

Abstract
Copolymer 1 (COP), a standardized mixture of synthetic polypeptides consisting of l-glutamic acid, l-lysine, l-alanine, and l-tyrosine, has beneficial effects in multiple sclerosis and experimental autoimmune encephalomyelitis. We selected a panel of 721 COP-reactive T cell lines (TCL) from the blood of COP-treated and untreated multiple sclerosis patients and from healthy donors by using the split-well cloning technique. All TCL selected with COP proliferated in response to COP but not to myelin basic protein (MBP). Conversely, 31 control TCL selected with MBP proliferated in response to MBP but not to COP. We used intracellular double-immunofluorescence flow cytometry for quantitative analysis of cytokine production (IL-4, IFN-γ) by the TCL. The majority of the COP-reactive TCL from untreated multiple sclerosis patients and normal donors predominantly produced IFN-γ and, accordingly, were classified as T helper 1 cells (TH1). In contrast, the majority of the COP-reactive TCL from COP-treated patients predominantly (but not exclusively) produced IL-4—i.e., were TH2 (P < 0.05 as assessed by using a suitable preference intensity index). Longitudinal analyses revealed that the cytokine profile of COP-reactive TCL tends to shift from TH1 to TH2 during treatment. Interestingly, although there was no proliferative cross-reaction, about 10% of the COP-reactive TCL responded to MBP by secretion of small amounts of IL-4 or IFN-γ, depending on the cytokine profile of the TCL. These results are consistent with a protective effect of COP-reactive TH2 cells. It is hypothesized that these cells are activated by COP in the periphery, migrate into the central nervous system, and produce immunomodulatory cytokines after local recognition of MBP.
Copolymer 1 (COP, glatiramer acetate, Copaxone) is a standardized mixture of synthetic polypeptides consisting of l-glutamic acid, l-lysine, l-alanine, and l-tyrosine with a defined molar residue ratio of 0.14:0.34:0.43:0.1 and an average molecular mass of 4,700–11,000 Da. COP has beneficial effects on the clinical course and magnetic resonance imaging (MRI)-defined brain lesions of patients with multiple sclerosis (MS) (1–4). Furthermore, COP has suppressive and protective effects in experimental autoimmune encephalomyelitis (EAE) induced by various encephalitogens in different species (5–9), but not in other experimental autoimmune models (10). On the basis of extensive in vitro and in vivo studies in EAE, it has been proposed that COP acts by two basic mechanisms, (i) competition with myelin basic protein (MBP) at the MHC and T cell antigen receptor (TCR) level (11–14), and (ii) induction of T helper 2 (TH2)-type regulatory T cells (13, 15, 16). Relatively little is presently known about the in vitro and in vivo effects of COP in the human immune system (14, 17–19).
In the present study we isolated and analyzed a large panel of human COP-reactive T cell lines (TCL) by using the split-well cloning protocol (20). Consistent with previously reported results in EAE animals (13), we found that treatment with COP induces a shift from a TH1-biased cytokine profile observed in COP-reactive TCL obtained from untreated MS patients and healthy donors, toward a TH2-biased profile observed in TCL obtained from COP-treated patients. Furthermore, 8–15% of the tested COP-reactive TCL responded to MBP by secretion of small amounts of either IL-4 (TH2 or TH0 lines) or IFN-γ (TH1 or TH0 lines), although none of our COP-reactive TCL proliferated in the presence of MBP. The results indicate that the therapeutic effect of COP in MS may be related to a cytokine shift of COP-reactive T cells from TH1 to TH2, and to a cross-reaction with MBP at the level of cytokine production.
Materials and Methods
Patients and Control Subjects.
Blood was drawn with informed consent from 26 MS patients and 4 healthy donors. At the time of first sampling, 15 patients were treated with COP (20 mg s.c. per day; Teva Pharma, Kirchzarten, Germany). Six of the untreated patients were later started on COP; from these patients, only the data before treatment were included in the statistical analysis. All donors were HLA-typed (Table 1).
Table 1.
Basic characteristics of MS patients and healthy donors (HD)
| No. | Initials | Sex | Age, yr | Duration of disease, yr* | EDSS*† | HLA-DR type |
|---|---|---|---|---|---|---|
| MS 1 | SSt | F | 35 | 9 | 4.0 | 2, 5 |
| MS 2 | HK | M | 34 | 8 | 1.0 | 3, 12 |
| MS 3 | AZ | M | 28 | 1 | 1.0 | 2, 7 |
| MS 4 | HM | F | 25 | 2 | 1.0 | 2 |
| MS 5 | SW | F | 40 | 11 | 3.5 | 2, 8 |
| MS 6 | CS | F | 42 | 4 | 1.0 | 4, 8 |
| MS 7 | GJ | F | 30 | 1 | 1.0 | 4, 6 |
| MS 8 | WHa | F | 43 | 1 | 2.5 | 2, 11 |
| MS 9 | HC | F | 32 | 2 | 2.5 | 11, 12 |
| MS 10 | SZ | F | 34 | 4 | 2.5 | 2, 7 |
| MS 11 | LN | F | 36 | 1 | 2.5 | 1, 2 |
| MS 12 | BG | F | 33 | 4 | 1.5 | 4, 7 |
| MS 13 | UB | F | 39 | 10 | 6.0 | 4, 7 |
| MS 14 | US | F | 35 | 2 | 1.0 | 7, 10 |
| MS 15 | SH | F | 31 | 3 | 1.0 | 3 |
| MS 16 | BS | M | 21 | 1 | 1.5 | 13, 14 |
| MS 17 | KR | F | 39 | 4 | 1.5 | 1, 7 |
| MS 18 | RD | M | 40 | 17 | 2.5 | 2 |
| MS 19 | RM | F | 34 | 7 | 1.5 | 7, 13 |
| MS 20 | IB | F | 48 | 20 | 4.0 | 7, 11 |
| MS 21 | MB | F | 34 | 1 | 1.0 | 2, 7 |
| MS 22 | MC | F | 30 | 3 | 5.0 | 3, 7 |
| MS 23 | BK | F | 43 | 5 | 1.0 | 2, 4 |
| MS 24 | RR | M | 28 | 2 | 4.0 | 2, 11 |
| MS 25 | RO | F | 33 | 2 | 3.5 | 7, 13 |
| MS 26 | AF | F | 18 | 2 | 1.0 | 10, 12 |
| HD 1 | WH | M | 30 | — | — | 2, 11 |
| HD 2 | CH | M | 31 | — | — | 11, 13 |
| HD 3 | CB | F | 29 | — | — | 4, 11 |
| HD 4 | VV | F | 34 | — | — | 4, 15 |
At the time of first sampling.
EDSS, expanded disability status scale (31).
Antigens.
COP (batch 242992997, average molecular mass 7,000 Da, and batch 242992899, average molecular mass 6,400 Da) was obtained from Teva Pharmaceutical Industries, Petah Tiqva, Israel. The two batches were cross-reactive with each other as assessed in a proliferation assay. MBP was purified from human brain by standard methods (21). Overlapping peptides covering the entire human MBP molecule were synthesized by using an automatic peptide synthesizer (431A; Applied Biosystems). The extracellular Ig-like domain of human myelin-oligodendrocyte glycoprotein (MOG), amino acids 1–125, was expressed in an Escherichia coli system as described before (22). As a recombinant control, rat S100β protein was expressed and prepared in the same way (23). Tetanus toxoid (TT) was kindly provided by Chiron Behring, Marburg, Germany. Tuberculin purified protein derivative (PPD; batch RT49) was purchased from Statens Serum Institut, Copenhagen.
Cell Culture and Isolation of TCL.
All cell cultures were performed in RPMI medium 1640 (GIBCO) supplemented with 5% pooled and heat-inactivated human AB serum (German Red Cross, Baden-Baden) containing 2 mM glutamine, 100 units/ml penicillin, 100 μg/ml streptomycin (all from GIBCO), and 20 μg/ml ciprofloxacin (Ciprobay; Bayer Vital, Leverkusen, Germany) and incubated at 37°C in an atmosphere of 5% CO2/95% air. Long-term TCL were selected from peripheral blood mononuclear cells (PBMC) by using the split-well cloning technique as previously described (20).
Proliferation Assay.
Antigen-specific proliferation was determined by [3H]thymidine incorporation at several restimulation (R) steps (minimum R2). Autologous PBMC x-irradiated with 40 Gy (Stabiloplan 2; Siemens, Erlangen, Germany) were used as antigen-presenting cells (APC). APC (1 × 105 per well) were preincubated for 1 h in the presence or absence of various antigens (COP, 50 μg/ml; MBP, 30 μg/ml; MBP peptides, 10 μg/ml; MOG, 15 μg/ml; S100, 15 μg/ml; TT, 2 μg/ml; PPD, 10 μg/ml) in 96-well round-bottom microtiter plates (Nunc) in duplicate. Phytohemagglutinin (PHA, 10 μg/ml; Sigma) was used as a maximal stimulus. TCL cells were pooled, kept on ice for 3–4 h, and added to each well. After 48 h, [methyl-3H]thymidine (0.2–0.5 μCi per well; Amersham Buchler, Braunschweig, Germany; 1 μCi = 37 kBq) was added for another 16–18 h. Cells were harvested and [3H]thymidine incorporation was measured with a direct β-counter (Matrix TM 96; Packard, Frankfurt). Note that this method yields only 20% of the counts obtained by standard liquid scintillation systems. Only TCL with a minimum of 300–500 absolute cpm and a minimum stimulation index (SI) of 3.0 (except TT-reactive TCL: SI ≥ 1.8) were taken into account. The median SI of the COP-reactive TCL was 44.7 (range 3.0–1,826).
The MHC restriction was determined by using blocking mAbs to HLA-DR (L243; American Type Culture Collection) or HLA-DQ (SPVL3; Immunotech, Marseille, France), which were preincubated with the APC for 45 min at a final concentration of 20 μg/ml before adding the antigens.
Cytokine Production.
Antigen-induced production of IL-4 and IFN-γ was measured by ELISA (Endogen, Woburn, MA), using the supernatants of the proliferation assay. For this purpose, aliquots of 100 μl were removed from each well just before labeling with [methyl-3H]thymidine. Cytokine concentrations at least 2 SD above background were considered positive. According to the manufacturer's manuals, the lower limit of detection (sensitivity) was <2 pg/ml IL-4 or IFN-γ.
Characterization of the Cytokine Profile by Intracellular Double-Fluorescence Flow Cytometry.
The cytokine profile of the TCL was analyzed 8–10 days after restimulation in the absence of viable APC. COP-reactive TCL cells were stimulated with phorbol 12-myristate 13-acetate (PMA, 2.5 μg/ml) and ionomycin (250 ng/ml; both from Sigma) for 3 h, the last 2 h in the presence of the glycoprotein secretion blocker monensin (2 nmol/ml; Sigma). The T cells were then washed with PBS, fixed with 4% paraformaldehyde (Merck), and permeabilized with 0.1% saponin/PBS (Sigma). The T cells were stained by using appropriate concentrations of mAbs directed against IL-4 [8D4–8, phycoerythrin (PE)-labeled; PharMingen] and IFN-γ [B27, fluorescein isothiocyanate (FITC)-labeled; PharMingen] or the corresponding isotype controls (mouse IgG1 PE-labeled, Becton Dickinson; mouse IgG1 FITC-labeled, Immunotech).
The cytokine profile was analyzed with a FACScan (Becton Dickinson). Data from 5,000 cells were accumulated and the results were analyzed as dot plots representing the relative fluorescence intensity (Fig. 1). On a dot plot showing forward and sideward scatter, lymphoid cells were gated for further analysis (Fig. 1). Note that for unknown reasons, dead cells stained positive with the anti-IL-4 mAb and had to be excluded by gating. To define the predominant cytokine profile of each TCL, the following algorithm was applied (cf. Fig. 1): Only cells positive for IFN-γ, IL-4, or both were considered “activated.” If a single-positive fraction (i) exceeded 50% of all “activated” cells, and (ii) was at least 20% higher than the other single-positive fraction, the line was defined “TH1” (IFN-γ) or “TH2” (IL-4). All other TCL were designated as “TH0.” TCL with less than 100 “activated” events were not taken into account. Five or more TCL per donor were considered representative.
Figure 1.

Cytokine profile of COP-reactive TCL analyzed by intracellular double-fluorescence flow cytometry. (Upper Left) Dot plot of scatter parameters. (Upper Right) Isotype controls of one representative TCL. (Lower) Cytokine profiles of three representative TCL. Dot-plot events in the single-positive and double-positive quadrants were added. They represent “activated” cells. The numbers represent the percentage of events in each quadrant relative to the total number of activated cells. TH1, TH0, and TH2 assignments were made according to the algorithm described in the text.
Phenotypical Characterization of TCL by Flow Cytometry.
TCL were stained with labeled mAbs directed against CD3 (UCHT1, FITC-labeled; DAKO), CD4 (RPA-T4, PE-labeled, PharMingen), and CD8 (DK25, FITC-labeled; DAKO) and the corresponding isotype controls described above. The TCR Vβ (variable region) repertoire was analyzed by using mAbs recognizing the following subfamilies: Vβ2, Vβ3, Vβ5.3, Vβ7, Vβ8, Vβ9, Vβ11, Vβ12, Vβ13.6, Vβ14, Vβ16, Vβ17, Vβ18, Vβ20, Vβ21.3, Vβ22, Vβ23 (all Immunotech), Vβ3.1, Vβ5a, Vβ6.7 (T-Cell Diagnostics, Woburn, MA), Vβ5b (T-Cell Sciences, Cambridge, MA), Vβ7.1 (Labgen, Frankfurt). mAbs and isotype controls (mouse IgG1, Becton Dickinson; mouse IgG2a and IgG2b, Cymbus, Chandlers Ford, U.K.) were visualized with an FITC-labeled goat anti-mouse IgG antibody (Jackson ImmunoResearch, West Grove, PA).
Statistical Analysis.
Each line was assigned one of three numbers, namely −1 for the TH1 lines, 0 for the TH0 lines, and +1 for the TH2 lines. For each individual donor, a “preference intensity index” (I) was calculated for the TCL isolated from the donor according to the formula:
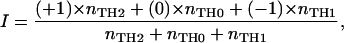 |
where nTH2, nTH0, and nTH1 denote the total number of TH2-, TH0-, and TH1-type TCL isolated from a given individual at a particular time. In this way, I is a metric variable expressing the proportion of TH1 TCL with respect to TH2 TCL independent of the absolute number of TCL obtained per donor. I varies between two extreme values, −1 (when all TCL are TH1) and +1 (when all TCL are TH2). For comparing the three groups (treated vs. untreated MS patients vs. healthy donors), a one-factorial analysis of covariance (ANCOVA) was applied with the age of the donors as covariate. If a significant group effect was observed, post hoc tests (tests with contrasts) were applied to identify pairs of groups with significant preference intensity differences. α = 0.05 was accepted as nominal level of significance and corrected for the post hoc tests according to the Bonferroni procedure to keep the type I error ≤ 0.05.
Results
Isolation and Characterization of COP-Reactive TCL.
We plated a total of 3,031 wells and isolated a panel of 721 COP-reactive TCL (23.8%). One hundred and sixty TCL were isolated from 1,054 plated wells (15.2%, untreated MS patients), 300 TCL from 1,155 plated wells (26.0%, COP-treated patients), and 90 lines from 330 plated wells (27.3%, healthy donors). In addition, 171 TCL could be established after onset of COP treatment from 492 plated wells (34.8%, previously untreated patients). All COP-reactive TCL showed a proliferative response to COP (mean SI 78.2, median SI 44.7, range 3.0–1,826), but none of the tested TCL proliferated significantly in response to MBP, MBP peptides, or any other antigens tested (MOG, S100β, and TT) (Fig. 2). Vice versa, 31 MBP-reactive TCL established from 2 untreated patients (25 lines) and 2 healthy donors (6 lines) proliferated in response to MBP (mean SI 223, median SI 109, range 5.3–1,044) but not to COP (Fig. 2). Interestingly, thus far, we have been unable to select MBP-specific TCL from 474 plated wells cultured from COP-treated patients, whereas TT-reactive TCL (16 lines) and PPD-reactive TCL (17 lines) could be easily isolated from both treated and untreated donors.
Figure 2.

Proliferative response of a representative COP-reactive and a MBP-reactive TCL. TCL were stimulated with COP, MBP, various control antigens (MOG, S-100β, and TT), or the T-cell mitogen phytohemagglutinin (PHA). There was no detectable cross-reaction between COP and MBP at the level of proliferation. Ag, antigen.
The complete phenotype, HLA restriction, and TCR usage were analyzed only in a subset of TCL. With the exception of two TCL which were composed of approximately equal numbers of CD4+ and CD8+ cells, the analyzed TCL (n = 40) were predominantly CD4+ (mean 87.4%, median 93.4%). The tested TCL (n = 12) were restricted by HLA-DR as assessed by 42–99% inhibition with an anti-HLA-DR mAb. TCR-Vβ expression (analyzed in 10 TCL) was heterogeneous.
Cross-Sectional Analysis: Effect of COP Treatment on the Cytokine Profile of COP-Reactive TCL.
We analyzed the cytokine profile of 693 of our COP-reactive TCL (150 TCL from untreated patients, 284 TCL from COP-treated patients, 90 TCL from healthy donors, 169 TCL from previously untreated patients after onset of COP treatment) by double-fluorescence flow cytometry at various restimulation steps (mostly R2 and R3). To minimize the influence of culture conditions, we strictly kept all TCL under identical conditions. Longitudinal comparisons of the cytokine profile of individual TCL at different restimulation steps showed that despite small fluctuations, the predominant cytokine profile remained stable: Of nine TCL that were followed up to five times (R2–R16), seven TCL strictly kept their predominant cytokine profile, whereas two TCL shifted from TH1 (R3) to TH0 (R8–R14).
To further validate this method, we assessed the COP-induced secretion of IFN-γ or IL-4 in a subset of 164 TCL by ELISA. Virtually all (98%) of the tested TCL showed a COP-induced cytokine response.
Fig. 3 shows the overall cytokine profiles of the complete panel of our COP-reactive TCL. Comparing the three groups (treated vs. untreated MS patients vs. healthy donors), the analysis of covariance revealed a significant effect [F(2, 29) = 10.26, significance of F = 0.001]. Although the donors were not age-matched, “age” as a covariate did not seem to play a role. The mean preference intensity index in COP-treated MS patients (I = +0.23) was significantly higher (indeed positive and thus skewed to TH2) than in untreated MS patients (I = −0.42) and untreated control subjects (I = −0.65) (tests with contrasts, P < 0.05). By pooling untreated patients and healthy donors and subsequently comparing the mean preference intensity of the combined group (I = −0.48) with the treated patients' group (I = +0.23) by ANCOVA, significant differences were observed [F (1, 29) = 20.40, significance of F < 0.0001]. The results suggest that treatment with COP induces a shift from TH1 (COP lines from untreated patients and healthy donors) toward TH2 (COP-treated MS patients). Clearly, the data do not allow us to decide whether this change occurs at the level of the cell population, individual cells, or both.
Figure 3.

Overview of the cytokine profiles of COP-reactive TCL of healthy donors (A), untreated MS patients (B), and COP-treated MS patients (C). Each column under patients' initials represents a panel of COP-reactive TCL isolated at one time point. Red indicates TH1, gray TH0, and green TH2. In C, the duration of treatment is indicated at the top. Large arrows indicate intraindividual longitudinal comparisons before and during COP treatment. Small arrows indicate intraindividual longitudinal comparisons after various times of COP treatment. (D) Comparison of the preference intensity indices I (calculated as described in the text) in untreated MS patients (left, ♦) and healthy controls (left, ▴), and COP-treated MS patients (right, ♦). I < 0 indicates a TH1 bias and I > 0 a TH2 bias, independent of the absolute number of TCL obtained per donor. Open squares represent mean preference intensity indices (±SD) of untreated donors (left) and COP-treated patients (right). Lines indicate intraindividual comparisons of six patients (only the data before treatment were included in the statistical analysis).
The TH2-inducing effect was specific for COP, as it was not seen with PPD- and TT-reactive TCL from two treated patients (one example is shown in Fig. 4). After 6 months of COP treatment, the COP-reactive TCL from the donor CS were either TH2 or TH0 (cf. Fig. 3), whereas 3 of 3 PPD- and 2 of 2 TT-reactive TCL were TH1 (Fig. 4). Moreover, while 14 of 15 COP-reactive TCL from the donor SH (after 15 months of treatment) were TH2 and 1 was TH0, all of this donor's 4 PPD- and 3 TT-reactive TCL were TH1.
Figure 4.

Cytokine profile of representative COP-, PPD- and TT-reactive TCL obtained from one COP-treated patient (month 6) and analyzed by intracellular double-fluorescence flow cytometry. The numbers represent the percentage of events in each quadrant relative to the total number of activated cells. TH1 and TH2 assignments were made according to the algorithm described in the text (cf. Fig. 1).
Longitudinal Analysis: Change of the Cytokine Profile of COP-Reactive TCL During Treatment of Individual Patients.
To further corroborate the results obtained by cross-sectional analysis, we investigated some patients longitudinally, that is, before and after various time periods of COP treatment (Fig. 3).
Cytokine profile before and after treatment.
TCL from one patient (GJ) had an unbiased cytokine profile before treatment (preference intensity I = −0.08), which almost completely shifted toward TH2 after 1 month of treatment (I = +0.92). After 3 months of treatment, the cytokine profile shifted back to TH0 (I = −0.10) (Fig. 3). Another patient, CS, kept a TH1-biased cytokine profile from before treatment (I = −0.38) during at least 2 months of treatment (I = −0.42). After 6 months of treatment, a slight shift toward TH2 was observed (I = +0.19). Four other patients (WHa, HC, SZ, and LN) showed no significant shift after 1–3 months of treatment.
To exclude that the longitudinal changes of the cytokine profile were related to the source of APC (autologous APC from untreated patients vs. autologous APC from treated patients), we compared five COP-reactive TCL from an untreated patient (GJ) restimulated with COP presented by (i) frozen APC that had been collected before treatment, and (ii) fresh autologous APC obtained after 4 weeks of treatment. The cytokine profile of these TCL were virtually identical (four lines were TH2, one line was TH1), independent of the source of APC (data not shown).
Cytokine profile during prolonged treatment.
Additional TCL could be obtained from individual patients after various intervals of prolonged treatment (Fig. 3). In patient RO, there was a trend for a shift from TH1 to TH2 (the preference intensity index I was −0.14 after 1 month and +0.42 after 6 months of treatment) which virtually shifted back to TH1 after 9 months of treatment (I = −0.10) (Fig. 3). Another patient (RR) showed the opposite trend during therapy. At month 1 and 2, the cytokine profile was biased toward TH2 (I = +0.67), at month 9, it was TH0-dominated (I = −0.13), whereas at month 12, the cytokine profile became TH1 (I = −0.48). The cytokine profiles of MC, BK, and AF remained TH2 up to 9 months of treatment.
Cytokine Secretion of COP-Reactive TCL After Cross-Stimulation with MBP.
As mentioned before, none of the tested human COP-reactive TCL showed a proliferative response when challenged with MBP, and vice versa. Previous observations in EAE demonstrated that COP-reactive TCL from mice treated with COP also did not proliferate in the presence of MBP, but some COP-reactive TCL produced IL-4 when stimulated with MBP (13). On the basis of these results, we tested whether human COP-reactive TCL can also be induced to cytokine secretion by stimulation with MBP. We found that, indeed, several COP-reactive TCL responded to MBP by secretion of low amounts of IL-4 or IFN-γ, depending on the predominant cytokine profile of the TCL. We tested 111 TCL for IL-4 secretion, and 53 TCL for IFN-γ secretion. Of these, 9/111 (8.1%) and 8/53 (15.1%) responded to MBP by significant (>2 SD above background) production of IL-4 or IFN-γ, irrespective of the source of the TCL (treated and untreated MS patients, healthy donors) (Fig. 5). Conversely, 1 of 7 tested MBP-specific TCL responded to COP by production of IL-4 (5.5 pg/ml vs. 0 pg/mg in the negative control). Furthermore, 2 of 12 tested COP-reactive TCL responded to MOG by production of IFN-γ (27.0 vs. 3.7 and 18.1 vs. 3.3 pg/ml, respectively).
Figure 5.

Proliferative response and cytokine production by two COP-reactive TCL. The left vertical axis denotes proliferation (gray bars), and the right vertical axis denotes cytokine secretion (black bars) measured by ELISA in supernatants of the same assay. Ag, antigen. (Upper) IL-4 secretion by a TH2 COP-reactive TCL. (Lower) IFN-γ secretion by a TH0 COP-reactive TCL. Asterisks denote cytokine levels greater than 2 SD above background. The lower limit of detection (sensitivity) of the cytokine ELISAs is <2 pg/ml IL-4 or IFN-γ.
Discussion
Our analysis of a large panel of COP-reactive human TCL revealed that the cytokine profile of the COP-reactive TCL tends to shift from TH1 before treatment to TH2 during treatment. Furthermore, about 10% of our COP-reactive TCL responded to MBP by cytokine secretion but not proliferation. Both observations are remarkably consistent with previously reported results in EAE (13, 15).
An important technical aspect of our study is that we analyzed the cytokine profiles of the COP-reactive TCL by intracellular double-fluorescence flow cytometry. This allowed us to precisely quantify the cytokine profile of each individual TCL and assign a preference intensity index to each patient, ranging on a continuous scale from −1 (100% TH1 lines) to +1 (100% TH2 lines). Parallel determinations with ELISA in a subset of TCL were consistent with the flow cytometry data. Although the observed cytokine shift was most obvious on cross-sectional analysis, our (limited) longitudinal data support the idea that a cytokine shift occurs during treatment. It is important to note, however, that the observed shift is a statistical phenomenon: individual patients showed a TH1- rather than TH2-biased cytokine profile despite prolonged treatment with COP. Furthermore, some COP-treated patients seem to shift back from a TH2 profile to a TH0 or even a TH1 profile. Yet, the cytokine data extend previous observations in COP-treated MS patients who showed increased levels of IL-10, transforming growth factor β (TGF-β), and IL-4 in peripheral blood cells (19). In our study, we selected one prototypical TH1 cytokine (IFN-γ) and one TH2 cytokine (IL-4). Assessment of additional cytokines, such as IL-5, -6, -10, -12, and -13, lymphotoxin, or TGF-β would probably not provide much additional information, and was simply not feasible, owing mainly to restrictions in cell numbers.
It will be interesting to establish in future studies whether the observed COP-induced TH2 shift is related to the clinical response. However, correlation with clinical outcome measures requires prospective studies with proper quantitative assessment of clinical scores and quantitative MRI data. An important question in this regard is whether the TH2 shift can help to differentiate between clinical “responders” and “nonresponders” to treatment. Furthermore, recent observations indicate that the beneficial effect of COP as detected by MRI requires several months to develop (3). Our data are consistent with such a delayed effect. It will also be interesting to see whether and to what extent the observed MRI effects are paralleled by the COP-induced TH2 shift.
The influence of COP treatment on the cytokine profile seems to be COP-specific, as it was not observed in PPD- or TT-reactive TCL obtained from same individuals. The mechanism of the COP-induced TH2 shift is unknown. As COP is applied s.c., it is likely that cutaneous Langerhans dendritic cells play an important role as local APC. In vitro data indicate that indeed, Langerhans cells can induce a TH2 response (24–26). As in other studies of antigen-induced cytokine responses, our data do not allow us to decide whether the treatment-related shift occurs at the level of single cells or cell population. Furthermore, it is presently unclear whether the s.c. route of application is essential for the COP-induced TH2 shift to occur. This question will probably be answered by the results of a clinical trial designed to compare the effects of s.c. and oral COP. In EAE, orally applied COP is clearly effective (16, 27).
About 10% of our human COP-reactive TCL respond to MBP by cytokine secretion but not proliferation. Again, this observation is consistent with recent observations in EAE (13): COP-reactive TCL isolated from (SJL/J × BALB/c)F1 mice immunized with COP exhibited a TH2 cytokine profile, secreting IL-4, IL-5, IL-6, and IL-10 but not IL-2 or IFN-γ in response to COP (13, 15). As in our study, some of the mouse COP-reactive T cells cross-reacted to MBP by cytokine secretion but not proliferation (13). The observed cross-reaction between COP and MBP at the cytokine level is apparently not unique to MBP but could also be observed with another myelin autoantigen, MOG, in a few TCL.
Regarding the possible mechanism of COP in vivo, it is known that COP binds efficiently to MHC class II molecules (11–13), and it competes with MBP at both the MHC class II and TCR levels (14, 17, 18). However, because it seems unlikely that significant amounts of COP can reach the central nervous system, these in vitro effects probably do not explain the clinical effects observed in vivo.
The following hypothetical scenario would accommodate both the previously reported EAE results and our observations in COP-treated patients: Chronic s.c. application of COP induces COP-reactive TH2 cells, which are able to cross the blood–brain barrier because they are activated (28). Inside the central nervous system, the COP-reactive T cells are confronted with products of myelin turnover presented by local APC (29). Some of the COP-reactive T cells react to MBP by secretion of protective cytokines such as IL-4. This might exert suppressive bystander effects on other inflammatory cells (13, 16, 30).
Acknowledgments
We are grateful to Drs. J. Haas and U. Augustin (Jewish Hospital, Berlin), Dr. N. König (Marianne-Strauß-Hospital, Berg, Germany), and Dr. C. Zimmermann (Institute for Clinical Neuroimmunology, University of Munich, Germany) for providing clinical samples. We thank Drs. E. Albert and S. Scholz (Department of Immunogenetics, University of Munich) for the HLA-typing and Dr. L. Jiang and Ms. M. Sölch for excellent technical assistance. This work was supported by Teva Pharma/Hoechst Marion Roussel. O.N. and H. Wiendl are postdoctoral fellows supported by the Deutsche Forschungsgemeinschaft. The Institute for Clinical Neuroimmunology is supported by the Hermann and Lilly Schilling Foundation.
Abbreviations
- APC
antigen-presenting cells
- COP
copolymer 1
- EAE
experimental autoimmune encephalomyelitis
- MBP
myelin basic protein
- MOG
myelin-oligodendrocyte glycoprotein
- MS
multiple sclerosis
- PE
phycoerythrin
- PPD
purified protein derivative
- SI
stimulation index
- TCL
T cell line(s)
- TCR
T cell antigen receptor
- TH0
TH1, and TH2, T helper type 0, 1, and 2 cells, respectively
- TT
tetanus toxoid
References
- 1.Bornstein M B, Miller A, Slagle S, Weitzman M, Crystal H, Drexler E, Keilson M, Merriam A, Wassertheil-Smoller S, Spada V, et al. N Engl J Med. 1987;317:408–414. doi: 10.1056/NEJM198708133170703. [DOI] [PubMed] [Google Scholar]
- 2.Johnson K P, Brooks B R, Cohen J A, Ford C C, Goldstein J, Lisak R P, Myers L W, Panitch H S, Rose J W, Schiffer R B, et al. The Copolymer 1 Multiple Sclerosis Study Group. Neurology. 1995;45:1268–1276. doi: 10.1212/wnl.45.7.1268. [DOI] [PubMed] [Google Scholar]
- 3.Johnson K P, Brooks B R, Cohen J A, Ford C C, Goldstein J, Lisak R P, Myers L W, Panitch H S, Rose J W, Schiffer R B, et al. The Copolymer 1 Multiple Sclerosis Study Group. Neurology. 1998;50:701–708. doi: 10.1212/wnl.50.3.701. [DOI] [PubMed] [Google Scholar]
- 4.Mancardi G L, Sardanelli F, Parodi R C, Melani E, Capello E, Inglese M, Ferrari A, Sormani M P, Ottonello C, Levrero F, et al. Neurology. 1998;50:1127–1133. doi: 10.1212/wnl.50.4.1127. [DOI] [PubMed] [Google Scholar]
- 5.Teitelbaum D, Meshorer A, Hirshfeld T, Arnon R, Sela M. Eur J Immunol. 1971;1:242–248. doi: 10.1002/eji.1830010406. [DOI] [PubMed] [Google Scholar]
- 6.Teitelbaum D, Webb C, Meshorer A, Arnon R, Sela M. Eur J Immunol. 1973;3:273–279. doi: 10.1002/eji.1830030505. [DOI] [PubMed] [Google Scholar]
- 7.Teitelbaum D, Webb C, Bree M, Meshorer A, Arnon R, Sela M. Clin Immunol Immunopathol. 1974;3:256–262. doi: 10.1016/0090-1229(74)90012-9. [DOI] [PubMed] [Google Scholar]
- 8.Teitelbaum D, Fridkis-Hareli M, Arnon R, Sela M. J Neuroimmunol. 1996;64:209–217. doi: 10.1016/0165-5728(95)00180-8. [DOI] [PubMed] [Google Scholar]
- 9.Ben-Nun A, Mendel I, Bakimer R, Fridkis-Hareli M, Teitelbaum D, Arnon R, Sela M, Kerlero de Rosbo N. J Neurol. 1996;243, Suppl. 1:S14–S22. doi: 10.1007/BF00873697. [DOI] [PubMed] [Google Scholar]
- 10.Arnon R. Immunol Lett. 1996;50:1–15. doi: 10.1016/0165-2478(96)02506-0. [DOI] [PubMed] [Google Scholar]
- 11.Fridkis-Hareli M, Teitelbaum D, Gurevich E, Pecht I, Brautbar C, Kwon O J, Brenner T, Arnon R, Sela M. Proc Natl Acad Sci USA. 1994;91:4872–4876. doi: 10.1073/pnas.91.11.4872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fridkis-Hareli M, Neveu J M, Robinson R A, Lane W S, Gauthier L, Wucherpfennig K W, Sela M, Strominger J L. J Immunol. 1999;162:4697–4704. [PubMed] [Google Scholar]
- 13.Aharoni R, Teitelbaum D, Sela M, Arnon R. J Neuroimmunol. 1998;91:135–146. doi: 10.1016/s0165-5728(98)00166-0. [DOI] [PubMed] [Google Scholar]
- 14.Aharoni R, Teitelbaum D, Arnon R, Sela M. Proc Natl Acad Sci USA. 1999;96:634–639. doi: 10.1073/pnas.96.2.634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aharoni R, Teitelbaum D, Sela M, Arnon R. Proc Natl Acad Sci USA. 1997;94:10821–10826. doi: 10.1073/pnas.94.20.10821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weiner H L. Proc Natl Acad Sci USA. 1999;96:3333–3335. doi: 10.1073/pnas.96.7.3333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Teitelbaum D, Milo R, Arnon R, Sela M. Proc Natl Acad Sci USA. 1992;89:137–141. doi: 10.1073/pnas.89.1.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Racke M K, Martin R, McFarland H, Fritz R B. J Neuroimmunol. 1992;37:75–84. doi: 10.1016/0165-5728(92)90157-g. [DOI] [PubMed] [Google Scholar]
- 19.Miller A, Shapiro S, Gershtein R, Kinarty A, Rawashdeh H, Honigman S, Lahat N. J Neuroimmunol. 1998;92:113–121. doi: 10.1016/s0165-5728(98)00191-x. [DOI] [PubMed] [Google Scholar]
- 20.Pette M, Fujita K, Kitze B, Whitaker J N, Albert E, Kappos L, Wekerle H. Neurology. 1990;40:1770–1776. doi: 10.1212/wnl.40.11.1770. [DOI] [PubMed] [Google Scholar]
- 21.Eylar E H, Kniskern P J, Jackson J J. Methods Enzymol. 1979;32B:323–341. [PubMed] [Google Scholar]
- 22.Brehm U, Piddlesden S J, Gardinier M V, Linington C. J Neuroimmunol. 1999;97:9–15. doi: 10.1016/s0165-5728(99)00010-7. [DOI] [PubMed] [Google Scholar]
- 23.Schmidt S, Linington C, Zipp F, Sotgiu S, De Waal Malefyt R, Wekerle H, Hohlfeld R. Brain. 1997;120:1437–1445. doi: 10.1093/brain/120.8.1437. [DOI] [PubMed] [Google Scholar]
- 24.Hauser C, Snapper C M, Ohara J, Paul W E, Katz S I. Eur J Immunol. 1989;19:245–251. doi: 10.1002/eji.1830190205. [DOI] [PubMed] [Google Scholar]
- 25.Simon J C, Cruz P D, Bergstresser P R, Tigelaar R E. J Immunol. 1990;145:2087–2091. [PubMed] [Google Scholar]
- 26.Chuang Y-H, Chiang B-L, Chou C-C, Hsieh K-H. Int Arch Allergy Immunol. 1996;111:366–371. doi: 10.1159/000237394. [DOI] [PubMed] [Google Scholar]
- 27.Teitelbaum D, Arnon R, Sela M. Proc Natl Acad Sci USA. 1999;96:3842–3847. doi: 10.1073/pnas.96.7.3842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wekerle H, Linington C, Lassmann H, Meyermann R. Trends Neurosci. 1986;9:271–277. [Google Scholar]
- 29.Krogsgaard M, Wucherpfennig K W, Canella B, Hansen B E, Svejgaard A, Pyrdol J, Ditzel H, Raine C, Engberg J, Fugger L. J Exp Med. 2000;191:1395–1412. doi: 10.1084/jem.191.8.1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hohlfeld R. Brain. 1997;120:865–916. doi: 10.1093/brain/120.5.865. [DOI] [PubMed] [Google Scholar]
- 31.Kurtzke J F. Neurology. 1983;33:1444–1452. doi: 10.1212/wnl.33.11.1444. [DOI] [PubMed] [Google Scholar]