

Abstract
Lipoproteins having a lipid-modified cysteine at the N-terminus are localized on either the inner or the outer membrane of Escherichia coli depending on the residue at position 2. Five Lol proteins involved in the sorting and membrane localization of lipoprotein are highly conserved in Gram-negative bacteria. We determined the crystal structures of a periplasmic chaperone, LolA, and an outer membrane lipoprotein receptor, LolB. Despite their dissimilar amino acid sequences, the structures of LolA and LolB are strikingly similar to each other. Both have a hydrophobic cavity consisting of an unclosed β barrel and an α-helical lid. The cavity represents a possible binding site for the lipid moiety of lipoproteins. Detailed structural differences between the two proteins provide significant insights into the molecular mechanisms underlying the energy-independent transfer of lipoproteins from LolA to LolB and from LolB to the outer membrane. Furthermore, the structures of both LolA and LolB determined from different crystal forms revealed the distinct structural dynamics regarding the association and dissociation of lipoproteins. The results are discussed in the context of the current model for the lipoprotein transfer from the inner to the outer membrane through a hydrophilic environment.
Keywords: crystal structure/lipoprotein localization/LolA/LolB/periplasm
Introduction
The transport of lipid-modified proteins between membranes through hydrophilic environments requires protein aceous factors. In eukaryotic cells, for example, Rab proteins involved in the vesicular transport system are transported between membranes after forming a complex with Rab GDP-dissociation inhibitor (Zerial and Huber, 1995; Novick and Zerial, 1997). Bacterial lipoproteins having a lipid-modified cysteine at the N-terminus are important components of the cell envelope and responsible for various cellular activities (Hayashi and Wu, 1990). Borrelia burgdorferi, the Lyme disease spirochaete, has >100 lipoproteins, some of which are localized on the cell surface through an as yet unknown mechanism and induce an immunoresponse of host cells (Fraser et al., 1997; Brightbill et al., 1999). Escherichia coli also has at least 90 lipoproteins (Masuda et al., 2002) on the periplasmic surface of either the inner or the outer membrane depending on the sorting signal located next to the N-terminal cysteine (Yamaguchi et al., 1988). Aspartate at this position functions as an inner membrane-specific signal, whereas other residues direct lipoproteins to the outer membrane (Yamaguchi et al., 1988; Terada et al., 2001).
Lipoproteins are synthesized as precursors with a signal peptide and then translocated across the inner membrane (Pugsley, 1993; Driessen et al., 2001). Subsequent processing to mature lipoproteins occurs sequentially on the periplasmic surface of the inner membrane, i.e. attachment of diglyceride through a thioether linkage to the cysteine residue located at the N-terminus of the mature region, cleavage of the signal peptide by a lipoprotein-specific signal peptidase and attachment of an acyl chain to the amino group of cysteine (Sankaran and Wu, 1994). Mature lipoproteins thus formed are then sorted and localized to the respective membranes by the Lol system. The LolCDE complex in the inner membrane belongs to the ABC (ATP-binding cassette) transporter superfamily and releases outer membrane-specific lipoproteins in an ATP-dependent manner (Yakushi et al., 2000), leading to the formation of a water-soluble complex with a periplasmic molecular chaperone, LolA (Matsuyama et al., 1995). The LolA–lipoprotein complex crosses the periplasm to the outer membrane, where a lipoprotein receptor, LolB, is present (Matsuyama et al., 1997). Upon interaction of the LolA–lipoprotein complex with LolB, lipoproteins are transferred from LolA to LolB and finally localized to the outer membrane. The depletion of any Lol protein is lethal for E.coli (Tajima et al., 1998; Tanaka et al., 2001; Narita et al., 2002).
Both LolA (Matsuyama et al., 1995) and LolB (Matsuyama et al., 1997) function as a monomeric form. The LolA–lipoprotein complex is stable both in vivo (Tanaka et al., 2001) and in vitro (Matsuyama et al., 1995, 1997) only in the absence of the LolB function, suggesting that the affinity for lipoproteins is lower for LolA than for LolB. The in vitro transfer of lipoproteins from the LolA–lipoprotein complex to LolB (Matsuyama et al., 1997) also suggests that the lipoprotein transfer occurs because of the affinity difference between the two proteins. The LolB–lipoprotein complex is also stable in the absence of the outer membrane (Matsuyama et al., 1997). These observations, taken together, indicate that not only lipoprotein binding but also rapid transfer of associated lipoproteins are common functions of LolA and LolB. Although non-functional LolA mutants have been isolated (Miyamoto et al., 2001, 2002), little is known about the structural basis of the lipoprotein trafficking mechanism. To clarify the molecular mechanisms of LolA and LolB, the crystal structures of the two proteins from E.coli were determined.
Results and discussion
Overall structure of LolA
We determined the crystal structure of a periplasmic chaperone, LolA, with two crystal forms, i.e. orthorhombic and trigonal forms (Takeda et al., 2003a). The structure of the orthorhombic form was solved by the multiple-wavelength anomalous dispersion (MAD) method, using a platinum derivative, and refined at 1.65 Å resolution. The structure of the trigonal form was solved by the molecular replacement method and refined at 1.9 Å resolution (see Materials and methods). The crystallographic data and refinement statistics for the two crystal forms are summarized in Table I. The structure of LolA is characterized by an 11-stranded antiparallel β-sheet (β1–β11) forming an unclosed β barrel and three α-helices (α1–α3) attached to the concave face of the sheet (Figure 1A and B). The long loop connecting β11 and β12 is located outside the β-sheet. The β12 strand forms a parallel β-sheet with β6. The strand order is 789[10][11]123456[12]. The inner surface of the β-sheet and three α-helices is highly hydrophobic (as discussed later in detail).
Table I. Data collection and refinement statistics.
| LolA orthorhombic form | LolA trigonal form | LolB monoclinic form | LolB hexagonal form | |
|---|---|---|---|---|
| Crystal data |
|
|
|
|
| Space group | I222 | P3221 | P21 | P6322 |
| Cell parameters | ||||
| a (Å) | 55.8 | 60.6 | 37.2 | 71.4 |
| b (Å) | 75.4 | 60.6 | 112.4 | 71.4 |
| c (Å) | 99.5 | 79.0 | 47.8 | 133.9 |
| β (°) |
— |
— |
111.4 |
— |
| Data collection |
|
|
|
|
| Resolution range (Å) | 40.0–1.65 (1.71–1.65) | 30–1.90 (1.97–1.90) | 30.0–1.90 (1.97–1.90) | 30.0–2.20 (2.28–2.20) |
| Redundancy | 15.2 | 9.1 | 3.1 | 19.9 |
| Completeness (%) | 93.2 (51.6) | 99.4 (99.5) | 97.9 (95.0) | 98.2 (93.4) |
|
Rsym (%) |
4.3 (23.6) |
3.9 (25.6) |
6.2 (27.9) |
7.3 (25.3) |
| Refinement |
|
|
|
|
| Protein residues | 177 | 174 | 354 | 177 |
| Heterogen molecules | 5 | 0 | 4 | 3 |
| Water molecules | 160 | 82 | 215 | 177 |
| Rwork (%) | 22.2 | 22.6 | 21.5 | 21.7 |
| Rfree (%) | 24.9 | 25.9 | 24.9 | 24.7 |
| R.m.s. deviations | ||||
| Bonds (Å) | 0.006 | 0.006 | 0.006 | 0.007 |
| Angle (°) | 1.3 | 1.2 | 1.3 | 1.2 |
| Coordinates error (Å) | 0.25 | 0.30 | 0.29 | 0.30 |
Values in parentheses refer to the highest resolution shell.
Rsym = ΣhklΣi|Ihkl,i – <Ihkl>|/ΣhklΣiIhkl,i.
Rwork = Σhkl|Fobs – Fcalc|/Σ|Fobs|.
Rfree was calculated with the 5% of the reflections not included for refinement as a test set.
R.m.s., root mean square. The coordinates error was calculated from a Luzzati plot using the test set of reflections.

Fig. 1. Crystal structures of Escherichia coli LolA. (A) The LolA molecule is shown as a ribbon model, in which α-helices (α1–α3) and β-strands (β1–β11) are shown in red and green, respectively. Loops and short 310 helices are shown in yellow and a long loop and β12 in gray. Residues which had been mutagenized (Miyamoto et al., 2001, 2002) are shown as a ball-and-stick model. (B) LolA shown in (A) was rotated by 90° around the horizontal axis. (C) Sequence alignment of LolA homologs. α and 310 helices are indicated by cylinders and β-strands by arrows. The color scheme is the same as that in (A) and (B). The mutagenized residues are indicated by red triangles. Aromatic residues surrounding the hydrophobic cavity (see text) are indicated by green triangles. Eco, Escherichia coli; Sty, Salmonella typhimurium; Ype, Yersinia pestis; Hin, Haemophilus influenzae; Aac, Actinobacillus actinomycetemcomitans; Pmu, Pasteurella multocida.
Overall structure of LolB
Because outer membrane receptor LolB is an insoluble lipoprotein, a soluble LolB mutant, mLolB, having alanine in place of cysteine at position 1 (Matsuyama et al., 1997) was used for crystallization. Two crystal forms, i.e. monoclinic and hexagonal forms (Takeda et al., 2003b), were obtained for mLolB lacking the N-terminal acyl chains. The structure of the monoclinic form was solved by the MAD method, using selenomethionyl mLolB, and refined at 1.9 Å resolution. The structure of the hexagonal form was solved by the molecular replacement method and refined at 2.2 Å resolution (see Materials and methods). The crystallographic data and refinement statistics for the two crystal forms are listed in Table I. Figure 2A and B show ribbon models of LolB. Strikingly, the molecular structure of LolB is very similar to that of LolA, despite the low sequence identity of 8% between these two proteins. The structure of LolB also comprises an antiparallel β-sheet (β1–β11) covered by three α-helices (α1–α3). The strand order is 789[10][11]123456. The inner surface of the β-sheet and three α-helices is also highly hydrophobic. The C-terminal long loop and β12 found in LolA do not exist in LolB. The results of detailed structural comparison of the two Lol proteins are discussed later.

Fig. 2. Crystal structures of Escherichia coli LolB. (A) The LolB molecule is shown as a ribbon model, in which α-helices (α1–α3) and β-strands (β1–β11) are shown in pink and blue, respectively. Loops and short 310 helices are shown in yellow. Leu68, a conserved hydrophobic residue at the protruding loop, is shown as a ball-and-stick model. (B) LolB shown in (A) was rotated by 90° around the horizontal axis. (C) Sequence alignment of LolB homologs. α and 310 helices are indicated by cylinders and β-strands by arrows. The color scheme is the same as that in (A) and (B). Residues forming the hydrophobic cavity (see text) are indicated by blue triangles. A conserved hydrophobic residue, Leu68 is indicated by a yellow triangle. Eco, Escherichia coli; Sty, Salmonella typhimurium; Ype, Yersinia pestis; Hin, Haemophilus influenzae; Aac, Actinobacillus actinomycetemcomitans; Pmu, Pasteurella multocida.
Structural differences between the two crystal forms of LolA
Proteins can take on various conformations to exhibit minimum energy in solution, and these conformations are closely related to their functions. However, a certain conformation is stabilized by the restriction of the crystal packing. Therefore, the crystal structures determined for more than two crystal forms provide useful information as to protein dynamics. The structures of LolA determined for the two crystal forms were compared. Although the overall architectures are essentially identical, some significant differences were found between the two structures. These differences are important for clarifying the molecular mechanisms of LolA functions. The two molecular structures were superposed, which was performed with the LSQKAB program (Kabsch, 1976) in the CCP4 software suite (CCP4, 1994), and it was indicated that the root-mean-square deviation of all Cα atoms is 1.67 Å. Large differences were observed in the three helices and flexible loop regions (Figure 3A). For example, the average deviation of the α2 helix is ∼1.85 Å (Figure 3A and B). In contrast, the β-sheet can be well superimposed with small deviations, except that the C-terminal part of the β3 strand and the N-terminal part of the β4 strand exhibit significant differences between the two structures because of the marked flexibility of the loop connecting the β3 and β4 strands (loop 3–4). One residue, Gly37, a completely conserved residue, exhibits a large deviation value of 2.3 Å (Figure 3A), even though this residue is located at the middle of β2. This residue seems to be critical for movement of the α2 helix and allows conformational changes of Phe90 in the α2 helix and Trp49 in the β3 strand, both of which are located close to Gly37. The temperature factors of the three α-helices are small in the two crystals. Nevertheless, the two crystal forms exhibited these structural differences, indicating that the α-helices of LolA are inherently mobile in solution.

Fig. 3. Structural differences between the two crystal forms of LolA. (A) The positional deviations of Cα atoms between the orthorhombic and trigonal crystals of LolA are indicated against the residue number. The color scheme corresponds to that in Figure 1A. Regions exhibiting large deviations are indicated. (B) Stereoview of the superposed orthorhombic (gray) and trigonal (green) crystal forms of LolA. (C) The cis peptide bond between Arg43 and Pro44 in the orthorhombic form of LolA. A sigma A-weighted Fobs – Fcalc omit map of these two residues is shown at the 3.5σ level as a green mesh, in which these residues were excluded from the map calculation. (D) Hydrogen bonds between Arg43 and the plugging helices (α1–α2) in the trigonal form of LolA. The structure of the orthorhombic form (gray) is superimposed for comparison. Dashed lines indicate hydrogen bonds within 3.3 Å between Arg43 and the α-helices.
Hydrophobic cavity of LolA is closed
It has been reported that the occurrence of X-Pro cis peptide bonds is ∼5% (Stewart et al., 1990; Jabs et al., 1999). In contrast, two of seven X-Pro bonds, Arg43–Pro44 and Gln53–Pro54, were found to have the cis conformation in LolA. These residues other than Gln53 are highly conserved among LolA homologs (Figure 1C). The side chain of Arg43 is oriented toward the interior of the molecule (Figure 1A and B) due to this cis peptide bond (Figure 3C). Although the conformation of Arg43 and its hydrogen bond pattern differ between the orthorhombic and trigonal crystals, the side chain N atoms of Arg43 are hydrogen bonded in both crystals to the main chain carbonyls of residues in the α1 and α2 helices, thereby causing the tight fixation of the helices to the β2 strand (Figure 3D). No other residue in LolA plays such a role.
The inner surface of the β-sheet and three α-helices consists of hydrophobic residues and forms a hollow cavity, which is separated from the bulk solvent (the probe radius of 1.4 Å was employed). The hydrophobic cavity of LolA is most likely the binding site of a lipoprotein’s acyl chain. The cavity is surrounded by the side chains of the aromatic residues, as indicated in Figure 4A. These aromatic residues are highly conserved among LolA homologs (Figure 1C, green triangles). Phe90 located in the α2 helix undergoes a hydrophobic interaction with Trp49 and Leu59 in the orthorhombic form, whereas that in the trigonal form interacts with Phe47 and Trp49. As a result, residues sealing the cavity differ between the orthorhombic (Figure 4B and C) and trigonal (Figure 4D and E) forms. In spite of these differences, the shape of the hydrophobic cavity does not differ significantly between the two crystals. Moreover, the hydrophobic cavities in both crystals remain disconnected from the bulk solvent by the three α-helices (α1–α3) fixed to β2. This ‘lid’ is expected to undergo opening and closing upon the accommodation and release of lipoproteins, respectively. The closed form of LolA is stabilized by the hydrogen bonds between Arg43 and residues in the α1 and α2 helices. It has been shown that LolA easily transfers associated lipoproteins to LolB and returns to the free form (Matsuyama et al., 1997). These observations indicate that the closed form of LolA is more stable than the lipoprotein-bound form. Disruption of the hydrogen bonds is necessary for the lid opening and may be catalyzed by the LolCDE complex at the expense of ATP energy.

Fig. 4. Hydrophobic cavity of LolA. (A) The molecular surface of LolA. Aromatic residues (Phe, Trp and Tyr) are shown in green and other hydrophobic residues (Ala, Ile, Leu, Met, Pro and Val) in light green. The plugging α-helices are shown as tubes. The view is the same as that in Figure 1A. (B and C) Side and top views of the internal hydrophobic cavities in the orthorhombic form of LolA. The probe radius is 1.4 Å. The side chains of residues involved in sealing the cavities (Phe47, Trp49, Leu59, Phe90 and Met91) are shown in red. (D and E) Side and top views of the internal hydrophobic cavities in the trigonal form of LolA. The side chains of residues involved in sealing the cavities (Phe47, Trp49, Phe90 and Tyr152) are shown in red.
A similar mechanism involving the movement of an α-helix for opening a binding site has been reported for BmrR, a transcription activator of a multidrug exporter, which binds various lipophilic drugs (Zheleznova et al., 1999). LolA also binds lipoproteins having acyl chains of various lengths and degrees of saturation (Fukuda et al., 2002). The presence of such a lid/lock binding mechanism has been supported by the results of analyses of defective LolA mutants (mutation positions indicated by red triangles in Figure 1C). These residues are found at the interface of the β-strands and the α-helical lid (Figure 1A and B). A mutant of LolA possessing glutamate in place of phenylalanine at position 47, F47E, cannot bind lipoproteins (Miyamoto et al., 2002). Additional hydrogen bonds may be formed between the β-sheet and the lid helices in the F47E mutant, making opening of the lid more difficult. The LolA (R43L) and (E144W) mutants bind lipoproteins but cannot transfer associated lipoproteins to LolB (Miyamoto et al., 2001; A.Miyamoto, S.Matsuyama and H.Tokuda, unpublished results). Hydrogen bonds between the β-sheet and lid helices cannot be formed in R43L and can be weakened by the introduction of a large hydrophobic residue in E144W. These mutations decrease the stability of the free form relative to that of the bound form, thereby causing the accumulation of the complex in the periplasm (Miyamoto et al., 2001, 2002).
Hydrophobic cavity of LolB is open
We obtained three structures for LolB with two crystal forms: the monoclinic form containing two molecules in an asymmetric unit; and the hexagonal form. The root-mean-square deviations of Cα atoms between the hexagonal crystal and two molecules, A and B, in the monoclinic crystal are 1.14 and 1.30 Å, respectively, whereas the deviation value is 0.80 Å between the two molecules in the monoclinic crystal. The largest differences are observed in loop regions between β-strands (Figure 5A). The deviation of helices is lower in LolB than in LolA, whereas that of the β-sheet is higher in LolB than in LolA. The large deviation of the LolB β-sheet was caused by polyethylene glycol 2000 monomethyl ether (PEGMME2000), which was used in the crystallization process. PEGMME2000 is exclusively present in the hydrophobic cavity of the hexagonal form (compare Figure 5C and D) and spreads out the β-sheet (Figure 5B). Although the size of the cavity approximately corresponds to the size of the C18 chains existing abundantly in the membrane, the 12 atoms of PEGMME2000 could be modeled from the observed electron density. This hydrophobic cavity is most likely an acyl chain binding site, in which LolB accommodates the acyl chain of lipoproteins like PEGMME2000 (Figure 6A). Residues forming the cavity are highly conserved (Figure 2C, blue triangles) and consist of mainly leucine and isoleucine, which have more flexible hydrophobic side chains than aromatic residues. In contrast, the hydrophobic cavity of LolA is formed from aromatic residues (Figure 4A). Such a difference in the cavity-forming residues is expected to contribute to the difference in the affinity for lipoproteins between LolA and LolB. Judging from the cavity size, only a single acyl chain can be accommodated in this cavity. The binding mode of the other two acyl chains remains to be elucidated.

Fig. 5. Structural deviation of LolB. (A) The positional deviations of Cα atoms between the monoclinic (molecule A) and hexagonal crystals of LolB are indicated against the residue number. The color scheme is the same as that in Figure 2A. In addition, the deviations between molecules A and B in the monoclinic form and between molecule B in the monoclinic crystal and the hexagonal crystal are plotted in black and gray, respectively. (B) Stereoview of the superposed three structures: molecules A (black) and B (gray) in the monoclinic crystal and the hexagonal crystal (blue). The β-sheet in the hexagonal crystal (blue) is spread out. (C) A sigma A-weighted Fobs – Fcalc omit map of the residues involved in formation of the hydrophobic cavities of molecule A in the monoclinic crystal is shown at the 3.0σ level as a blue mesh. These residues were excluded from the map calculation. The view is the same as that in Figure 2A. (D) A sigma A-weighted Fobs – Fcalc omit map of the residues involved in formation of the hydrophobic cavity in the hexagonal crystal is shown at the 3.0σ level as a blue mesh. A sigma A-weighted Fobs – Fcalc omit map of the probable PEGMME2000 molecule is also shown at the 2.5σ level as a red mesh. These maps were calculated separately and superimposed.

Fig. 6. Hydrophobic cavity of LolB. (A) The molecular surface of LolB in the hexagonal crystal. Aromatic residues (Phe, Trp and Tyr) are shown in blue and other hydrophobic residues (Ala, Ile, Leu, Met, Pro and Val) in light blue. The α-helices are shown as tubes. The 12 atoms of the PEGMME2000 molecule constructed from the observed electron density are shown as a CPK model in orange. The additional six atoms (C13–C18) shown in beige were modeled into this PEGMME2000 molecule in order to visualize the C18 acyl chain bound to a lipoprotein. (B and C) Side and top views of the internal hydrophobic cavities of molecule A in the monoclinic crystal. The probe radius is 1.4 Å. The side chains of residues involved in sealing the inner cavities (Leu64, Leu73 and Met107) are shown in red. (D and E) Side and top views of the hydrophobic cavity of molecule B in the monoclinic crystal. The entrance of the pore is indicated by an arrow in (E). (F and G) Side and top views of the internal hydrophobic cavities of the hexagonal form. The surface of the cavity is drawn transparently. The PEGMME2000 molecule is shown as a CPK model.
The connectivity of the hydrophobic cavity to the bulk solvent significantly differs between the two structures of the LolB monoclinic crystal. The cavity of molecule A is sealed by the side chains of three residues, Leu64, Leu73 and Met107 (Figure 6B and C), whereas that of molecule B is connected to the solvent region (Figure 6D and E). The difference is caused by the conformational change of the side chain of Leu64. The χ1 values of this residue are 75.3 and –75.6° for molecules A and B, respectively. Other features near the entrance of the hydrophobic cavity are essentially the same. Therefore, the cavity seems to expand spontaneously to the solvent region. The hydrophobic cavity of the hexagonal form is completely open to the bulk solvent because of the existence of PEGMME2000 (Figure 6F and G). Taken together, these results indicate that LolB spontaneously binds hydrophobic compounds such as PEGMME2000 present in solution and expands its β-strands.
The hydrophobic loop and the N-terminal region of LolB
A hydrophobic leucine residue (Leu68) is located at the tip of the loop between β3 and β4, while this loop protrudes into the solvent region (Figure 2B). This leucine (or isoleucine) is conserved among LolB homologs (Figure 2C, yellow triangle). A neighboring residue, Pro67, is also hydrophobic. The loop containing these two hydrophobic residues may function when LolB transfers lipoproteins to the outer membrane. On the other hand, LolA has no such protruding loop, so that unfavorable backward transfer of lipoproteins to the inner membrane is unlikely to occur.
Residues Ala1–Pro9 of LolB are invisible in the electron density maps of the two crystal forms due to the disorder of the structure. Wild-type LolB is anchored on the outer membrane by three acyl chains which are covalently bound at the N-terminal Cys1. The N-terminal regions of LolB homologs are enriched with glycine and proline residues (Figure 2C, yellow letters), suggesting that the formation of a secondary structure is unfavorable. Flexibility of the N-terminal loop of LolB may be important when lipoproteins are transferred from LolB to the outer membrane.
Structural comparison between LolA and LolB
Because of their high structural similarity, LolA and LolB are likely to belong to the same structural family and to have the same ancestor protein. The LolA and LolB structures were well superimposed using the DALI server (Holm and Sander, 1995), with a root-mean-square deviation of 3.2 Å over 134 Cα atoms and a Z-score of 10.6. The superimposed structures also revealed some remarkable differences between them. The α1 helix near the N-terminus protrudes out of the β-sheet further in LolB than in LolA. The α2 helix of LolB is located on the surface of the molecule and exposed to the solvent region, whereas that of LolA is located at the center of the molecule. The α3 helices of LolA and LolB are almost perpendicularly oriented to each other when the core β barrels are superimposed. Arg43 of LolA functions as a lock disconnecting its hydrophobic cavity from the solvent region, whereas no residue plays such a role in LolB.
It has been suggested that LolB has higher affinity for lipoproteins than LolA (Matsuyama et al., 1997; Miyamoto et al., 2001; Tanaka et al., 2001). The structures of LolA and LolB revealed here indicate that both the tightly fixed lid and the hydrophobic cavity formed by aromatic residues are responsible for the lower affinity of LolA. It is also expected that the free form of LolA is more stable than the lipoprotein bound form, presumably causing the ejection of bound lipoproteins. On the other hand, not only free LolB but also PEGMME2000-bound LolB can be crystallized. These differences in the properties of LolA and LolB are essential for the vectorial transport of lipoproteins. Differences in the electrostatic properties of LolA (pI = ∼6) and LolB (pI = ∼9) determined on isoelectric focusing may also be important for the transient association of the two proteins.
Structural comparison with other proteins
The LolA and LolB structures were compared with other protein structures using the DALI server (Holm and Sander, 1995). Proteins with β-barrel structures, such as lipocalin family proteins (Banaszak et al., 1994), have a completely closed β-barrel structure, although these proteins also bind various hydrophobic molecules. Other lipid-binding proteins with β-strands, such as Rho GDP-dissociation inhibitor (Gosser et al., 1997) and phosphatidylethanolamine-binding protein (Serre et al., 1998), exhibit completely different connectivity from LolA or LolB. The structures of the MLN64-START domain protein (Tsujishita and Hurley, 2000) and phosphatidylinositol transfer protein (Yoder et al., 2001; Schouten et al., 2002) are probably the most similar, in which an unclosed β-barrel structure and several helices form a hydrophobic tunnel that binds cholesterol and phospholipid, respectively. The hydrophobic loop found in LolB also exists in these proteins, in which the loops are assumed to be involved in lipid loading and unloading. However, both the arrangement of helices and the number of β-strands are substantially different from those in LolA and LolB. Moreover, there is no significant sequence similarity between the two Lol proteins and these proteins. In the periplasmic space of Gram-negative bacteria, substrate-binding proteins collect particular substrates with high affinity and transfer them to ABC transporters. These proteins commonly have two domains (Boos and Eppler, 2001). Although LolA is a periplasmic protein, the structures of periplasmic binding proteins are significantly different from those of LolA and LolB.
Lipoprotein translocation mechanism
The structures of LolA and LolB shown here together with our previous observations further extend our understanding of the mechanism underlying Lol system-dependent lipoprotein transfer, as summarized below and in Figure 7.
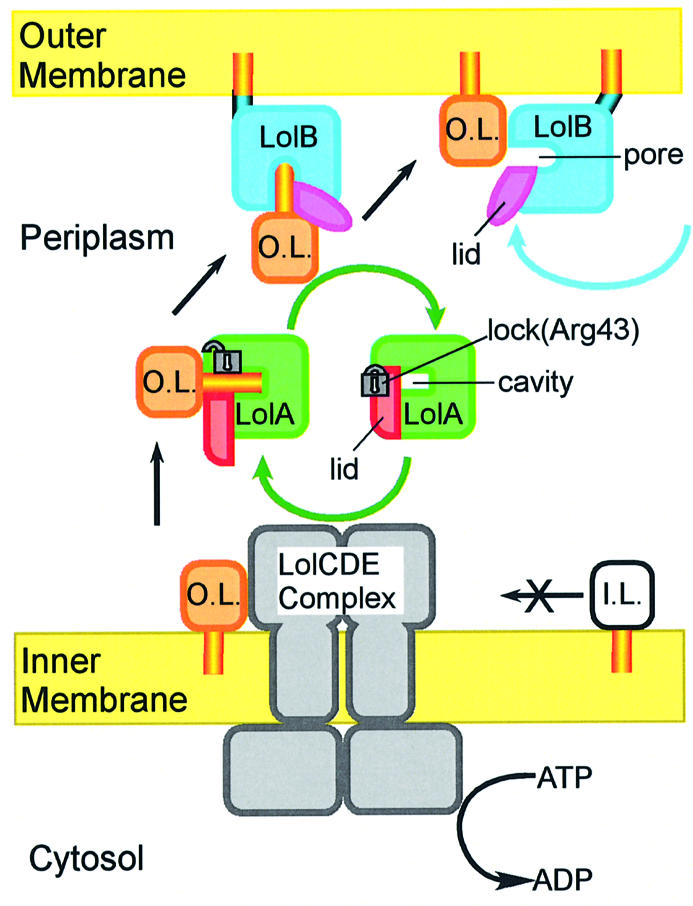
Fig. 7. Schematic representation of the outer membrane localization of lipoproteins mediated by the Lol system. I.L., inner membrane lipoproteins that have Asp at position 2; O.L., outer membrane lipoproteins that have a residue other than Asp at position 2.
(i) When the LolCDE complex interacts with outer membrane-specific lipoproteins, LolD releases the ATP energy on the cytoplasmic side of the membrane (Masuda et al., 2002). This energy is transferred from LolD to LolC/LolE and utilized to release lipoproteins from the outer leaflet of the membrane, leading to the formation of a water-soluble LolA–lipoprotein complex in the periplasm. This requires the opening of the LolA lid through disruption of the hydrogen bonds between Arg43 and the lid helices of LolA. The ATP energy released on the cytoplasmic side of membranes is thus utilized on the periplasmic side of the membrane to form a rather unstable LolA–lipoprotein complex. Inner membrane-specific lipo proteins are not recognized by LolCDE and therefore remain in the inner membrane (Masuda et al., 2002).
(ii) When the LolA–lipoprotein complex interacts with LolB, lipoproteins are transferred from LolA to LolB according to the affinity difference between their hydrophobic cavities for lipoproteins. LolA turns into a more stable free form upon the release of lipoproteins and then catalyzes the next lipoprotein release reaction cycle.
(iii) LolB anchored to the outer membrane swings through the flexible N-terminal region toward the outer membrane and then transfers the associated lipoproteins to the inner leaflet of the outer membrane. Lipoproteins are stably anchored to the outer membrane through three acyl chains.
The sequences of all Lol proteins are highly conserved among Gram-negative bacteria, and most bacteria have lipoproteins in their envelopes. The lipoprotein transfer mechanism involving the very similar hydrophobic cavities of LolA and LolB should also be conserved among Gram-negative bacteria and may provide insights into other traffic systems for hydrophobic proteins or lipids.
Materials and methods
Expression and purification of LolA and LolB
Wild-type LolA was overexpressed and purified as described previously (Matsuyama et al., 1995). A water-soluble LolB mutant, mLolB, was overexpressed and purified as described previously (Matsuyama et al., 1997). Selenomethionyl mLolB was overexpressed in E.coli P4X8 (Hfr P4X λ–metB1) grown at 37°C in minimal medium containing selenomethionine and purified as described for mLolB.
Crystallographic studies of LolA
LolA was concentrated to ∼20 mg/ml in 10 mM Tris–HCl pH 7.4. Two crystal forms of LolA were obtained by the hanging drop vapor diffusion method. Orthorhombic crystals were grown in 15% (w/v) PEG8000, 80 mM sodium cacodylate pH 6.5, 0.15 M zinc acetate and 20% (v/v) glycerol at 4°C. Trigonal crystals were obtained in a solution comprising 15% (w/v) PEG1500, 20 mM Tris–HCl pH 8.0 and 20% (v/v) glycerol at 20°C. A platinum derivative of an orthorhombic crystal was prepared by soaking a crystal in a reservoir solution containing 2 mM K2PtBr4 for 5 days. For diffraction data collection at a cryogenic temperature, all orthorhombic crystals were picked up with a nylon loop (Hampton Research) and rapidly cooled with liquid ethane. The orthorhombic crystals belong to space group I222, and the unit-cell dimensions are a = 55.8, b = 75.4 and c = 99.5 Å. The trigonal crystals belong to space group P3121 (or P3221) with unit-cell dimensions of a = b = 60.6 and c = 79.0 Å. Multi-wavelength anomalous diffraction data for the platinum derivative of an orthorhombic crystal were collected for one crystal. Initial phases up to 2.5 Å resolution were calculated with the SOLVE program (Terwilliger and Berendzen, 1999) and gave an overall figure of merit of 0.52. After density modification with the RESOLVE program (Terwilliger, 2000), the overall figure of merit was improved to 0.57. Details of the crystallization, data collection and phase determination will be reported elsewhere (Takeda et al., 2003a). The structural model was constructed into the electron density map using the XtalView program (McRee, 1999). The structure was refined by the simulated annealing method with the CNS program (Brunger et al., 1998) against the native data. Five per cent of the observed reflections were omitted from the refinement and used for calculating the Rfree factor. The final structural model exhibits Rfree and Rwork values of 0.249 and 0.222, respectively. The final structure does not include the amino acid residues (27–31) corresponding to the flexible loop region. The structure of the trigonal form was solved by the molecular replacement method with the CNS program using the molecular structure of the orthorhombic form of LolA as a search model. The correct space group was determined to be P3221. The final Rfree and Rwork factors for the model at 1.9 Å resolution are 0.259 and 0.226, respectively.
Crystallographic studies of LolB
LolB was concentrated to ∼20 mg/ml in 10 mM Tris–HCl pH 8.0. Two different crystal forms were obtained by the hanging drop vapor diffusion method. Monoclinic crystals were grown in 30% (w/v) PEGMME2000, 0.1 mM sodium acetate pH 4.6 and 0.2 M ammonium sulfate at 20°C. Monoclinic crystals of selenomethionylated protein were obtained in 30% (w/v) PEGMME2000, 0.1 mM sodium acetate pH 4.6 and 0.2 M cesium sulfate at 20°C. For diffraction experiments at a cryogenic temperature, all crystals were picked up with a nylon loop (Hampton Research) and rapidly cooled with liquid ethane without any cryo-protectant. The crystals belong to space group P21 with two molecules in an asymmetric unit with unit-cell dimensions of a = 37.2, b = 112.4, c = 47.8 Å and β = 111.4°. The initial phases were calculated by the MAD method with the selenomethionylated derivative using four-wavelength data sets. All sites of the 10 selenium atoms were determined with the SOLVE program. The overall figure of merit at 2.5 Å resolution was 0.50. Subsequent solvent flattening was performed using the RESOLVE program with an overall figure of merit 0.55. Details of the crystallization, data collection and phase determination will be reported elsewhere (Takeda et al., 2003b). The final model structure at 1.9 Å resolution exhibits Rfree and Rwork values of 0.249 and 0.215, respectively. Least-square superpositioning of the two molecules in the asymmetric unit gave the root-mean-square deviation value of 0.80 Å. The hexagonal crystals of LolB were also grown in 30% (w/v) PEGMME2000, 0.1 mM sodium acetate pH 4.6, 0.2 M ammonium sulfate and 0.15 M sodium iodide at 20°C. The crystals belong to space group P6322 with unit-cell dimensions of a = b = 71.4 and c = 133.9 Å. The structure was solved by the molecular replacement method with the CNS program using the molecular structure of the monoclinic form of LolB as a search model. The final Rfree and Rwork factors for the model at 2.2 Å resolution are 0.247 and 0.217, respectively. All the diffraction data were collected at the BL38B1 and BL44B2 beamlines of SPring-8 and processed with the HKL2000 package (Otwinowski and Minor, 1997). Ramachandran plots obtained for all structural models using the PROCHECK program (Laskowski et al., 1993) indicated that all residues are in the most favored or additional allowed regions, i.e. there are none in the generously allowed or disallowed regions. Figures were prepared using the MolScript (Kraulis, 1991), XtalView (McRee, 1999) and WebLabViewerPro (Accelrys, San Diego, CA, USA) programs.
Coordinates
The coordinates for LolA (orthorhombic and trigonal forms) and LolB (monoclinic and hexagonal forms) have been deposited in the Protein Data Bank under accession Nos 1IWL, 1UA8, 1IWM and 1IWN, respectively.
Acknowledgments
Acknowledgements
We wish to express our gratitude to Drs H.Tanida, S.Y.Park, S.Adachi and T.Hikima for their help in the data collection at SPring-8. This work was supported by grants for the ‘Research for the Future’ Program from the Japan Society for the Promotion of Science (97L00501) and for the National Project on Protein Structural and Functional Analyses from the Ministry of Education, Culture, Sports, Science and Technology.
References
- Banaszak L., Winter,N., Xu,Z., Bernlohr,D.A., Cowan,S. and Jones,T.A. (1994) Lipid-binding proteins: a family of fatty acid and retinoid transport proteins. Adv. Protein Chem., 45, 89–151. [DOI] [PubMed] [Google Scholar]
- Boos W. and Eppler,T. (2001) Prokaryotic binding protein-dependent ABC transporters. In Winkelmann,G. (ed.), Microbial Transport Systems. Wiley-VCH, Weinheim, Germany, pp. 77–114. [Google Scholar]
- Brightbill H.D et al. (1999) Host defense mechanisms triggered by microbial lipoproteins through Toll-like receptors. Science, 285, 732–739. [DOI] [PubMed] [Google Scholar]
- Brunger A.T. et al. (1998) Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D, 54, 905–921. [DOI] [PubMed] [Google Scholar]
- CCP4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D, 50, 760–763. [DOI] [PubMed] [Google Scholar]
- Driessen A.J.M., Manting,E.H. and van der Does,C. (2001) The structural basis of protein targeting and translocation in bacteria. Nat. Struct. Biol., 8, 492–498. [DOI] [PubMed] [Google Scholar]
- Fraser C.M. et al. (1997) Genomic sequence of a Lyme disease spirochaete, Borrelia burgdorferi. Nature, 390, 580–586. [DOI] [PubMed] [Google Scholar]
- Fukuda A., Matsuyama,S., Hara,T., Nakayama,J., Nagasawa,H. and Tokuda,H. (2002) Aminoacylation of the N-terminal cysteine is essential for Lol-dependent release of lipoproteins from membranes but does not depend on lipoprotein sorting signals. J. Biol. Chem., 277, 43512–43518. [DOI] [PubMed] [Google Scholar]
- Gosser Y.Q., Nomanbhoy,T.K., Aghazadeh,B., Manor,D., Combs,C., Cerione,R.A. and Rosen,M.K. (1997) C-terminal binding domain of Rho GDP-dissociation directs N-terminal inhibitory peptide to GTPases. Nature, 387, 814–819. [DOI] [PubMed] [Google Scholar]
- Hayashi S. and Wu,H.C. (1990) Lipoproteins in bacteria. J. Bioenerg. Biomembr., 22, 451–471. [DOI] [PubMed] [Google Scholar]
- Holm L. and Sander,C. (1995) DALI: a network tool for protein-structure comparison. Trends Biochem. Sci., 20, 478–480. [DOI] [PubMed] [Google Scholar]
- Jabs A., Weiss,M.S. and Hilgenfeld,R. (1999) Non-proline cis peptide bonds in proteins. J. Mol. Biol., 286, 291–304. [DOI] [PubMed] [Google Scholar]
- Kabsch W. (1976) A solution for the best rotation to relate two sets of vectors. Acta Crystallogr. A, 32, 922–923. [Google Scholar]
- Kraulis P.J. (1991) MOLSCRIPT: a program to produce both detail and schematic plots of protein structures. J. Appl. Crystallogr., 24, 946–950. [Google Scholar]
- Laskowski R.A., MacArthur,M.W. and Thornton,J.M. (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr., 26, 283–291. [Google Scholar]
- Masuda K., Matsuyama,S. and Tokuda,H. (2002) Elucidation of the function of lipoprotein-sorting signals that determine membrane localization. Proc. Natl Acad. Sci. USA, 99, 7390–7395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuyama S., Tajima,T. and Tokuda,H. (1995) A novel periplasmic carrier protein involved in the sorting and transport of Escherichia coli lipoproteins destined for the outer membrane. EMBO J., 14, 3365–3372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuyama S., Yokota,N. and Tokuda,H. (1997) A novel outer membrane lipoprotein, LolB (HemM), involved in the LolA (p20)-dependent localization of lipoproteins to the outer membrane of Escherichia coli. EMBO J., 16, 6947–6955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McRee D.E. (1999) Practical Protein Crystallography, 2nd edn. Academic Press, San Diego, CA. [Google Scholar]
- Miyamoto A., Matsuyama,S. and Tokuda,H. (2001) Mutant of LolA, a lipoprotein-specific molecular chaperone of Escherichia coli, defective transfer of lipoprotein to LolB. Biochem. Biophys. Res. Commun., 287, 1125–1128. [DOI] [PubMed] [Google Scholar]
- Miyamoto A., Matsuyama,S. and Tokuda,H. (2002) Dominant negative mutant of a lipoprotein-specific molecular chaperone, LolA, tightly associates with LolCDE. FEBS Lett., 528, 193–196. [DOI] [PubMed] [Google Scholar]
- Narita S., Tanaka,K., Matsuyama,S. and Tokuda,H. (2002) Disruption of lolCDE, encoding an ATP-binding cassette transporter, is lethal for Escherichia coli and prevents release of lipoproteins from the inner membrane. J. Bacteriol., 184, 1417–1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novick P. and Zerial,M. (1997) The diversity of Rab proteins in vesicle transport. Curr. Opin. Cell Biol., 9, 496–504. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z. and Minor,W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol., 276, 307–326. [DOI] [PubMed] [Google Scholar]
- Pugsley A.P. (1993) The complete general secretory pathway in Gram-negative bacteria. Microbiol. Rev., 57, 50–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankaran K. and Wu,H.C. (1994) Lipid modification of bacterial prolipoprotein. J. Biol. Chem., 269, 19701–19706. [PubMed] [Google Scholar]
- Schouten A., Agianian,B., Westerman,J., Kroon,J., Wirtz,K.W. and Gros,P. (2002) Structure of apo-phosphatidylinositol transfer protein α provides insight into membrane association. EMBO J., 21, 2117–2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serre L., Vallee,B., Bureaud,N., Schoentgen,F. and Zelwer,C. (1998) Crystal structure of the phosphatidylethanolamine-binding protein from bovine brain: a novel structural class of phospholipid-binding proteins. Structure, 6, 1255–1265. [DOI] [PubMed] [Google Scholar]
- Stewart D.E., Sarkar,A. and Wampler,J.E. (1990) Occurrence and role of cis peptide bonds in protein structures. J. Mol. Biol., 214, 253–260. [DOI] [PubMed] [Google Scholar]
- Tajima T., Yokota,N., Matsuyama,S. and Tokuda,H. (1998) Genetic analyses of the in vivo function of LolA, a periplasmic chaperone involved in the outer membrane localization of Escherichia coli lipoproteins. FEBS Lett., 439, 51–54. [DOI] [PubMed] [Google Scholar]
- Takeda K., Miyatake,H., Yokota,N., Matsuyama,S., Tokuda,H. and Miki,K. (2003a) A practical phasing procedure of MAD method without the Aid of XAFS measurements: successful solution in the structure determination of the outer membrane lipoprotein carrier LolA. Acta Crystallogr. D, in press. [DOI] [PubMed] [Google Scholar]
- Takeda K., Miyatake,H., Yokota,N., Matsuyama,S., Tokuda,H. and Miki,K. (2003b) Crystallization and preliminary crystallographic study of the outer membrane lipoprotein receptor LolB, a member of the lipoprotein localization factors. Acta Crystallogr. D, 59, in press. [DOI] [PubMed] [Google Scholar]
- Tanaka K., Matsuyama,S. and Tokuda,H. (2001) Deletion of lolB encoding an outer membrane lipoprotein is lethal for Escherichia coli and causes the accumulation of lipoprotein localization intermediates in the periplasm. J. Bacteriol., 183, 6538–6542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terada M., Kuroda,T., Matsuyama,S. and Tokuda,H. (2001) Lipoprotein-sorting signals evaluated as the LolA-dependent release of lipoproteins from the cytoplasmic membrane of Escherichia coli. J. Biol. Chem., 276, 47690–47694. [DOI] [PubMed] [Google Scholar]
- Terwilliger T.C. (2000) Maximum-likelihood density modification. Acta Crystallogr. D, 56, 965–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger T.C. and Berendzen,J. (1999) Automated MAD and MIR structure solution. Acta Crystallogr. D, 55, 849–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujishita Y. and Hurley,J.H. (2000) Structure and lipid transport mechanism of a StAR-related domain. Nat. Struct. Biol., 7, 408–414. [DOI] [PubMed] [Google Scholar]
- Yakushi T., Masuda,K., Narita,S., Matsuyama,S. and Tokuda,H. (2000) A new ABC transporter mediating the detachment of lipid-modified proteins from membranes. Nat. Cell Biol., 2, 212–218. [DOI] [PubMed] [Google Scholar]
- Yamaguchi K., Yu,F. and Inouye,M. (1988) A signal amino acid determinant of the membrane localization of lipoproteins in E.coli. Cell, 53, 423–432. [DOI] [PubMed] [Google Scholar]
- Yoder M.D., Thomas,L.M., Tremblay,J.M., Oliver,R.L., Yarbrough,L.R. and Helmkamp,G.M.Jr (2001) Structure of a multifunctional protein, mammalian phosphatidylinositol transfer protein complexed with phosphatidylcholine. J. Biol. Chem., 276, 9246–9252. [DOI] [PubMed] [Google Scholar]
- Zerial M. and Huber,L.A. (1995) Guidebook to the Small GTPases. Oxford University Press, Oxford, UK. [Google Scholar]
- Zheleznova E.E., Markham,P.N., Neyfakh,A.A. and Brennan,R.G. (1999) Structural basis of multidrug recognition by BmrR, a transcription activator of a multidrug transporter. Cell, 96, 353–362. [DOI] [PubMed] [Google Scholar]