and Howley,P.M. (1997) The bovine papillomavirus E6 protein binds to the LD motif repeats of paxillin and blocks its interaction with vinculin and the focal adhesion kinase. J. Biol. Chem., 272, 33373–33376. [DOI] [PubMed] [Google Scholar]
- Turner C.E. (2000) Paxillin and focal adhesion signalling. Nat. Cell Biol., 2, E231–E236. [DOI] [PubMed] [Google Scholar]
- Wang K.K. (2000) Calpain and caspase: can you tell the difference? Trends Neurosci., 23, 20–26. [DOI] [PubMed] [Google Scholar]
- Wang K.K.W. and Yuen,P.-w. (1999) Calpain: Pharmacology and Toxicology of a Cellular Protease. Taylor & Francis, London, UK. [Google Scholar]
- Wang K.K. et al. (1996) An α-mercaptoacrylic acid derivative is a selective nonpeptide cell-permeable calpain inhibitor and is neuroprotective. Proc. Natl Acad. Sci. USA, 93, 6687–6692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widmann C., Gerwins,P., Johnson,N.L., Jarpe,M.B. and Johnson,G.L. (1998) MEK kinase 1, a substrate for DEVD-directed caspases, is involved in genotoxin-induced apoptosis. Mol. Cell. Biol., 18, 2416–2429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada K.M. and Miyamoto,S. (1995) Integrin transmembrane signaling and cytoskeletal control. Curr. Opin. Cell Biol., 7, 681–689. [DOI] [PubMed] [Google Scholar]
- Yujiri T., Sather,S., Fanger,G.R. and Johnson,G.L. (1998) Role of MEKK1 in cell survival and activation of JNK and ERK pathways defined by targeted gene disruption. Science, 282, 1911–1914. [DOI] [PubMed] [Google Scholar]
- Yujiri T., Fanger,G.R., Garrington,T.P., Schlesinger,T.K., Gibson,S. and Johnson,G.L. (1999) MEK kinase 1 (MEKK1) transduces c-Jun NH2-terminal kinase activation in response to changes in the microtubule cytoskeleton. J. Biol. Chem., 274, 12605–12610. [DOI] [PubMed] [Google Scholar]
- Yujiri T. et al. (2000) MEK kinase 1 gene disruption alters cell migration and c-Jun NH2-terminal kinase regulation but does not cause a measurable defect in NF-κB activation. Proc. Natl Acad. Sci. USA, 97, 7272–7277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yujiri T., Nawata,R., Takahashi,T., Sato,Y., Tanizawa,Y., Kitamura,T. and Oka,Y. (2003) MEK kinase 1 interacts with focal adhesion kinase and regulates insulin receptor substrate-1 expression. J. Biol. Chem., 278, 3846–3851. [DOI] [PubMed] [Google Scholar]