

Abstract
Fungal sterol glucosyltransferases, which synthesize sterol glucoside (SG), contain a GRAM domain as well as a pleckstrin homology and a catalytic domain. The GRAM domain is suggested to play a role in membrane traffic and pathogenesis, but its significance in any biological processes has never been experimentally demonstrated. We describe herein that sterol glucosyltransferase (Ugt51/Paz4) is essential for pexophagy (peroxisome degradation), but not for macroautophagy in the methylotrophic yeast Pichia pastoris. By expressing truncated forms of this protein, we determined the individual contributions of each of these domains to pexophagy. During micropexophagy, the glucosyltransferase was associated with a recently identified membrane structure: the micropexophagic apparatus. A single amino acid substitution within the GRAM domain abolished this association as well as micropexophagy. This result shows that GRAM is essential for proper protein association with its target membrane. In contrast, deletion of the catalytic domain did not impair protein localization, but abolished pexophagy, suggesting that SG synthesis is required for this process.
Keywords: autophagy/PAZ4/pexophagy/Pichia pastoris/UGT51
Introduction
Genome-wide analyses have identified an enormous body of information on the primary structures of proteins and their motifs, but the actual roles of many of these modules and motifs remain to be elucidated. One such module is the recently identified GRAM domain, which occurs in the following three representative protein families: glucosyltransferases, Rab-like GTPase activators, and myotubularins (Doerks et al., 2000). This domain consists of ∼70 amino acid residues and is predicted to form four β-strands and one α-helix. Proteins harbouring the GRAM domain are widespread throughout the eukaryotes. Since many GRAM-domain proteins harbour additional lipid-binding motifs, e.g. FYVE or pleckstrin homology (PH) domains, they are thought to be associated with intracellular membranes. The physiological and biochemical functions of GRAM domains, however, have not been determined at either the cellular or molecular levels. Evidence for the physiological importance of the GRAM domain arose from patients suffering X-linked myotubular myopathy, some of which harbour point mutations within the GRAM domain of the myotubularin gene (de Gouyon et al., 1997).
Here, we report an investigation of GRAM-domain function using the fungal Ugt51/Paz4 sterol glucosyltransferase (UDP-glucose:sterol glucosyltransferase; EC 2.4.1.173). This enzyme catalyses the biosynthesis of sterol glucoside (SG), a minor membrane lipid present in many eukaryotes (Warnecke et al., 1999; Sakaki et al., 2001). The first sterol glucosyltransferase was initially purified from the oat (Warnecke and Heinz, 1994), and subsequently genes and cDNAs coding for the enzyme were cloned from plants, fungi, and Dictyostelium discoideum (Warnecke et al., 1997, 1999). The sterol glucosyltransferases from different yeast species display relatively narrow substrate specificities with respect to both activated sugar and glycosyl acceptor (Warnecke et al., 1999). In addition to the catalytic domain at the C-terminus, these sterol glucosyltransferases possess both a GRAM domain and a PH domain at their N-termini (Doerks et al., 2000). The presence of the PH domain supports the idea that these proteins are membrane associated, since the PH domain exhibits lipid-binding activity (Lemmon and Ferguson, 2000). Genetic and physiological analyses of the GRAM domain and/or sterol glucosyltransferases, however, have been hampered by the lack of any known physiological phenotype resulting from the loss of these enzymes and their products, SGs, until quite recently. SG was suggested to act as a sugar donor for an initial step in plant cellulose biosynthesis (Peng et al., 2002), and a sterol glucosyltransferase deletion mutant of the phytopathogenic fungus Colletotrichum gloeosporioides showed reduced virulence (Kim et al., 2002).
The present work will demonstrate that the sterol glucosyltransferase is essential for peroxisome degradation in Pichia pastoris. Peroxisome degradation in vacuoles, termed pexophagy, represents one aspect of the metabolic regulation of organelle turnover and intracellular membrane dynamics (Sakai and Subramani, 2000). The methylotrophic yeast P.pastoris is unique in that it has two morphologically distinct modes of pexophagy, i.e. macropexophagy and micropexophagy (Figure 1) (Tuttle and Dunn, 1995). When P.pastoris grows on methanol as the sole carbon source, peroxisomes proliferate that contain several enzymes, e.g. alcohol oxidase (Aox), required for methanol utilization. Shifting the carbon source to glucose or ethanol induces a shift towards other metabolic pathways. Under these conditions the peroxisomes become superfluous and are transported to lysosomes/vacuoles where they are degraded by lipases and proteases. During micropexophagy, which is induced by a glucose shift, the vacuole itself undergoes sequential morphological changes and incorporates the peroxisomes as a cluster (Figure 1A). First, the vacuole contacts a peroxisome cluster and starts to form lobes around the peroxisomes. In the meantime, a novel membrane structure involved in micropexophagy, called the micropexophagic apparatus (MIPA), is formed and attaches to the peroxisome cluster (H.Mukaiyama, M.Baba, M.Osumi, S.Aoyagi, N.Kato, Y.Ohsumi and Y.Sakai, submitted for publication). Subsequently, the peroxisomes and the MIPA are sealed by the vacuolar membrane lobes (Figure 1A). The function of the MIPA in this process is still unclear, though it is probably required for sequestration of peroxisomes from the cytosol by vacuolar membranes (H.Mukaiyama, M.Baba, M.Osumi, S.Aoyagi, N.Kato, Y.Ohsumi and Y.Sakai, submitted for publication).

Fig. 1. Overview of peroxisome sequestration via two distinct modes of pexophagy. V, vacuole; p, peroxisome cluster; MIPA, micropexophagic apparatus; MPP, macropexophagosome. (A) Micropexophagy. The vacuole itself engulfs a peroxisome cluster. At the initial stage (Stage 0), spherical vacuoles and peroxisome clusters are observed. Subsequently, the organelles initiate contact (Stage 1a), and the vacuolar membrane elongates to form lobes around the peroxisomes (Stage 1b). This lobe formation often results in fragmentation of the vacuole. Meanwhile, the MIPA is formed and attaches to the peroxisome cluster. At this stage, the MIPA is typically observed as a dot or cup-shaped image by fluorescence microscopy (Figures 8 and 9). After the vacuolar lobes envelop the MIPA (Stage 1c), the vacuole sequesters the peroxisomes (Stage 2). The MIPA is thought to ensure this peroxisome sequestration step. Finally, the peroxisomes are lysed in the vacuole (Stage 3). (B) Macropexophagy. A newly synthesized macropexophagosome first envelops a single peroxisome within the cluster and subsequently fuses with the vacuolar membrane, resulting in the release of the peroxisome surrounded by the inner membrane of the macropexophagosome into the vacuole.
A shift from methanol to ethanol, however, leads to macropexophagy, wherein a double-membrane structure, designated the macropexophagosome, enwraps a peroxisome and subsequently fuses with the vacuolar membrane, delivering the peroxisome to the inner side of the lytic organelle (Figure 1B).
Chemical and gene-tagging mutagenesis has been used to isolate P.pastoris mutants defective in micropexophagy (Yuan et al., 1997; Stromhaug et al., 2001; Mukaiyama et al., 2002). Subsequently, about twenty genes essential for this process were identified and designated as PAZ (pexophagy Zeocin-resistant mutant) or GSA (glucose-induced selective autophagy) genes (Stromhaug et al., 2001; Mukaiyama et al., 2002).
Several of these PAZ/GSA genes from P.pastoris are also involved in two related autophagic pathways, which have mainly been studied in Saccharomyces cerevisiae. Starvation-induced macroautophagy is a bulk degradation system responsible for removal of cytosolic components and organelles. The constitutive cytosol-to-vacuole targeting (Cvt) pathway transports aminopeptidase I (API) to the vacuole (Klionsky and Emr, 2000). Both pathways have several features in common with the two modes of pexophagy. First, the target cargos are degraded in the vacuole, and secondly, de novo membrane formation occurs in both cases (Tuttle and Dunn, 1995; Klionsky and Ohsumi, 1999; H.Mukaiyama, M.Baba, M.Osumi, S.Aoyagi, N.Kato, Y.Ohsumi and Y.Sakai, submitted for publication). Thus, it is not surprising that genes required for macroautophagy (APG/AUT) and the Cvt pathway (CVT) (Klionsky and Ohsumi, 1999) may have homologous counterparts in P.pastoris (PAZ/GSA) (Stromhaug et al., 2001; Mukaiyama et al., 2002). This has also been demonstrated by our recent finding that at least four PAZ gene products that contribute to the formation of the MIPA are homologous to S.cerevisiae APG gene products (H.Mukaiyama, M.Baba, M.Osumi, S.Aoyagi, N.Kato, Y.Ohsumi and Y.Sakai, submitted for publication). In addition, all the tested APG genes were found to be required for pexophagy in S.cerevisiae (Hutchins et al., 1999). In this study, we first identify Ugt51/Paz4 as a factor selectively involved in pexophagy, but not in starvation-induced macroautophagy in P.pastoris, and reveal that the GRAM domain of Ugt51 has a critical role during pexophagy.
Results
Analysis of micropexophagy-defective P.pastoris mutants generated by gene-tagging mutagenesis revealed that one of the genes affected by this method, PAZ4, is identical to the recently cloned sterol glucosyltransferase gene UGT51 (Warnecke et al., 1999; Mukaiyama et al., 2002). As a result, we generated a targeted ugt51/paz4 null-mutant and observed its ability to execute pexophagy.
Ugt51 is required for micropexophagy and macropexophagy
The UGT51-encoding open reading frame (ORF) was deleted from the P.pastoris genome, with confirmation by Southern analysis (data not shown). The resultant ugt51Δ mutant and the wild-type strain were studied with regard to pexophagy by an Aox-degradation assay, which allows visualization of residual peroxisomal matrix protein Aox activity following pexophagy to yield a quantitative index of non-degraded peroxisomes (Mukaiyama et al., 2002). As shown in Figure 2A, the wild-type strain formed white colonies, indicating complete degradation of Aox during micropexophagy and macropexophagy (after a methanol-to-glucose or ethanol shift, respectively). In contrast, ugt51Δ colonies were deep purple as a result of persisting Aox activity, indicating a deficiency in pexophagy. The Aox degradation defect of the ugt51Δ strain was also confirmed by immunoblot analysis using anti-Aox antibody (Figure 2B). The Aox band disappeared 9 h after the shift to glucose in the wild-type strain, but persisted in the ugt51Δ strain. Taken together with the results from the fluorescence studies shown below (Figure 6), we concluded that UGT51 is essential for both micropexophagy and macropexophagy.
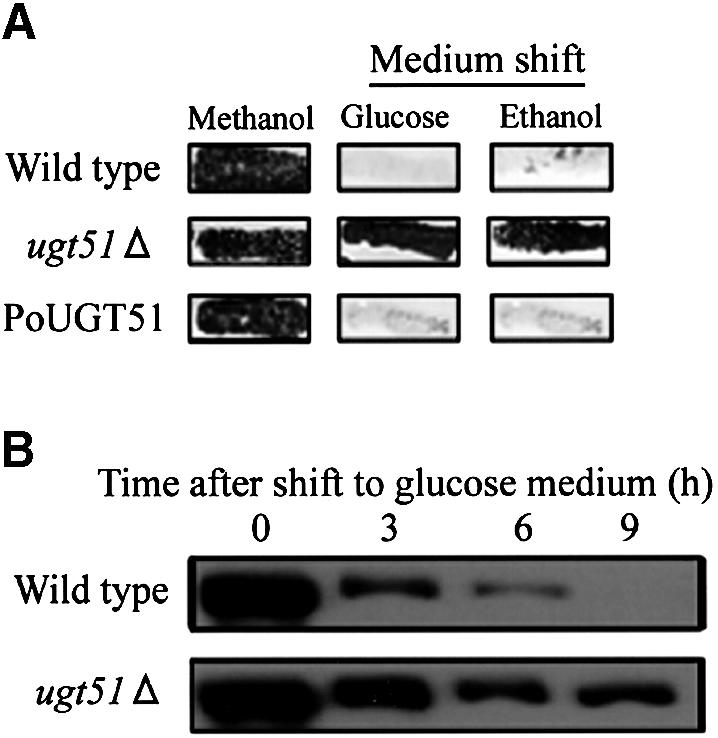
Fig. 2. Deletion of UGT51 leads to impaired degradation of the peroxisomal marker enzyme Aox. (A) Pichia pastoris cells were grown in methanol medium for 2 days and replica-plated to either a nylon membrane filter attached on a glucose medium plate or to an ethanol medium plate. Purple represents the persistence of the peroxisomal protein Aox detected by its activity staining. In wild-type cells, Aox was degraded following the carbon source shift to glucose (micropexophagy) or ethanol (macropexophagy). In contrast, both mechanisms were impaired in the ugt51Δ mutant (second line). Re-introduction of the UGT51 gene with its original promoter in the ugt51Δ strain restored peroxisome degradation (PoUGT51) (third line). (B) Wild-type and ugt51Δ cells were transferred to glucose medium, and harvested after the indicated times. Protein extracts were then subjected to immunoblot analysis using anti-Aox antibody. The decrease in the intensity of the Aox band was blocked in the ugt51Δ strain.

Fig. 6. The final stage of peroxisome sequestration is inhibited in the ugt51Δ cells. Wild-type and ugt51Δ cells expressing GFP fused with the peroxisome targeting signal PTS1 were transferred from methanol to glucose or ethanol medium to induce micropexophagy or macropexophagy, respectively. Images of vacuolar membrane staining with FM4-64 are presented in the left panels, and superimposed images with FM4-64 (red) and GFP–PTS1 signal (peroxisomal matrix, green) are shown in the right panels. (A and B) In the wild-type strain, GFP- fluorescence appeared diffusely throughout the vacuolar lumen 3 h after transfer to glucose (A, Stage 3) or ethanol medium (B) indicating normal procession of pexophagy. (C) In ugt51Δ cells, deeply invaginated vacuoles enwrapping intact peroxisomes were observed following a 3 h incubation in glucose medium (Stage 1c), but the GFP signal never diffused throughout the vacuole. (D) Longer incubation in glucose led to a decrease in the number of cells with peroxisome-sealing vacuolar lobes, and instead rounded vacuoles adjacent to peroxisome clusters represented the majority of observed structures. (E) The morphology of vacuole and peroxisome did not change for 3 h after transfer of ugt51Δ cells from methanol to ethanol.
Since UGT51 encodes a sterol glucosyltransferase, the quantity of its reaction product, SG, was compared between the wild-type and the ugt51Δ strain. As expected, SG was not detectable by thin-layer chromatography (TLC) of lipid extracts from the ugt51Δ strain, while it was present in very low quantities in wild-type extracts (Figure 3A). Therefore, we measured sterol glucosyltransferase activity by a more sensitive in vitro enzyme assay using radiolabelled substrates (Figure 3B). In wild-type cells, enzyme activity was detected, whereas it was absent in the ugt51Δ mutant, confirming that the deletion of UGT51 indeed completely abolished SG biosynthesis (Figure 3B).

Fig. 3. The catalytic domain, but not the PH or GRAM domains, is essential for SG biosynthesis by Ugt51. Methanol-grown P.pastoris cells were shifted to glucose medium and harvested after 2 h. (A) Total lipids were extracted and subjected to thin-layer chromatography. Low levels of SG were detected in wild-type cells, whereas the knock-out strain ugt51Δ was completely devoid of this lipid. The expression of GFP-tagged Ugt51 variants under the control of the strong AOX1 promoter led to the accumulation of considerable amounts of SG, even in strains expressing Ugt51 devoid of the GRAM or PH domain (strains GFP–UGT51, GFP–UGT51-PHΔ and GFP–UGT51-GRAMΔ). Only the deletion of the catalytic domain diminished SG synthesis (strain GFP–UGT51-CATΔ). (B) In vitro enzyme assays were performed to detect the sterol glucosyltransferase activity. Wild-type cells displayed low, but significant activity, which was absent in ugt51Δ cells. The catalytic activity in strain GFP–UGT51 was 30× higher than that of the wild type due to the strong expression by the AOX1 promoter.
The GRAM, PH and catalytic domains of Ugt51 are essential for micropexophagy
To elucidate the role of Ugt51 and SG biosynthesis in peroxisome degradation, different fragments of sterol glucosyltransferase were expressed in ugt51Δ cells. The resulting strains were evaluated for their ability to synthesize SG and to execute pexophagy. In order to investigate the localization of the domain-deleted enzymes, green fluorescent protein (GFP) was fused to the N-terminus of each full-length Ugt51 protein, and the constructs were then expressed under the control of the Ugt51 promoter (strain PoGFP–UGT51) or the strong AOX1 promoter (strain GFP–UGT51). The latter promoter was also used to express GFP-fusions with fragments of Ugt51 lacking either the PH, the GRAM or the catalytic domain (strains GFP–UGT51-PHΔ, GFP–UGT51-GRAMΔ and GFP–UGT51-CATΔ) (Figure 4B).

Fig. 4. Functional domains of the sterol glucosyltransferase Ugt51. (A) Schematic drawing of the annotated domains within Ugt51. The amino acid numbers at the borders of each domain are indicated. (B) Various Ugt51 variants were expressed in ugt51Δ cells under the control of either its original promoter (indicated by the prefix Po) or the strong AOX1 promoter (without indication). Resulting strains are grouped and a short-hand designation is given on the left. Their corresponding expression products are depicted schematically in the middle. Persistence of Aox is shown on the right panel. The strains that harbour the GRAM or catalytic domain-mutated Ugt51 retain Aox activity (strains GFP–UGT51-GRAMΔ, GFP–UGT51-Y642P and GFP–UGT51-CATΔ). The PH-domain deletion displayed impaired pexophagy when the enzyme was expressed under its original promoter (strain PoUGT51-PHΔ), but not when it was expressed under the AOX1 promoter (strain GFP–UGT51-PHΔ). The overview of the results for each strain is also shown in the right-hand columns. NT, not tested.
Analysis of the lipid composition of the various transformants by TLC revealed that all chimeric proteins were able to synthesize SG, except for the one that lacked the catalytic domain (Figure 3A). This shows that the fusion with GFP did not hamper SG synthesis and that neither the GRAM nor the PH domain was essential for in vivo enzymatic activity. SG synthesis in the strains expressing the truncated GFP-fusion proteins was even higher than that of the wild type, possibly due to the stronger AOX1 promoter.
In order to assign a functional role in micropexophagy to each domain of Ugt51, Aox-degradation assays were carried out (Figure 4B). Expression of the complete proteins Ugt51 or GFP–Ugt51 rescued the impairment of micropexophagy in the ugt51Δ strain regardless of the promoter used for expression (Figure 4B). In contrast, Ugt51 lacking the PH domain controlled by the original promoter could not restore micropexophagy. When overexpressed under the control of the strong AOX1 promoter, however, it could rescue the phenotype of the ugt51Δ strain (Figure 4B). The difference between the complementing activities of the two PH domain-deleted Ugt51 expressions is discussed below. GFP–Ugt51 derivatives lacking the GRAM domain or the catalytic domain did not complement the phenotype, even when expressed from the AOX1 promoter, showing that both of these domains are indispensable for Ugt51 function in micropexophagy. SG synthesis seems to be required but not sufficient for establishing micropexophagy, so most likely cooperation between the GRAM and PH domains of Ugt51 along with SG synthesis is needed.
In some patients with X-linked myotubular myopathy, the leucine residue at position 84 of myotubularin was found to be mutated to proline (de Gouyon et al., 1997). This residue is located within the α-helix of the GRAM domain. Computational analysis showed that the amino acid in Ugt51 corresponding to the site of the myotubularin mutation, Y642, also resides within the GRAM domain α-helix (Doerks et al., 2000). Strikingly, mutation of this single amino acid to proline resulted in the loss of micropexophagy in P.pastoris (Figure 4B), confirming the importance of this α-helix in GRAM domain functioning.
Peripheral association of Ugt51 with intracellular membranes
Since Ugt51 harbours a lipid-binding PH domain, it is likely to be associated to intracellular membranes. When micropexophagy was induced in P.pastoris cells by a carbon-source shift from methanol to glucose, the GFP–Ugt51 fusion protein was detected in a membrane fraction (the pellet after 100 000 g centrifugation of a cell-free homogenate) (Figure 5). After treatment of this membrane fraction with 1 M KCl or 0.1 M sodium carbonate followed by recentrifugation, GFP–Ugt51 was present in the soluble fraction, indicating peripheral binding to a membrane compartment (Figure 5). In contrast, treating the pellet with 1% Triton X-100 did not release GFP–Ugt51 from the membrane into the soluble fraction. Another membrane-associated factor in micropexophagy, termed Paz16, was extracted by either the KCl or Triton X-100 treatment (Y.Ano and Y.Sakai, unpublished data). The difference in the extractability indicates the peculiar properties of Ugt51 in membrane association.

Fig. 5. Membrane association of wild-type and variant Ugt51 proteins. Pichia pastoris ugt51Δ cells expressing different GFP-tagged Ugt51 fusion proteins were transferred from methanol to glucose medium and harvested after 1 h. The cells were lysed and the homogenates were fractionated by centrifugation at 100 000 g. The pellets (P1) and supernatant fractions (S1) were subjected to immunoblot analysis using anti-GFP antibody. In addition, the pellet fraction was treated with 1% Triton X-100 (TX-100), 1 M KCl; or 0.1 M Na2CO3, pH 11.0. After subsequent re-centrifugation, the resulting supernatants (S) and pellets (P) were also subjected to immunoblot analysis. Note that only Ugt51 lacking the PH domain was relocalized to the soluble fraction following Triton X-100 treatment. On the other hand, Paz16-CFP, another Paz protein, could be extracted to the soluble fraction by treatment with 1% Triton X-100 or 1 M KCl.
Next, the other GFP–Ugt51 fusion proteins were analysed in the same manner. Deletion of either the PH, GRAM or catalytic domains did not affect the properties in membrane association of Ugt51 (Figure 5), suggesting membrane association of Ugt51 is dependent upon the combined action of the three domains, or that another unidentified region is required for membrane association. Since the fusion proteins tested here were expressed under the control of the AOX1 promoter, it is possible that alterations in Ugt51 membrane association resulting from the lack of one single domain might have been complemented by strong overexpression of the protein.
GFP–Ugt51 lacking the PH domain was partially released from the membrane fraction following Triton X-100 treatment, while the other variants were retained in the membrane pellet (Figure 5), suggesting that the PH domain is involved in targeting or maintenance of the protein in a certain membrane fraction insoluble to Triton X-100.
Characterization of pexophagy in the ugt51Δ strain using fluorescently labelled cells
Previously, we developed a fluorescence assay to determine the stage at which each paz mutant strain interfered with micropexophagy, yielding key information on the function of PAZ gene products (Sakai et al., 1998). To extend these studies to Ugt51, we expressed GFP tagged with the peroxisome targeting signal 1 (GFP–PTS1) to label the peroxisomal matrix in the ugt51Δ and the wild-type strain. The resultant transformants were grown in the presence of the red dye FM4-64 to stain the vacuolar membrane.
When wild-type methanol-grown cells were shifted to glucose medium to induce micropexophagy, every step of the micropexophagic process could be observed, including the final stage (Stage 3; see Figure 6), characterized by diffuse GFP–PTS1 fluorescence throughout the vacuolar lumen (Figure 6A). In contrast, ugt51Δ cells showed deeply invaginated vacuoles wrapping the peroxisomes (Figure 6C, Stage 1c). Though some of the opposing tips in the vacuolar membrane lobes remained in contact with each other, longer incubation in glucose medium decreased the number of the cells with the peroxisome-sealing vacuolar lobes, and instead rounded vacuoles adjacent to peroxisome clusters represented the majority of the observed structures (Figure 6D). Thus, peroxisome clusters were not completely engulfed by vacuoles, and were never degraded. From these results we assume that the ugt51Δ strain is deficient for a reaction immediately prior to peroxisome sequestration (Stage 2) (Mukaiyama et al., 2002).
Regarding macropexophagy, the results were much the same. Three hours following a methanol-to-ethanol shift, diffuse GFP–PTS1 fluorescence could be observed in the wild-type vacuolar lumen, without significant vacuolar morphological changes (Figure 6B), a typical profile for the final stage of normal macropexophagy (Sakai et al., 1998). In contrast, GFP–PTS1 fluorescence was not observed in the vacuoles of the mutant strain (Figure 6E). These microscopic observations were consistent with the results of the Aox-degradation assay (Figure 2A), confirming that strain ugt51Δ is incapable of carrying out both micro- and macro-pexophagy.
Ugt51 is dispensable for starvation-induced macroautophagy in P.pastoris
To study starvation-induced macroautophagy in P.pastoris, a light-microscopic investigation of the macroautophagic bodies was carried out as described previously (Takeshige et al., 1992). When macroautophagy normally proceeds in glucose medium lacking nitrogen source, accumulating macroautophagic bodies can be detected within vacuoles following the addition of the proteinase inhibitor phenylmethylsulfonyl fluoride (PMSF). Both wild-type and ugt51Δ cells displayed one or two black granules in their vacuolar lumen after 8 h incubation in the medium (Figure 7A). In ugt51Δ pep4 prb1 cells lacking vacuolar peptidases, a similar granule structure was seen even in the absence of PMSF (data not shown). In contrast, no such granules were found in the presence of PMSF in a gsa11/paz7 strain that is defective in macroautophagy (GSA11/PAZ7 is homologous to S.cerevisiae APG2) (Stromhaug et al., 2001; Mukaiyama et al., 2002). This indicates that the ugt51Δ strain is capable of normal transport of autophagosomes to the vacuole, unlike macropexophagy-defective PAZ/APG mutants. For confirmation, cell viability after nitrogen starvation was used as an additional measure of macroautophagic ability, as impairment in this process halts cellular protein turnover leading to reduce viability in nitrogen-limited media (Tsukada and Ohsumi, 1993). After incubation for 10 days in glucose medium without nitrogen, the ugt51Δ strain retained a reasonable survival rate at approximately half of that of wild-type cells (Figure 7B). In contrast, the same treatment greatly diminished the viability of the gsa11/paz7 and gsa7/paz12 (GSA7/PAZ12 is homologous to APG7) (Yuan et al., 1999; Mukaiyama et al., 2002) cells to <10% of the wild-type strain (Figure 7B).
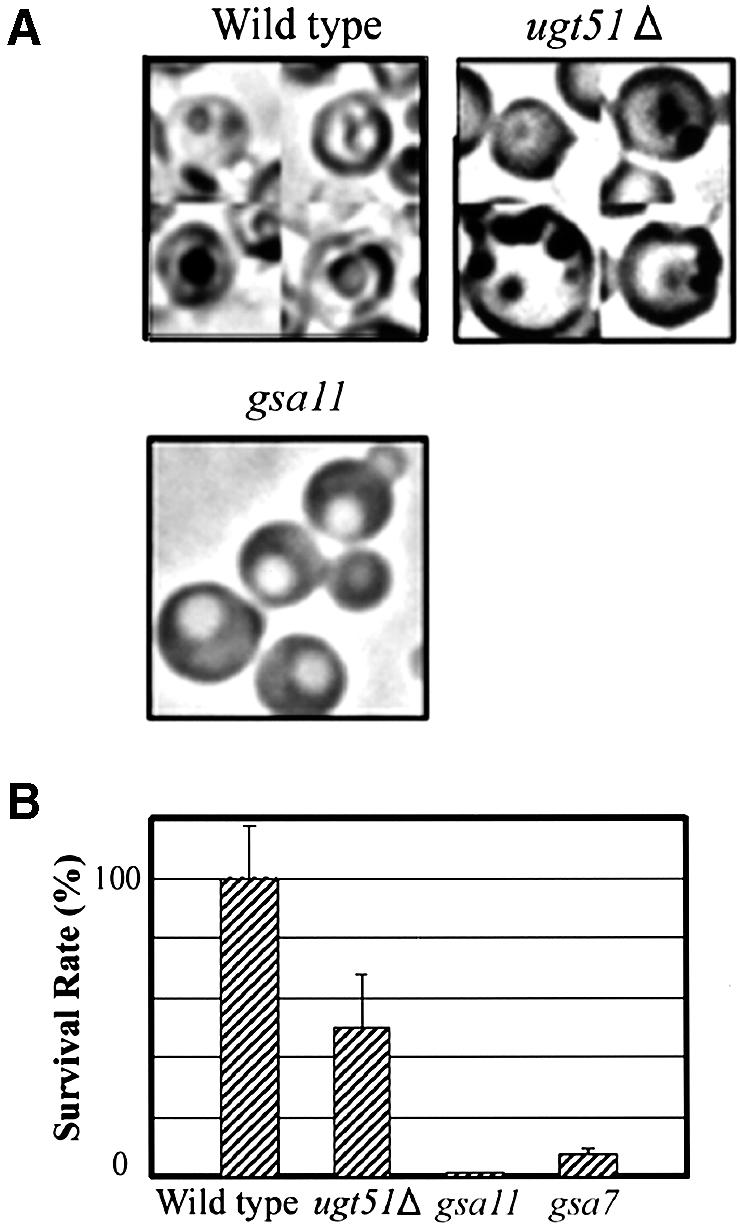
Fig. 7. Ugt51 is not involved in starvation-induced macroautophagy. (A) Light microscopy of P.pastoris wild-type, ugt51Δ, and starvation-induced macroautophagy-deficient gsa11/paz7 cells after 8 h incubation in glucose medium lacking nitrogen. In wild-type and ugt51Δ cells, black granules representing macroautophagic bodies were visible in the vacuolar lumen, but not in the gsa11 cells. (B) Cell viability was measured after 10 day incubation in glucose medium lacking nitrogen. The value (%) represents colony forming ability with reference to the wild type. Both gsa7 and gsa11 cells are macroautophagy deficient.
These results clearly demonstrate that the deletion of sterol glucosyltransferase in P.pastoris does not cause any detectable defects in starvation-induced macroautophagy. This specific function of Ugt51 on pexophagy is unique since the other previously known factors essential for pexophagy were also involved in the other autophagic pathways (see Discussion) (Hutchins et al., 1999; Kim et al., 1999, 2001; Noda et al., 2000; Stromhaug et al., 2001; Wang et al., 2001).
The GRAM domain of Ugt51 is essential for association with the MIPA
As Ugt51 was found to be a peripheral membrane protein (Figure 5), it is plausible that Ugt51 may associate with the MIPA. To examine this possibility, the intracellular localization of Ugt51 was investigated with GFP–Ugt51-expressing cells whose vacuoles had been labelled with FM4-64.
After peroxisome induction in methanol medium, GFP–Ugt51 had a diffuse and spotty localization throughout the cytoplasm (Figure 8A). One to three spots were observed per cell, and the majority of the structures were in proximity to the vacuole. A subsequent shift to glucose medium led to a gradual change in GFP–Ugt51 distribution. After 90 min, the spots had completely disappeared and been replaced by a new cup-shaped pattern that contacted the invaginating vacuolar membrane at the tips of both lobes (Figure 8B).
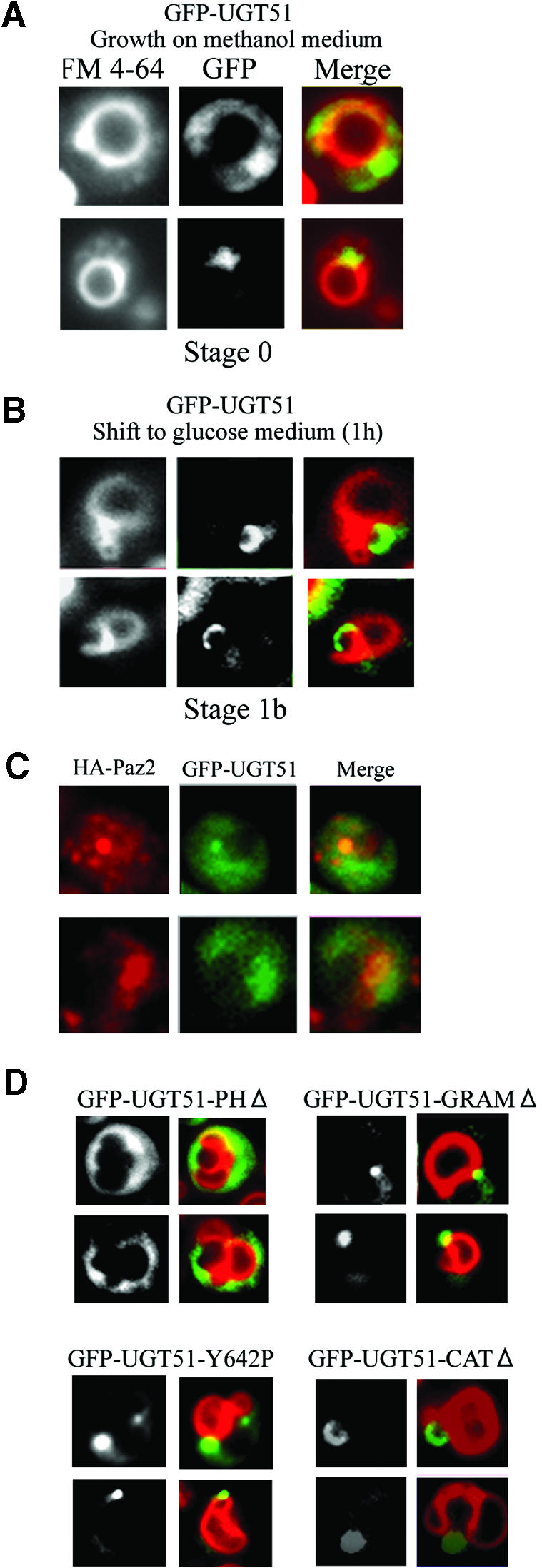
Fig. 8. Association of GFP–Ugt51 with the MIPA requires the GRAM domain. (A) When GFP–UGT51 cells were grown on methanol medium, a concentrated fluorescence could be observed in the proximity of the vacuoles. (B) When shifted to glucose medium to induce micropexophagy, the GFP signal pattern changed into a cup-shaped structure. (C) Immunofluorescence microscopy of P.pastoris cells expressing GFP–Paz4 and HA-Paz2 under micropexophagic conditions. Left panels show the HA-Paz2 signal, the middle panels GFP–Paz4 signal, and the right panels the merged images. The two signals coexisted in a dot (upper panels) or cup-shaped (lower panels) region, which indicated that Ugt51 localizes to the MIPA. (D) Strains expressing different truncated GFP–Ugt51 variants were studied under micropexophagic conditions. Left panels show the GFP images and right panels the respective images merged with FM4-64 fluorescence. The cup-shaped fluorescence pattern was observed in cells expressing Ugt51 lacking either the PH or the catalytic domain (strains GFP–UGT51-PHΔ and GFP–UGT51-CATΔ), but never in cells expressing GRAM domain-impaired Ugt51 (strains GFP–UGT51-GRAMΔ and GFP–UGT51-Y642P).
The localization pattern of GFP–Ugt51 in this micropexophagic process was identical to that observed with GFP–Paz2, the only known protein present in MIPA (H.Mukaiyama, M.Baba, M.Osumi, S.Aoyagi, N.Kato, Y.Ohsumi and Y.Sakai, submitted for publication). In order to confirm the colocalization of Ugt51 and Paz2, we performed immunofluorescence microscopy that detects GFP–Ugt51 and haemagglutinin (HA)-tagged Paz2 (Figure 8C). Most of the two signals converged in a dot or cup-like structure, indicating colocalization of the two proteins. Therefore we concluded that Paz4 was located on MIPA during micropexophagy.
Finally, we carried out fluorescence studies on the cytological effects of expressing truncated GFP–Ugt51 derivatives after induction of micropexophagy. In ugt51Δ cells harbouring a GFP–Ugt51 construct lacking the catalytic domain, the distribution of the GFP signal was similar to that of cells expressing full-length GFP–Ugt51 (Figure 8D). Cells expressing a GFP–Ugt51 construct lacking the PH domain also localized to the MIPA, but with the lower specificity, since the GFP signal was observed to be associated with other subcellular compartments. In contrast, both proteins with mutant GRAM domains, GFP–Ugt51-GRAMΔ and GFP–Ugt51-Y642P, were concentrated in perivacuolar, spot structures and never formed the characteristic cup-like structure (Figure 8D). From these results, we conclude that the association of Ugt51 to the MIPA depends on the presence of a functional GRAM domain.
Discussion
This study demonstrates that the UGT51/PAZ4 sterol glucosyltransferase gene is essential for both modes of peroxisome degradation in P.pastoris, micro- and macro-pexophagy. The UGT51 gene was also dispensable not only for the starvation-induced macroautophagy but also for the Cvt pathway in S.cerevisiae (T.Noda and Y.Sakai, unpublished data). Therefore, this gene is unique in that it is essential for peroxisome degradation, but not for macroautophagy or the Cvt pathway. Since fungal sterol glucosyltransferase is an enzyme consisting of different modules, the GRAM, PH and catalytic domains, we sought to determine a role for each of these domains in pexophagy.
Several membrane-bound proteins contain PH domains that exhibit affinity for specific phosphoinositides (Lemmon and Ferguson, 2000). It has been reported that a tyrosine kinase is recruited to detergent-insoluble, glycosphingolipid-enriched microdomains (DIGs) through its PH domain (Woods et al., 2001). Hence, it is likely that the PH domain of Ugt51 also regulates the association of Ugt51 with particular membranes or membrane domains. We found that expression of Ugt51-PHΔ under its original promoter (strain PoUGT51-PHΔ) did not restore pexophagy to ugt51Δ cells (Figure 4B), though the defect could be complemented by overexpression of the truncated enzyme as in strain GFP–Ugt51-PHΔ. This truncated form of Ugt51 localized to the membrane fraction as did the full-length protein, but was no longer restricted to the Triton X-100-insoluble compartment (Figure 5). Thus, the PH domain of Ugt51 is required for proper delivery to its target membranes, and, unless the expression level of the protein is artificially increased, correct localization is critical for proper functioning of Ugt51 in pexophagy.
GRAM domains were first identified by computational analyses of proteins from fungi, plants and animals (Doerks et al., 2000), but so far no function has been assigned to this widely distributed protein motif. Here, we present the first evidence that it is indispensable for proper delivery of Ugt51 to its target membrane, the MIPA (Figures 4B and 8D). The putative structure of the GRAM domain is that of four β-strands followed by one α-helix. This structure is believed to be physiologically important, as some corresponding point mutations were found within myotubularin gene of X-linked myotubular myopathy patients (de Gouyon et al., 1997; Laporte et al., 1997). One of the observed mutations found in the patient with milder symptoms is the replacement of leucine 84 with proline within the putative α-helix of the GRAM domain. We assume that the site-directed mutagenesis we carried out, replacing the corresponding amino acid of Ugt51 with proline (GFP–Ugt51-Y642P), affected the structure of the Ugt51 GRAM domain in the same way as did the L84P myotubularin mutation in these patients.
Through the present study, we obtained some insights into the putative function of SG in pexophagy. The Pichia strain expressing GFP–Ugt51 lacking the catalytic region was blocked in micropexophagy (Figure 4B), though the enzyme was still associated with the MIPA (Figure 8D). While this result shows the necessity of the Ugt51 catalytic activity for micropexophagy, we also found the pexophagy impairment in strain GFP–UGT51-GRAMΔ with normal SG synthesis ability (Figures 3A and 4B). Several trials to replenish SG in the ugt51Δ cells by addition of SG to the culture medium were not able to complement their pexophagic impairment. Even the application of ergosterol-biosynthesis inhibitors, e.g. ketoconazole or lovastatin, to enhance the SG uptake (Lorenz and Parks, 1990) did not lead to recovery of pexophagic competence (data not shown). These results together, indicate that SG synthesis alone was not sufficient for Ugt51 function in micropexophagy. Rather, it seems plausible that pexophagic processes may require biosynthesis of SG within the MIPA, which is accomplished by the specific localization of Ugt51.
Our result that Ugt51 is localized in a Triton X-100-insoluble membrane fraction (Figure 5) suggests that this protein may reside in a certain membrane domain. Interestingly, the transfer of glucose or lactose to another neutral lipid, ceramide, was reported to be necessary for Src tyrosine kinase localization in DIGs in a human cultured cell line, while it was not involved in the condensation of other components in the DIGs (Inokuchi et al., 2000). Similarly, SG synthesis on the specific membrane domain, e.g. in the MIPA, might help recruitment of certain factors necessary for pexophagy.
Although many questions regarding the membrane association of Ugt51 and the molecular mechanisms of pexophagy remain to be elucidated, we have herein provided the first evidence regarding the function of GRAM domains, specifically their involvement in targeting of proteins to certain membranes. In addition, this study suggests that the biosynthesis of the lipid SG plays a specific role in peroxisome degradation. The systems used during the course of these studies provide a sound experimental framework to dissect the roles played by proteins and lipids at the molecular level.
Materials and methods
Strains and media
Table I summarizes the yeast strains used in this study. We used the complete medium YPD (1% yeast extract, 2% peptone and 2% glucose) or synthetic dextrose minimal medium [0.67% yeast nitrogen base without amino acids (YNB; Difco), 2% glucose and auxotrophic amino acids if necessary] to proliferate the cells. Variants of the minimal medium, as described in the text, contained either 0.5% methanol or 0.5% ethanol instead of glucose as the carbon source. Peroxisomes were induced on methanol medium, and subsequently pexophagy was initiated by a shift to glucose medium (micropexophagy) or ethanol medium (macropexophagy). The cells were shifted to glucose medium lacking nitrogen (0.17% YNB without amino acids and nitrogen, and 2% glucose) to measure macroautophagy competence.
Table I. Pichia pastoris strains used in this study.
| Strain | Designation | Genotype (explanation, plasmid used for transformation) | Reference |
|---|---|---|---|
| PPY12 | wild-type | arg4 his4 (parental strain for UGT51 deletion) | Sakai et al. (1998) |
| STW1 | GFP–PTS1WT | PPY12 his4::PAOX1GFP-PTS1-HIS4 (GFP targeted to peroxisomes) | Sakai et al. (1998) |
| PZR7 | paz7Δ | STW1 paz7::ZeocinR | Mukaiyama et al. (2002) |
| PZR12 | paz12Δ | STW1 paz12::ZeocinR | Mukaiyama et al. (2002) |
| PZO101 | ugt51Δ | PPY12 ugt51::ZeocinR (sterol glucosyltransferase KO-strain) | This study |
| PZO102 | PoUGT51 | PZO101 his4::PUGT51UGT51-HIS4 (pEAE) | This study |
| PZO103 | GFP-PTS1ugt51Δ | PZO101 his4::PAOX1GFP-PTS1-HIS4 (pTW51) | This study |
| PZO104 | ugt51Δpep4prb1 | SMD1163 (pep4 prb1 his4) his4::pTW51 ugt51::Zeocin | This study |
| PZO105 | PoUGT51-PHΔ | PZO101 his4::PUGT51UGT51-PHΔ -HIS4 (pPZ4dPH) | This study |
| PZO200 | PoGFP-UGT51 | PZO101 his4::PUGT51GFP-UGT51-HIS4 (pGPZ43) | This study |
| PZO201 | GFP–UGT51 | PZO101 his4::PAOX1GFP-UGT51-HIS4 (pGPZ42) | This study |
| PZO202 | GFP–UGT51-PHΔ | PZO101 his4::PAOX1GFP-UGT51-PHΔ-HIS4 (pGPZ4dPH) | This study |
| PZO203 | GFP–UGT51-GRAMΔ | PZO101 his4::PAOX1GFP-UGT51-GRAMΔ-HIS4 (pGPZ4dGRAM) | This study |
| PZO204 | GFP–UGT51-CATΔ | PZO101 his4::PAOX1GFP-UGT51-CATΔ-HIS4 (pGPZ4dCT) | This study |
| PZO205 | GFP–UGT51-Y642P | PZO101 his4::PAOX1GFP-UGT51-Y642P-HIS4 (pGPZ4Y642P) | This study |
| PZO206 | GFP–UGT51/HA-Paz2 | PZO201 arg4::PPAZ2HA-PAZ2-ARG4 | This study |
DNA manipulation
All the DNA primers noted hereafter are listed in Table II.
Table II. Oligonucleotide primers used in this study.
| Primer name | DNA sequence |
|---|---|
| FM12 | AAAATGCATGATATCTTCTGTCGGGTCTACCCATAC |
| FM13 | GGAATGCATCCCTTCAGAACCCCCCTTAGAG |
| FM14 | AAAGCATGCATTTTGTGTAGCTTTTCTTTTTTTTTTC |
| FM15 | AAAGCATGCGATATCAAAAGAGGCGTTGAAC |
| UGTORFu | ACCATGTCACAACTCAGACCCAG |
| UGTORFd | TTACACTTCAAACCATGATC |
| NhePaz4u | CTAGCTAGCTCACAACTCAGACCCAGAGAT |
| Paz4d | CGAGCTCGCTTATTTGGCACCAGTACTTG |
| BamGFP5 | CGGGATCCATGAGTAAAGGAGAAGAACTTTTT |
| GFP3Nhe | CTAGCTAGCTTTGTATAGTTCATCCATGCCAT |
| PHfw | TTTCTTCCGAAAAGAGACTCCACTAAGAATAGCGGTGGTCATGTCACC |
| PHrv | GGTGACATGACCACCGCTATTCTTAGTGGAGTCTCTTTTCGGAAGAAATG |
| GRAMfw | GAAAGAGAAGTTGCACAGTCCAGAGGCTTTAGATTCGGGTACTCTGGG |
| GRAMrv | CCCAGAGTACCCGAATCTAAAGCCTCTGGACTGTGCAACTTCTCTTTC |
| Y642Pfw | ATTATAAACATTCTCAATATCGGAAGGTGGAAGTATCATAGTTGTGTTTCTG |
| Y642Prv | CACAAACACAACTATGATACTTCCACCTTCCGATATTGAGAATGTTTATAAT |
| Paz4tr765 | GAGGAGGTACAAATTTGTGCTATAAATCGATGG |
UGT51 deletion. To construct the ugt51Δ strain, the entire ORF of PAZ4/UGT51 (DDBJ/EMBL/GenBank accession No. AF091397) (Warnecke et al., 1999) was replaced by the Sh ble gene, which confers resistance to Zeocin™ (Invitrogen). The 0.5 kb stretches homologous to the 3′- and 5′-noncoding regions of UGT51 were amplified by PCR with a genomic fragment of P.pastoris DNA and the following primers: FM12 and FM13 for the 5′-region and FM14 and FM15 for the 3′-region. The two PCR fragments were cloned into pGEM-T vector (Promega), resulting in pPCR1 and pPCR2. The Sh ble gene was excised by BamHI–EcoRV from the pGAPZC vector (Invitrogen) and cloned into the pGEM3zf(+) vector treated with BamHI–HincII, yielding pGZ. The 3′-region of UGT51 was excised by SphI from pPCR2 and cloned into pGZ (SphI cut), yielding pGZT. The 5′-region of UGT51 was excised by EcoRV–HincII from pPCR1 and cloned into pGZT (Ecl136II cut), resulting in pGZTP. This plasmid was digested by EcoRI–EcoRV and introduced into P.pastoris strain PPY12. Zeocin™-resistant colonies were selected and correct replacement of UGT51 was confirmed by Southern blot analysis. The selected strain was designated as strain ugt51Δ (PZO101).
UGT51 expression. A P.pastoris genomic fragment, which was previously cloned into pUC19 (Warnecke et al., 1999), was digested with EaeI, and the resulting 5.5 kb fragment containing UGT51 and its flanking DNA sequences (including its promoter) were cloned into the NotI-treated vector pPIC3.5 (Invitrogen). The resulting construct, designated pEAE, was digested with SalI, and introduced into the ugt51Δ strain PZO101, resulting in strain PoUGT51 (PZO102).
GFP–UGT51 expression. The ORF of UGT51 was PCR amplified with the primers UGTORFu and UGTORFd (Table II), and this fragment was cloned into vector pPIC3.5, yielding pC3.5-1. The ORF was also amplified with primers NhePaz4u and Paz4d, and cloned into pGEM-T vector, giving pPAZ41. The GFP sequence was PCR amplified with pTW51 (Sakai et al., 1998) as the template and primers BamGFP5 and GFP3Nhe. The amplified fragment was then cloned into the pGEM-T vector, giving pGFP1. The sequence containing the GFP-coding region was excised from pGFP1 with SpeI–NheI and cloned into SpeI–NheI-digested pPAZ41, yielding pGPZ41. The fragment containing GFP and the 3.0 kb UGT51 ORF was excised from pGPZ41 by BamHI digestion. This fragment was ligated with the BamHI fragment from pC3.5-1. The resultant plasmid pGPZ42 was digested with StuI and introduced into strain ugt51Δ (PZO101), yielding strain GFP–UGT51 (PZO201). The entire sequence of the GFP–UGT51 ORF in pGPZ41 was also excised with SacI and cloned into pHM100 (Mukaiyama et al., 2002). The resultant pGPZ43 was digested with StuI and introduced into strain ugt51Δ, yielding strain PoGFP-UGT51 (PZO200). For the immunofluorescence microscopy, HA-tagged Paz2 (Mukaiyama et al., 2002) was introduced to PZO201, which yielded strain PZO206.
Construction of Ugt51 derivatives with domain-deletion or point mutation. In order to generate PH- and GRAM-domain mutations, we used the QuikChange™ Site-Directed Mutagenesis Kit (Stratagene), which applies inverse PCR with pGPZ42 as the template. We deleted or mutated each domain of Ugt51 using the following primers: PHfw and PHrv for PH-domain deletion; GRAMfw and GRAMrv for GRAM-domain deletion; Y642Pfw and Y642Prv for GRAM-domain Y642P mutation. The resulting plasmids were named according to the mutation introduced: PH-domain deletion, pGPZ4dPH; GRAM-domain deletion, pGPZ4dGRAM; and GRAM-domain Y642P mutation, pGPZ4Y642P. The catalytic domain-deleted GFP–Ugt51 was PCR amplified with primers BamGFP5 and Paz4tr765. The amplified fragment was digested with BamHI and cloned into BamHI–SnaBI-treated pPIC3K vector (Invitrogen) to generate pGPZ4dCT. All of the constructed plasmids were linearized with StuI and introduced into strain ugt51Δ (PZO101), resulting in strains GFP–UGT51-PHΔ (PZO202), GFP–UGT51-GRAMΔ (PZO203), GFP–UGT-CATΔ (PZO204) and GFP–UGT51-Y642P (PZO205). The construct that contains the PH-deleted Ugt51 ORF under its original promoter was the ligation product of AatII–NotI-cleaved pGPZ4dPH and pEAE. Introduction of the linearized construct in ugt51Δ (PZO101) resulted in PoUGT51-PHΔ (PZO105).
Aox degradation assay
Cells were grown on a methanol plate for 2 days, and transferred to a nylon membrane attached on a glucose plate for 12 h to induce micropexophagy or on an ethanol plate for 48 h to induce macropexophagy. The in situ activity staining of Aox was described previously (Sakai et al., 1998). Immunoblot analysis of Aox degradation was performed as described previously (Mukaiyama et al., 2002).
Lipid analyses and in vitro activity assay of Ugt51
Lipid extraction and analyses as well as the in vitro determination of sterol glucosyltransferase activity were performed as described previously (Warnecke et al., 1999).
Cell viability test
The cells were first grown on YPD to stationary phase and subsequently transferred to glucose medium lacking nitrogen at OD610 = 0.1. The culture was incubated with vigorous shaking at 28°C for 10 days, and the diluted culture was plated on YPD plates. The number of colonies formed after 2 day incubation was counted.
Membrane association
Strains expressing GFP-tagged Ugt51 were grown in methanol medium for 15 h from a starting OD610 = 0.5, and shifted to glucose medium for 1 h. One thousand OD610 units of the cells were harvested and spheroplasted as described previously (Faber et al., 1998). The cells were then osmotically lysed at 4°C in 10 ml of buffer A (0.2 M sorbitol, 50 mM potassium acetate, 2 mM EDTA, 20 mM HEPES–KOH pH 6.8, 1 mM DTT, 20 µg/ml PMSF, 0.5 µg/ml leupeptin and 0.7 µg/ml pepstatin A). After 10 min centrifugation at 3000 g, the supernatant was retrieved and further centrifuged at 100 000 g for 1 h. An aliquot of the pellet equivalent to 30 OD units of cells was mixed with 1 ml of the following buffers: 0.1 M sodium carbonate pH 11 supplemented with 0.2 M sorbitol, 1 M potassium chloride solution, or 1% Triton X-100 in buffer A. The mixtures were kept at 25°C for 10 min and again centrifuged at 100 000 g for 1 h. The supernatant fractions were precipitated by tetrachloroacetic acid and the pellets were resuspended in Laemmli sample buffer (Ausubel et al., 1987). The persistent pellet fractions were also solublized in Laemmli sample buffer. All samples were subjected to SDS–PAGE and subsequently to immunoblot analysis using 1:3000 diluted rabbit anti-GFP antiserum (Molecular Probes) as a primary antibody and 1:10 000 diluted goat anti-rabbit IgG antibody conjugated to horseradish peroxidase (Amersham Pharmacia Biotech) as a secondary antibody. The Enhanced Chemiluminescent detection system (Amersham Pharmacia Biotech) was employed to visualize the GFP signal.
Microscopic analyses
The observation of peroxisome-vacuole dynamics was performed as described previously (Sakai et al., 1998). To visualize the localization of GFP–Ugt51 or its derivatives, cells were labelled with 0.93 µg/ml FM 4-64 (Molecular Probes) during 12 h incubation in methanol medium with a starting OD610 = 0.5. Subsequently, the cells were shifted to glucose or ethanol medium, and studied under an IX70 fluorescence microscope (Olympus) equipped with an XF52 filter set (Omega Optical Inc.). Immunofluorescence microscopy was performed according to a standard protocol (Pringle et al., 1991) except that the pH of fixation buffer was adjusted to 8.2 to preserve GFP signal. The GFP signal was directly acquired, while Paz2 localization was visualized with anti-HA antibody (Roche) as primary antibody and tetramethylrhodamine isothiocyanate (TRITC)-conjugated anti-mouse IgG antibody (Southern Biotechnology Associates) as secondary antibody. Images were acquired with a Sensys™ Charged Coupled Device camera (Photometrics) and analysed on MetaMorph imaging software (Universal Imaging Corporation).
Acknowledgments
Acknowledgements
We would like to thank Professor Y.Ohsumi (National Institute for Basic Biology) for his helpful discussion and Professor S.Subramani (University of California San Diego) for generously providing us with P.pastoris strains and reagents. The authors would like to thank Dr H.Yurimoto, Y.Ano, and other members of the Laboratory of Microbial Biotechnology for helpful advice and technical assistance. This research was supported in part by Grant-in-Aid for Scientific Research (S) 13854008 and on Priority Areas 12146202 to Y.S. from the Ministry of Education, Science, Sports, and Culture of Japan, and the NIBB Cooperative Research Program (1-107), and by a ‘dynamic bio’ project from the Ministry of Economy, Trade and Industry. This work was also supported by the Deutsche Forschungsgemeinschaft, Sonderforschungsbereich 470.
References
- Ausubel F.M., Brent,R., Kingston,R.E., Moore,D.D., Seidman,J.G., Smith,J.A. and Struhl,K. (eds) (1987) Current Protocols in Molecular Biology. Greene Publishing Associates and Wiley-Interscience, New York, NY. [Google Scholar]
- de Gouyon B.M., Zhao,W., Laporte,J., Mandel,J.L., Metzenberg,A. and Herman,G.E. (1997) Characterization of mutations in the myotubularin gene in twenty six patients with X-linked myotubular myopathy. Hum. Mol. Genet., 6, 1499–1504. [DOI] [PubMed] [Google Scholar]
- Doerks T., Strauss,M., Brendel,M. and Bork,P. (2000) GRAM, a novel domain in glucosyltransferases, myotubularins and other putative membrane-associated proteins. Trends Biochem. Sci., 25, 483–485. [DOI] [PubMed] [Google Scholar]
- Faber K.M., Elgersma,Y., Heyman,J.A., Koller,A., Luers,G.H., Nuttley,W.M., Terlecky,S.R., Wenzel,T.J. and Subramani,S. (1998) Use of Pichia pastoris as a model eukaryotic system. In Higgins,D.R. and Cregg,J.M. (eds), Pichia Protocols, Vol. 103. Humana Press, Totowa, NJ, pp. 121–147. [DOI] [PubMed] [Google Scholar]
- Hutchins M.U., Veenhuis,M. and Klionsky,D.J. (1999) Peroxisome degradation in Saccharomyces cerevisiae is dependent on machinary of macroautophagy and the Cvt pathway. J. Cell Sci., 112, 4079–4087. [DOI] [PubMed] [Google Scholar]
- Inokuchi J.I., Uemura,S., Kabayama,K. and Igarashi,Y. (2000) Glycosphingolipid deficiency affects functional microdomain formation in Lewis lung carcinoma cells. Glycoconj. J., 17, 239–245. [DOI] [PubMed] [Google Scholar]
- Kim J., Dalton,V.M., Eggerton,K.P., Scott,S.V. and Klionsky,D.J. (1999) Apg7p/Cvt2p is required for the cytoplasm-to-vacuole targeting, macroautophagy, and peroxisome degradation pathways. Mol. Biol. Cell, 10, 1337–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J. et al. (2001) Cvt9/gsa9 functions in sequestering selective cytosolic cargo destined for the vacuole. J. Cell Biol., 153, 381–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y.K., Wang,Y., Liu,Z.M. and Kolattukudy,P.E. (2002) Identific ation of a hard surface contact-induced gene in Colletotrichum gloeosporioides conidia as a sterol glycosyl transferase, a novel fungal virulence factor. Plant J., 30, 177–187. [DOI] [PubMed] [Google Scholar]
- Klionsky D.J. and Emr,S. (2000) Autophagy as a regulated pathway of cellular degradaton. Science, 290, 1717–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky D.J. and Ohsumi,Y. (1999) Vacuolar import of proteins and organelles from the cytoplasm. Annu. Rev. Cell Dev. Biol., 15, 1–32. [DOI] [PubMed] [Google Scholar]
- Laporte J. et al. (1997) Mutations in the MTM1 gene implicated in X-linked myotubular myopathy. Hum. Mol. Genet., 6, 1505–1512. [DOI] [PubMed] [Google Scholar]
- Lemmon M.A. and Ferguson,K.M., (2000) Pleckstrin homology domains: phosphoinositide-regulated membrane tethers. In Cockcroft,S. (ed.), Biology of Phosphoinositides, Vol. 27. Oxford University Press, London, UK, pp. 131–165. [Google Scholar]
- Lorenz R.T. and Parks,L.W. (1990) Effects of lovastatin (mevinolin) on sterol levels and on activity of azoles in Saccharomyces cerevisiae. Antimicrob. Agents Chemother., 34, 1660–1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukaiyama H., Oku,M., Samizo,T., Hammond,A.T., Glick,B.S., Kato,N. and Sakai,Y. (2002) Paz2 and 13 other PAZ gene products regulate vacuolar engulfment of peroxisomes during micropexophagy. Genes Cells, 7, 75–90. [DOI] [PubMed] [Google Scholar]
- Noda T., Kim,J., Huang,W.-P., Baba,M., Tokunaga,C., Ohsumi,Y. and Klionsky,D.J. (2000) Apg9p/Cvt7p is an integral membrane protein required for transport vesicle formation in the Cvt and autophagy pathways. J. Cell Biol., 148, 465–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng L., Kawagoe,Y., Hogan,P. and Delmer,D. (2002) Sitosterol-β-glucoside as primer for cellulose synthesis in plants. Science, 295, 147–150. [DOI] [PubMed] [Google Scholar]
- Pringle J.R., Adams,A.E., Drubin,D.G. and Haarer,B.K. (1991) Immunofluorescence methods for yeast. Methods Enzymol., 194, 565–602. [DOI] [PubMed] [Google Scholar]
- Sakai Y. and Subramani,S. (2000) Environmental response of yeast peroxisomes. Aspects of organelle assembly and degradation. Cell Biochem. Biophys., 32, 51–61. [DOI] [PubMed] [Google Scholar]
- Sakai Y., Koller,A., Rangell,L.K., Keller,G.A. and Subramani,S. (1998) Peroxisome degradation by microautophagy in Pichia pastoris: identification of specific steps and morphological intermediates. J. Cell Biol., 141, 625–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaki T., Zahringer,U., Warnecke,D.C., Fahl,A., Knogge,W. and Heinz,E. (2001) Sterol glycosides and cerebrosides accumulate in Pichia pastoris, Rhynchosporium secalis and other fungi under normal conditions or under heat shock and ethanol stress. Yeast, 18, 679–695. [DOI] [PubMed] [Google Scholar]
- Stromhaug P.E., Bevan,A. and Dunn,W.A.,Jr. (2001) GSA11 encodes a unique 208-kDa protein required for pexophagy and autophagy in Pichia pastoris. J. Biol. Chem., 276, 42422–42435. [DOI] [PubMed] [Google Scholar]
- Takeshige K., Baba,M., Tsuboi,S., Noda,T. and Ohsumi,Y. (1992) Autophagy in yeast demonstrated with proteinase-deficient mutants and conditions for its induction. J. Cell Biol., 119, 301–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukada M. and Ohsumi,Y. (1993) Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Lett., 333, 169–174. [DOI] [PubMed] [Google Scholar]
- Tuttle D.L. and Dunn,W.A. (1995) Divergent modes of autophagy in the methylotrophic yeast Pichia pastoris.J. Cell Sci., 108, 25–35. [DOI] [PubMed] [Google Scholar]
- Wang C.W., Kim,J., Huang,W.P., Abeliovich,H., Stromhaug,P.E., Dunn,W.A.,Jr. and Klionsky,D.J. (2001) Apg2 is a novel protein required for the cytoplasm to vacuole targeting, autophagy, and pexophagy pathways. J. Biol. Chem., 276, 30442–30451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warnecke D.C. and Heinz,E. (1994) Purification of a membrane-bound UDP-glucose:sterol β-d-glucosyltransferase based on its solubility in diethyl ether. Plant Physiol., 105, 1067–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warnecke D.C., Baltrusch,M., Buck,F., Wolter,F.P. and Heinz,E. (1997) UDP-glucose:sterol glucosyltransferase: cloning and functional expression in Escherichia coli. Plant Mol. Biol., 35, 597–603. [DOI] [PubMed] [Google Scholar]
- Warnecke D., Erdmann,R., Fahl,A., Hube,B., Muller,F., Zank,T., Zahringer,U. and Heinz,E. (1999) Cloning and functional expression of UGT genes encoding sterol glucosyltransferases from Saccharomyces cerevisiae, Candida albicans, Pichia pastoris, and Dictyostelium discoideum. J. Biol. Chem., 274, 13048–13059. [DOI] [PubMed] [Google Scholar]
- Woods M.L., Kivens,W.J., Adelsman,M.A., Qiu,Y., August,A. and Shimizu,Y. (2001) A novel function for the Tec family tyrosine kinase Itk in activation of β1 integrins by the T-cell receptor. EMBO J., 20, 1232–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan W., Tuttle,D.L., Shi,Y.J., Ralph,G.S. and Dunn,W.A.J. (1997) Glucose-induced microautophagy in Pichia pastoris requires the α-subunit of phosphofructokinase. J. Cell Sci., 110, 1935–1945. [DOI] [PubMed] [Google Scholar]
- Yuan W., Stromhaug,P.E. and Dunn,W.A.J. (1999) Glucose-induced autophagy of peroxisomes in Pichia pastoris requires a unique E1-like protein. Mol. Biol. Cell, 10, 1353–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]