

Abstract
Bradykinin (BK) or kallikreins activate B2 receptors (R) which couple Gαi and Gαq proteins to release arachidonic acid (AA) and elevate [Ca2+]i. Thrombin cleaves the protease-activated-receptor-1 (PAR1) that couples Gαi, Gαq and Gα12/13 proteins. In CHO cells stably transfected with human B2R, thrombin liberated little AA, but it significantly potentiated AA release by B2R agonists. We explored mechanisms of cooperativity between constitutively expressed PAR1 and B2R. We also examined human endothelial cells expressing both Rs constitutively. The PAR1 agonist hexapeptide (TRAP) was as effective as thrombin. Inhibitors of components of Gαi, Gαq and Gα12/13 signaling pathways, and a PKCα inhibitor, Gö6976 blocked potentiation while phorbol, an activator, enhanced it. Several inhibitors, including a RhoA kinase inhibitor, a [Ca2+]i antagonist, and an inositol-(1,3,4)-trisphosphate R antagonist, reduced mobilization of [Ca2+]i by thrombin and blocked potentiation of AA release by B2R agonists. Because either a non-selective inhibitor (isotetrandrine) of phospholipase A2 (PLA2) or a Ca2+-dependent PLA2 inhibitor abolished potentiation of AA release by thrombin, while a Ca2+-independent PLA2 inhibitor did not, we concluded that the mechanism involves Ca2+-dependent PLA2 activation. Both thrombin and TRAP modified activation and phosphorylation of the B2R induced by BK. In lower concentrations they enhanced it, while higher concentrations inhibited phosphorylation and diminished B2R activation. Protection of the N-terminal Ser1-Phe2 bond of TRAP by an aminopeptidase inhibitor made this peptide much more active than the unprotected agonist. Thus, PAR1 activation enhances AA release by B2R agonists through signal transduction pathway.
Keywords: PAR1, TRAP, protein kinases, potentiation, Ca2+, kallikrein
Abbreviations: PAR1, protease activated receptor type 1; TRAP, thrombin receptor activator peptide; Kallikrein, kallikrein of human plasma; BK, bradykinin; thr, thrombin; BEL, bromo-enol lactone; DAG, diacylglycerol; cPLA2 inhib, Ca2+-dependent phospholipase A2 inhibitor; IP3, inositol-(1,3,4)-trisphosphate; 2-APB, 2-aminoethoxydiphenyl borate; TMB-8,8-(N, N, diethylamino) octoyl-3,4,5-trimethoxybenzoate; Y27362, Rho kinase inhibitor; RGS10, regulator of G protein signaling type 10; ⊣ inhibition
INTRODUCTION
Thrombin, the potent physiologic activator of platelet aggregation, is a major factor in myocardial infarction and other thrombotic processes (15, 22, 32). In addition to promoting coagulation, thrombin plays a central role in the cellular response to injury or inflammation and wound repair. Thrombin initiates many of these complex cellular events by cleaving the Arg41-Ser42 bond of the protease-activated receptor-1 (PAR1) to expose a new N-terminus. This terminus then acts as a tethered ligand (13, 38) and activates PAR1. Various guanine nucleotide-binding (G) protein α-subunits can then couple with PAR1. When coupled to Gαq protein PAR1 generates diacylglycerol (DAG) and inositol-(1,4,5) trisphosphate (IP3), which in turn binds to IP3 Rs on the endoplasmic reticulum to trigger intracellular calcium [Ca2+]i release. Interaction of IP3 R with transient R potential channel 1 (TRPC1) is required to activate store depletion-induced Ca2+ entry (21, 27, 31). In addition to Gαq, the stimulated PAR1 couples Gαi/0 (14, 25, 37) and Gα12/13 proteins (12, 17, 27). Activation of the Gαi/o protein leads to release of the prostaglandin precursor, arachidonic acid (AA).
Activation of B2Rs for bradykinin (BK) can cause among others hypotension, bronchoconstriction, pain, or inflammation (3, 16) and releases vascular mediators such as NO, or prostaglandins, and others (3, 8–11). The PAR1-4 group of thrombin and trypsin Rs, as their name indicates, were thought to be unique protease-activated Rs. However, we showed that the human BK B2R can be activated by certain proteases as well (20). Kallikreins and some other human or bacterial serine proteases, including cathepsin G or endoarginase C, can activate B2 BK Rs directly (19, 20). Furthermore, activation of the ubiquitous B2R by BK or proteolytic enzymes (20) recruits both Gαi and Gαq proteins, as indicated by the release of AA and elevation of [Ca2+]i (18, 26).
Burch and Axelrod found that BK and thrombin synergized prostaglandin synthesis in fibroblasts, and after prolonged stimulation, thrombin amplified responses to BK (5–7). After we discovered that kallikrein and other proteases directly activate the BK B2Rs (19, 20), we used cultured cells to determine if thrombin would enhance AA release by kallikrein via B2R and if PAR1 activation potentiates kallikrein via signal transduction pathways. We established that the two proteases acted on a single R, either PAR1 or B2 because we obtained the same results with peptide agonists specific for the individual Rs. We used inhibitors of the major factors in signal transduction to point out the steps in the potentiation process.
MATERIALS AND METHODS
Chinese Hamster Ovary (CHO) cells were purchased from American Type Culture Collection (Rockville, MD). HPAE cells were obtained from Clonetics (Walkersville, MD). The cDNA encoding the human BK B2R was donated by Dr. K. Jarnigan, Syntex Co (Palo Alto, CA), the human regulator of G protein signaling type 10 (RGS10) cDNA was from the University of Missouri-Rolla (Rolla, MO). Mammalian expression vectors pcDNA3 was from Invitrogen (San Diego, CA); lipofectin and geneticin (G418) were from Gibco BRL (Gaithersburg, MD). [5,6,8,9,11,12,14,15-3H(N)]-arachidonic acid (AA); 100 C1/mmol) and [3H]BK were purchased from American Radiolabeled Chemicals (St. Louis, MO). The fura-2/AM, anti-GFP polyclonal antibodies were obtained from Molecular Probes (Eugene, OR), and D-valyl-leucyl-arginyl-aminomethyl-coumarine (D-Val-Leu-Arg-AMC) was from Enzyme Systems Products (Livermore, CA). Bromo-enol lactone (BEL) was obtained from Cayman Chemical Company (Ann Arbor, MI) and cPLA2 inhibitor (N-{(2S,4R)-4-(biphenyl-2-ylmethyl-isobutyl-amino)-1-[2-(2,4-difluorobenzoyl)-benzoyl]-pyrrolidin-2-ylmethyl}-3-[4-(2,4-dioxothiazolidin-5-ylidenemethyl)-phenyl]acrylamide, HCl), Gö6976, Y27632, H-89 dihydrochloride, 2-aminoethoxydiphenyl borate (2-APB) and 8-(N, N, diethylamino) octoyl 3,4,5-trimethoxybenzoate (TMB-8) from Calbiochem (La Jolla, CA). Human thrombin and human plasma kallikrein from Enzyme Research Lab (South Bend, IN), BK, porcine pancreatic kallikrein, thermolysin, trypsin, genistein, culture media, penicillin, phorbol-12-myristate 13-acetate (PMA), thrombin R activator peptide (TRAP) and scrambled TRAP (scTRAP), amastatin and other peptides and chemicals were purchased from Sigma Chemical (St Louis, MO). HOE 140 was a gift from Dr. B.A. Schölkens, Novartis Co., (Frankfurt, Germany). The Pro-Q diamond phosphoprotein gel stain kit was from Molecular Probes Inc. (Eugene, OR).
Cell Culture and Transfection
CHO cells were grown in Ham s F-12 culture medium supplemented with L-glutamine, penicillin-streptomycin, HEPES buffer and 10% fetal bovine serum (FBS). One day prior to transfection, cells were seeded into 60 mm dishes at 30–40 % confluence. The human B2R and human B2R C-terminally tagged with the green fluorescent protein (B2-GFPct) were inserted into a pCDNA3 vector, and CHO cells were transfected with human BK B2R (CHO/B2), the B2-GFPct (CHO/B2-GFPct), or co-transfected with human BK B2R and RGS10 (CHO/B2+RGS10) using lipofectin. After 2 h incubation at 37°C, cells were thoroughly washed with DMEM or Ham s F-12 medium, and 2 ml of the same medium containing geneticin (G418) was added to start the selection of stably B2 or tagged-B2R transfected cells. Blasticidin was added to select the RGS10 co-transfected cells. Different clones were selected and propagated using cloning rings. HPAE cells, which constitutively express BK B2Rs, were grown to confluence (4–6 passages).
[3H]BK saturation binding assay
To select the transfected clone with the highest expression of B2 or B2-GFPct Rs and to quantitate the effect of thrombin on BK binding to its R, we used a [3H]BK saturation binding assay (20). We found that both human BK B2 and B2-GFPct were expressed on CHO plasma membrane; we estimated the B2R to be 5.8 x 104 per cell and the B2-GFPct to be 1.4 x 105 per cell.
[Ca2+]i mobilization
We assessed the effectiveness of specific [Ca2+]i release inhibitors by measuring [Ca2+]i levels in CHO/B2 cells with a microspectrofluorometer (PTI Deltascan, Princeton, NJ) coupled to an inverted microscope and the Ca2+ sensitive fluorescent dye, fura-2/AM (20). The excitation wavelengths were 340 and 380 nm, and the emission wavelength was 510 nm. The signals are represented in figures as a ratio of bound/free Ca2+, F340/F380.
[3H]AA release
Endothelial and transfected CHO cells were grown to confluence in 6 well dishes. The medium was replaced with 1 ml of Ham s F12 medium containing 0.5 μCi/ml of [3H]AA, and cells were incubated for 16 h at 37°C. The labeled cells were then serum starved for 3 h prior to each assay. After washing with incubation medium (Ham s F12 medium plus 0.1% bovine serum albumin, BSA), they were incubated for 30 min at 37°C with either medium alone, 1 – 50 nM BK, or enzymes with or without the B2R blocker, HOE 140 (0.5 μM). The medium was removed and its [3H]AA content was measured by scintillation counting. The amount of released [3H]AA was expressed after subtracting the background (spontaneous release) (19). The experiments were carried out routinely in triplicate.
Thrombin and kallikrein assay
A putative direct effect of TRAP on human plasma kallikrein activity and inactivation of thrombin by DFP were followed fluorometrically by measuring cleavage of D-Val-Leu-Arg-AMC substrate by the two enzymes (1, 36). Plasma kallikrein (100 nM) was pre-incubated with increasing concentrations of TRAP from 1 μM to 300 μM for 15 min. Fifty μl of kallikrein solution was then mixed with 445 μl of 0.1 M Tris-HCl (pH 8.0). Addition of 5 μl of AMC substrate to final concentration of 0.1 mM initiated the reaction. The liberated fluorogenic coumarin derivative was measured in a spectrofluorometer with excitation at 380 nm and emission at 460 nm wavelength; the increase in fluorescence was monitored with a recording spectrofluorometer. We used thrombin irreversibly inhibited by DFP as an additional control.
Human BK B2 immunoprecipitation and phosphorylation
CHO/B2-GFPct cultures in 6-well culture dishes were treated with 1 μM BK in the presence or absence of increasing concentrations of thrombin (3 nM - 1 μM) for 30 min at 37°C. Cells were washed with ice-cold PBS and lysed with 0.4 ml 0.5% DOC buffer (pH 7.5) containing 1% NP-40, 0.1% SDS, 1 mM PMSF, 50 mM Tris, 150 mM NaCl and 10 μl protease inhibitor mixture. After shaking for 10 min at 4°C, the lysates were sonicated, then centrifuged at 16,000g for 15 min at 4°C. Supernatants were collected and diluted with 390 μl of 50 mM Tris buffer (pH 7.5) containing 150 mM NaCl and protease inhibitors. Samples were then incubated with 1 μg rabbit anti-GFP antibody overnight at 4°C. B2-GFPct R immune complexes were precipitated with protein A-Sepharose beads (Sigma, St. Louis, MO) at 4°C for 2 h. The beads were then washed five times with lysis buffer, and the precipitated proteins were eluted by boiling the beads in sample buffer (80 mM Tris [pH 6.8], 3% SDS, 15% glycerol, 0.01% bromophenol blue, 5% DTT). Proteins were then separated on a 4–20 % gradient SDS-PAGE gel. The phosphorylated R was selectively stained with a Pro-Q diamond phosphoprotein gel stain kit, and its 555/580 nm excitation/emission maxima was detected with a visible-light-scanning instrument (Molecular Imager FX pro plus, Bio Rad). Density of the band was quantified using Labworks software, and data were expressed as the ratio of phosphorylated protein density/total protein density.
Statistical analysis
Means and standard errors were calculated for all experiments, and statistical significance of differences between means was tested by a paired t-Test (Microsoft Excel).
RESULTS
Thrombin enhances [3H] arachidonic acid (AA) release by BK B2R agonists
BK or kallikrein released [3H]AA from labeled CHO cells that were stably transfected to express human B2R (CHO/B2). Although these cells express thrombin PAR1 constitutively, direct activation of PAR1 by 10 nM thrombin for 30 min at 37°C released relatively little labeled AA. If the amount released spontaneously was taken as baseline to equal 1.0, the value for AA was 1.76 ± 0.4 fold over the baseline. Figure 1a shows that the amount of AA released by thrombin was unaffected by 0.5 μM HOE 140, an inhibitor of the BK B2R. However, when thrombin activated PAR1 first, subsequent stimulation of B2R with porcine pancreatic kallikrein nearly doubled [3H]AA release from 6.4 ± 1.5 to 12.6 ± 2. Similar results were obtained with human plasma kallikrein (5.9 ± 0.8 to 11.0 ± 0.7) and BK (6.3 ± 1.7 to 11.8 ± 2.6), where thrombin activated cells reacted to the agonists with a significant increase in release of AA. HOE 140 (0.5 μM) completely blocked the effects of thrombin receptor activation. We consistently recorded thrombin potentiation of B2 agonists in numerous experiments: n = 21 for BK (1 nM) and n = 17 for tissue kallikrein (1–10 nM). HPAE cells that constitutively express both receptors confirmed positive cooperativity (p < 0.01) between PAR1 and BK B2Rs (Fig 1a, right panel). Ten nM human thrombin increased basal release of AA by endothelial cells slightly (1.4 ± 0.1) but also potentiated the effect of BK and plasma kallikrein on the BK B2R significantly.
Fig 1. PAR1 activation by human thrombin and TRAP potentiates BK B2 R agonists.
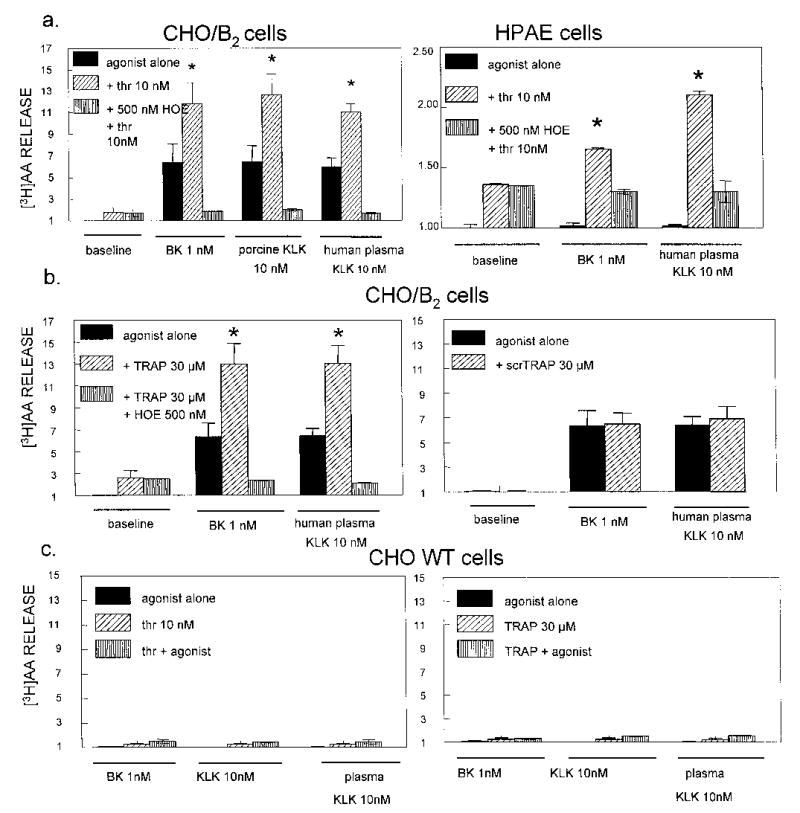
CHO cells transfected with human B2Rs (CHO/B2) and human pulmonary artery endothelial (HPAE) cells were incubated 30 min at 37°C with 1 nM BK, 10 nM human plasma kallikrein (KLK) or porcine tissue KLK in the presence or absence of 10 nM human thrombin (panel a), 30 μM TRAP or 30 μM scrambled TRAP (scrTRAP: [3H]AA release is expressed on the ordinate in relative units. Panel b). Thrombin and TRAP enhanced [3H]AA release by B2 R agonists; this response was blocked by the B2 antagonist, HOE 140. Solid columns = agonist alone; diagonal lines = combination of B2 R agonist and PAR1 agonist; vertical lines = combination of B2 R agonist and PAR1 agonist in the presence of 0.5 μM HOE 140. All data are means ± S.E.M. of 3–5 separate determinations. * indicates P < 0.05 vs control agonist alone.
Panel c: Thrombin and TRAP did not potentiate wild type (WT) CHO cells that express PAR1 but lack B2R. Solid column = agonist alone; diagonal lines = PAR1 agonist alone; vertical lines = combination of B2R agonist and PAR1 agonist. Data are means ± S.E.M. from 3–5 separate experiments.
The synthetic thrombin R activating peptide SFLLRN (TRAP) is composed of the first six amino acids of the new N-terminal tethered ligand that is revealed when thrombin cleaves PAR1. This peptide functions as a PAR1 agonist that activates the R independently of thrombin and proteolysis (12, 38). TRAP was used as a control for two reasons: it is specific for the PAR1 and it ensures that enhanced B2 agonist activity results from PAR1 activation, not from thrombin acting on B2R (Fig 1b). Like thrombin, TRAP (30 μM) released trace amounts of [3H]AA from CHO/B2 cells (2.4 ± 0.8), and the B2R blocker, HOE 140 (2.4 ± 0.1), did not affect it. TRAP doubled [3H]AA released by either plasma kallikrein (6.4 ± 1.5 to 13.0 ± 2) or BK (6.3 ± 1.7 to 12.9 ± 2.0), a significant elevation over control cells blocked by HOE 140. In separate experiments in vitro, TRAP did not inhibit substrate hydrolysis by kallikrein (not shown). Reaction to the PAR1 peptide agonist was specific, because 30 μM of a scrambled TRAP peptide (scrTRAP, LFRSLN) neither increased basal [3H]AA nor potentiated [3H]AA release induced by kallikrein or BK (Fig 1b, right panel). As an additional control, thrombin inhibited by 1 mM DFP failed to activate PAR1 and did not modify [3H]AA release by B2 R agonists. Together these data show that activation of the thrombin PAR1 leads to a positive cooperativity with BK R agonists resulting in increased receptor-mediated activities.
We did additional experiments with wild type (WT) CHO cells that have native PAR1 Rs but lack the B2 Rs. Fig 1c shows that treatment of WT CHO cells with BK, porcine pancreatic and human plasma kallikreins did not release [3H]AA. Basal release of [3H]AA induced by 10 nM thrombin (1.2 ± 0.7) or TRAP peptide (1.2 ± 0.2) was not significantly increased by added BK, tissue kallikrein, or plasma kallikrein in three separate experiments. These data confirm that expression of both receptors are required for receptor-to-receptor crosstalk and the positive cooperativity reflected by enhanced release of labeled AA.
Inhibitors of PKCα block potentiation
In order to determine the mechanisms of thrombin potentiation, we used selective inhibitors of G proteins (Gαi and Gαq) signaling pathways, phospholipases (PLA2, PLCβ) and protein kinases that regulate G protein coupled R activity. Because the PKCα subtype phosphorylates transient receptor potential Cl (TRPC1) channel and regulates store-operated Ca2+ entry in endothelial cells upon PAR1 activation (2), we inhibited PKCα to see if it could block potentiation of AA release by thrombin. CHO/B2 cells were pretreated for 30 min with either a non-specific PKC inhibitor, calphostin C (100 nM) or a specific inhibitor of PKCα, Gö6976 (100 nM) (30). PKC is not essential for regulation of the B2R activity (4), and calphostin C and Gö6976 did not inhibit AA release by either kallikrein or BK significantly (Fig 2). Addition of either calphostin C or Gö6976 inhibited thrombin potentiation without significantly affecting basal B2R activity, because it was only slightly less than in untreated cells (Fig 2a). Thrombin-potentiated AA release by BK was significantly reduced by calphostin C (7.3 ± 0.9) and Gö6976 (9.4 ± 1.1) compared to control cells (15.9 ± 1.4). The thrombin-enhanced response to kallikrein (KLK) was similarly blocked by calphostin C (4.1 ± 0.3) or Gö6976 (6.1 ± 0.9) v. control (10.2 ± 1.1) (Fig 2b). Gö6976 also inhibited the potentiation by TRAP (30 μM) and reduced the augmentation of either BK (15.1 ± 1.3 v. 9.7 ± 1.2) or kallikrein (10.3 ± 0.8 v. 5.5 ± 0.8) responses. Phorbol 12-myristate 13-acetate (PMA, 100 nM) had opposite effects, it enhanced thrombin potentiation of both BK (from 15.9 ± 1.4 in controls to 21.2 ± 1.3) and kallikrein (from 10.2 ± 1.1 to 16.3 ± 1.2) (Fig 2 ).
Fig 2. Inhibitors of PKCα blocked thrombin-induced potentiation.
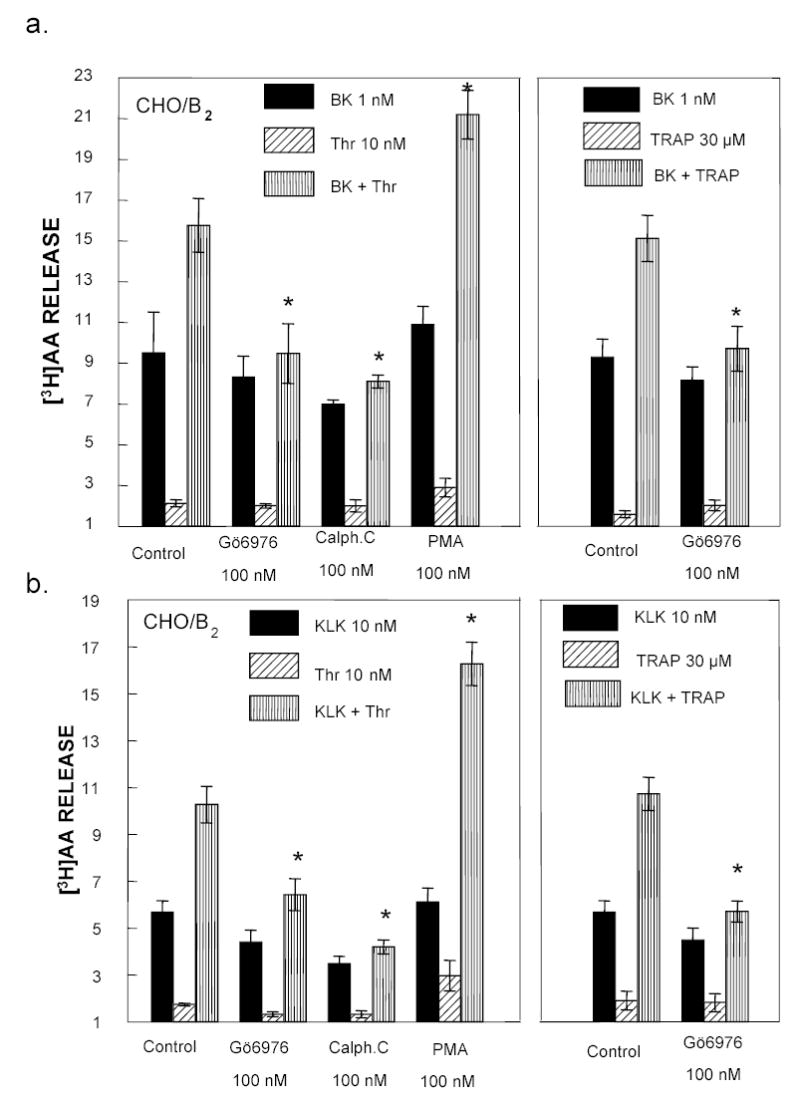
CHO/B2 cells were incubated with 100 nM Gö6976, calphostin C (Calph.C) or phorbol 12-myristate 13-acetate (PMA) for 30 min. Then 1 nM BK (panel a) or 10 nM KLK (panel b) was added in the presence or absence of 10 nM human thrombin or 30 μM TRAP. Ordinate as in Fig 1. Solid column = agonist alone; diagonal lines = PAR1 agonist alone; vertical lines = combination B2R agonist and PAR1 agonist. Data are means ± S.E.M from 3–5 separate experiments. * P < 0.05 vs control untreated cells. Phorbol enhanced the potentiation by PAR1 agonists, but PKC inhibitors blocked it.
Because different signaling pathways resulting from PAR1 activation might be involved in enhancement of the BK B2R agonists, we applied additional inhibitors of serine/threonine kinases: a PKG inhibitor, (RQIKIWFQNRRMKWKKLRKKKKH (0.5 μM), a PKA inhibitor, H-89 dihydrochloride, (0.5 μM), an IP3 kinase inhibitor, wortmannin (0.5 μM) and a tyrosine kinase inhibitor, genistein (100 μM). None of these agents significantly modified the thrombin potentiation of BK on the B2R (Table 1), indicating that it does not require PKG, PKA, IP3 kinase or tyrosine kinase. Methyl-β-cyclodextrin can be used to deplete cholesterol from cell membranes and lipid rafts. Since pretreatment of CHO/B2 cells with 5 mM of methyl-β-cyclodextrin for 30 min at 37°C had no effect, it is highly unlikely that thrombin potentiates B2R by heterodimerization with PAR1 (Table 1).
Table I.
Negative Inhibitors of B2R Potentiation
| [3H]AA release (relative units; basal release = 1) (n = 3)
|
||
|---|---|---|
| Treatment | BK | BK + thrombin |
| Vehicle | 6.8 ± 0.8 | 11.8 ± 0.7 |
| Tyr kinase inhibitor genistein 100 μM | 6.8 ± 0.4 | 11.6 ± 0.6 |
| PKG inhib. 0.5 μM (RQIKIWFQNRRMKWKKLRKKKKH) | 6.7 ± 0.3 | 11.7 ± 0.5 |
| PKA inhib. 0.5 μM (H-89, dihydrochloride) | 8.6 ± 0.4 | 11.7 ± 0.5 |
| IP3 kinase inhibitor wortmannin 0.5 μM | 6.5 ± 0.6 | 11.3 ± 0.9 |
| Methyl-β-cyclodextrin 5 mM | 6.9 ± 0.6 | 12.0 ± 0.8 |
Role of Gα12/13 in potentiation
Inhibition of PKCα, a downstream effector of Gαq, blocked potentiation of B2R agonists by thrombin. Stimulation of Gαq generates DAG and IP3, and IP3 then binds to IP3 R in the endoplasmic reticulum to trigger [Ca2+]i release. The interaction of IP3R with TRPC1 channels activates store depletion-induced Ca2+ entry (21, 27, 31). Together with DAG, Ca2+ mobilized from intracellular stores activates PKCα. Fig. 3 shows that activation of the IP3R and the subsequent [Ca2+]i release are required for the thrombin potentiation phenomenon. CHO/B2 cells were pretreated for 30 min with either 75 μM of 2-APB to inhibit the R of IP3 or 20 μM of TMB-8, a [Ca2+]i antagonist. 2-APB blocked the IP3R dependent [Ca2+]i mobilization (Fig 3a) and the thrombin-induced potentiation of B2 agonists (4.7 ± 0.6 compared to 9.9 ± 0.8) (Fig 3b). Potentiation by BK was also reduced in cells pretreated with the [Ca2+]i antagonist TMB-8 (7 ± 0.6 compared to 9.9 ± 0.8), a weaker inhibitor of Ca2+ flux (fig 3a).
Fig 3. Role of Gα12/13 in potentiation.
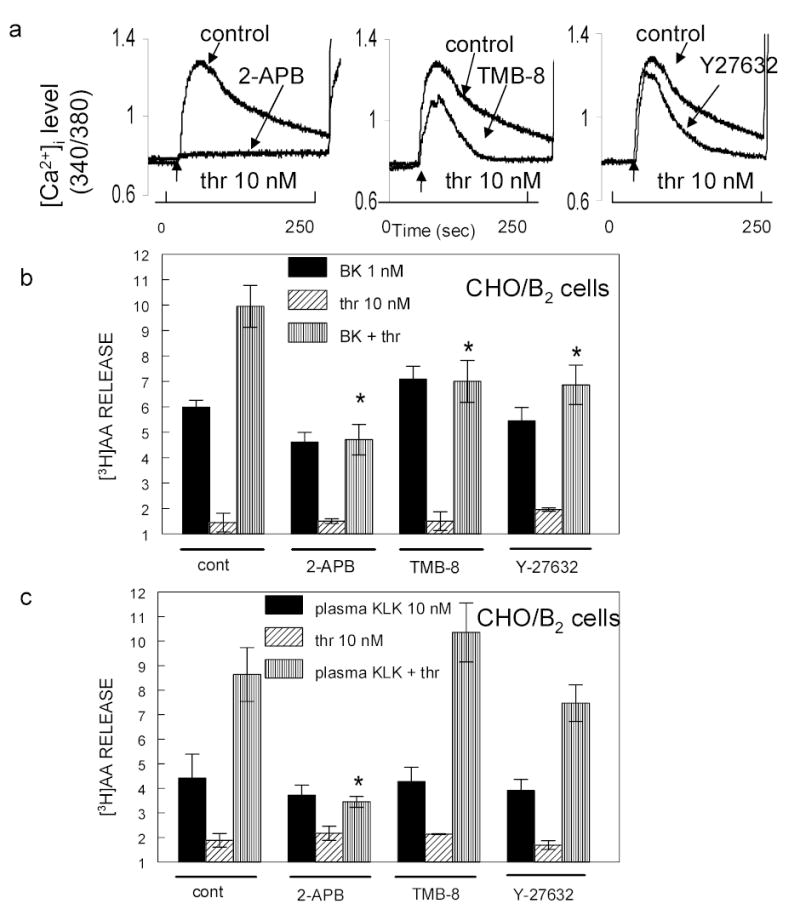
Panel a: Inhibition of thrombin-induced [Ca2+]i release. Fura-2AM loaded CHO/B2 cells were incubated with 75 μM 2-APB, 20 μM TMB-8 or 10 μM Y27632 for 30 min to inhibit IP3 R, [Ca2+]i release or RhoA kinase. Cells were then exposed to 10 nM thrombin. Abscissa: time in seconds. Ordinate: relative [Ca2+]i level. Experiments were repeated three times with similar results. Panels b and c: [3H]AA loaded CHO/B2 cells were treated with 75 μM 2-APB, 20 μM TMB-8 or 10 μM Y27632 for 30 min at 37 ºC and then exposed to 1 nM BK (panel b) or 10 nM plasma kallikrein (panel c) with or without 10 nM thrombin. Solid column = agonist alone; diagonal lines = PAR1 agonist alone; vertical lines = combination B2R agonist and PAR1 agonist. Data are means ± S.E.M. from 3–5 separate experiments. * P < 0.03 comparing B2R agonists in thrombin stimulated control cells. 2-APB blocks Ca2+ entry into cells, the two other agents reduced it. Inhibitors abolished or reduced potentiation by thrombin. TMB-8 blocked potentiation of BK but not that of kallikrein.
PAR1 agonists can activate other heterotrimeric G-proteins, the Gαi (14, 25, 37) and the Gα12/13 (13, 17, 27) that initiate cascading downstream processes. The released α subunit of G12/13 activates P115 Rho guanine nucleotide exchange factor (p115RhoGEF) and thereby also Rho. In order to determine whether thrombin potentiates BK effects through the Gα12/13 pathway as well, we pretreated CHO/B2 cells for 30 min with 10 μM Y27632, a Rho kinase inhibitor. This inhibitor decreased extracellular Ca2+ entry into thrombin-activated cells (Fig 3a) and consequently, lessened the potentiation of B2 agonist (6.8 ± 0.7 v. untreated cells 9.95 ± 0.8). It follows that the mechanism of potentiation at the B2R requires Rho acting via IP3 R to induce [Ca2+]i release and activate the subsequent signaling cascade. Based on our experiments with inhibitors, thrombin appears to potentiate plasma kallikrein differently than BK. Even though 2-APB completely blocked the thrombin-induced potentiation of kallikrein just as for BK, neither TMB-8 nor the RhoA kinase blocker, Y27632, blocked it (Fig 3c). These data thus suggest that influencing the R of IP3 is a focal point of interaction between the B2R and PAR1 signaling pathway. Additional experiments indicated that kallikrein and BK regulate [Ca2+]i mobilization through different mechanisms (D. Biyashev, unpublished observations).
Thrombin enhances cytosolic PLA2 activation
Among the different isoforms of PLA2, cytosolic PLA2 (cPLA2) is a major factor for receptor-regulated release of AA from membrane phospholipids (24). The activity of cPLA2 is Ca2+-dependent, and increased [Ca2+]i triggers translocation from the cytosol to plasma membranes (34, 35) to enhance its activity. Because the thrombin potentiation effect requires mobilization of [Ca2+]i, we investigated the role of cPLA2. We pre-incubated CHO/B2 cells for 30 min with 20 μM of isotetrandrine, a non-selective inhibitor that blocks both Ca2+-dependent and Ca2+-independent PLA2s. When both Ca2+-dependent and Ca2+-independent PLA2s were inhibited, basal release of [3H]AA by 1 nM BK or by 10 nM plasma kallikrein was blunted (2.5 ± 0.1 v 5.9 ± 0.3 and 3.9 ± 0.5 v 7.4 ± 0.9), as was potentiation of these agonists by thrombin. A more specific inhibitor of cPLA2 (20 nM; see Materials) similarly reduced the effects of B2 R agonists or plasma kallikrein on untreated cells as well as their potentiation by thrombin. In cells where cPLA2 was inhibited, release of AA fell to approximately half that in uninhibited cells. We used bromo-enol lactone (BEL), a specific Ca2+-independent PLA2 (iPLA2) inhibitor, as a control. Figure 4 shows that BEL (20 μM) after 30 min preequilibration inhibited the release of [3H]AA by BK by 32% (4.0 ± 0.5 v 5.9 ± 0.3), but not that of plasma kallikrein (7.16 ± 1.0 v 13.5 ± 1.0). However, the relative potentiation by thrombin of either B2R agonist was not abolished by BEL. Even though the release of AA by BK in the presence of thrombin decreased, the ratio of [3H]AA released by BK in presence of thrombin (7.9 ± 0.7) over that released in absence of thrombin (4.0 ± 0.5) was similar to the ratio in control cells (9.9 ± 0.8 over 5.9 ± 0.3) (Fig. 4).
Fig 4. Thrombin enhances cytosolic PLA2 (cPLA2) activation by B2 agonists to release AA.
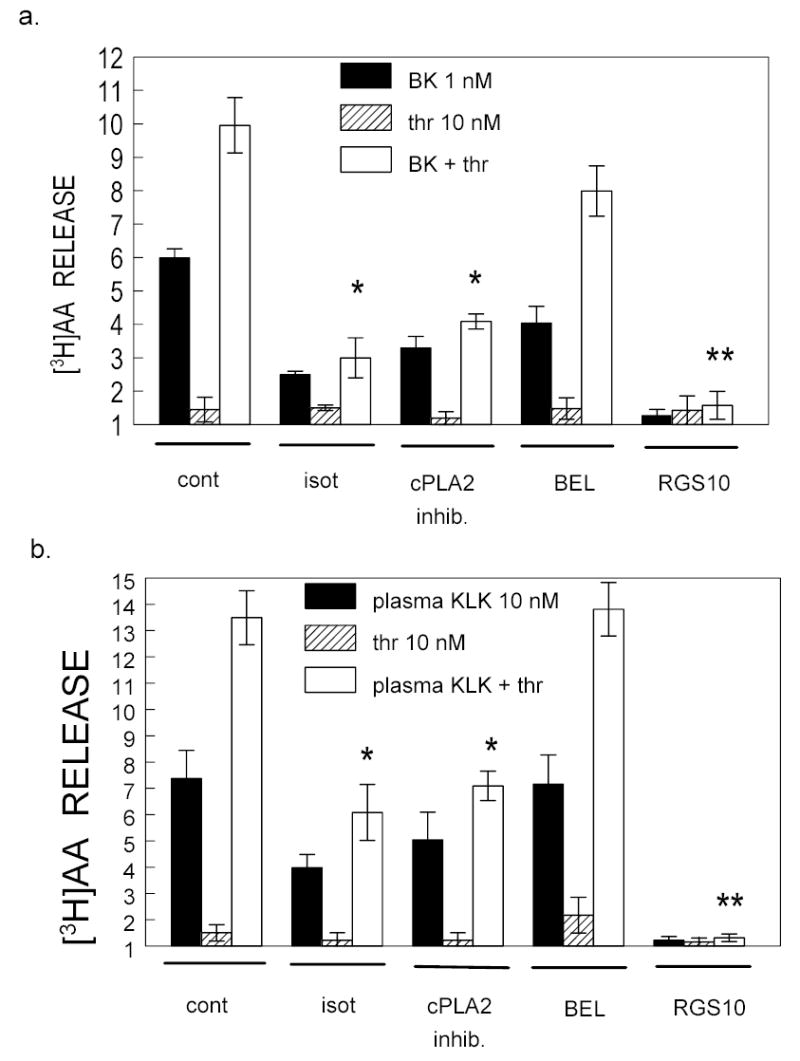
CHO/B2 cells were treated with 20 μM isotetrandrine (ISO), cPLA2inhib (100 nM), a specific cPLA2, inhibitor, or with 20 μM bromoenol lactone (BEL) for 30 min prior to incubation with agonists for a further 30 min. Solid bars = BK 1 nM (panel a) or plasma kallikrein 10 nM (panel b); diagonal lined bars = thrombin 10 nM; Open bars = thrombin + B2 receptor agonist. Data are means ± S.E.M. from 3–4 separate experiments. * P < 0.01 and ** P < 0.005 indicate significant differences with untreated cells stimulated with B2R agonists and thrombin. The cPLA2 inhibitors reduced AA release, abolished potentiation (*), but BEL did not. The expression of RGS10, a negative modulator of Gαi/o, completely blocked B2R agonists even in presence of thrombin.
Effect of transfected Gαi negative regulator
We repeated our thrombin potentiation experiments in CHO/B2 cells that were co-transfected with a regulator of G protein signaling type 10 (RGS10), which negatively modulates Gαi/o signaling upon Rs activation. Figure 4 shows that expression of RGS10 completely blocked B2R agonists, even in the presence of thrombin. RGS10 serves as a GTPase-activating protein for Gαi/o, but it may also regulate the Gαq subunit (33). Data from these experiments suggest that thrombin mobilizes [Ca2+]i to activate the Ca2+ dependent cPLA2 and thus potentiates the activity of BK B2 R agonists as reflected by a doubling of AA release.
Thrombin modifies the phosphorylation of the BK R
To determine how PAR1 agonists augment B2 agonists, we investigated their actions on B2R phosphorylation. We used 1 μM BK to detect the marked phosphorylated R better. Figs 5a and 6a show changes in [3H]AA release from CHO/B2 cells. The response to BK alone was taken as 100 % and plotted it vs. results from increased concentrations of PAR1 agonists. Thrombin potentiated either 1 μM or 1 nM BK, but the response was more prominent at the lower BK concentration (Fig 5a). Fig 5b shows the precipitation of B2R with antibodies to GFP in CHO/B2-GFP transfected cells. CHO/B2-GFPct cells were incubated for 30 min with 1 μM BK (37°C), and increasing concentrations of thrombin were added. We established that the GFP tag attached to the C terminus of the B2R affected neither release AA nor mobilization of [Ca2+]i, (19). Furthermore, this manipulation did not influence potentiation by thrombin (data not shown). The phosphorylated R was quantified and normalized to the total protein level using the ratio of phosphorylated protein density/total protein density (Fig 5b). The increased basal phosphorylation of the receptor following BK challenge (4.3 ± 0.5-fold) was taken as 100 %. Fig 5b shows how B2R phosphorylation changed with increasing amounts of thrombin. In absence of added BK, thrombin did not phosphorylate the BK B2R (data not shown), but it enhanced BK-induced phosphorylation of B2-GFPct R in a concentration-dependent manner, reaching a maximum of 166 % at 30 nM. Higher concentrations of thrombin decreased it. The phosphorylation of B2R that thrombin augmented was inhibited in cells pretreated with either 75 μM of 2-APB or 100 nM of Gö6976 (Fig 5b).
Fig 5. Effect of thrombin on stimulated B2R phosphorylation.
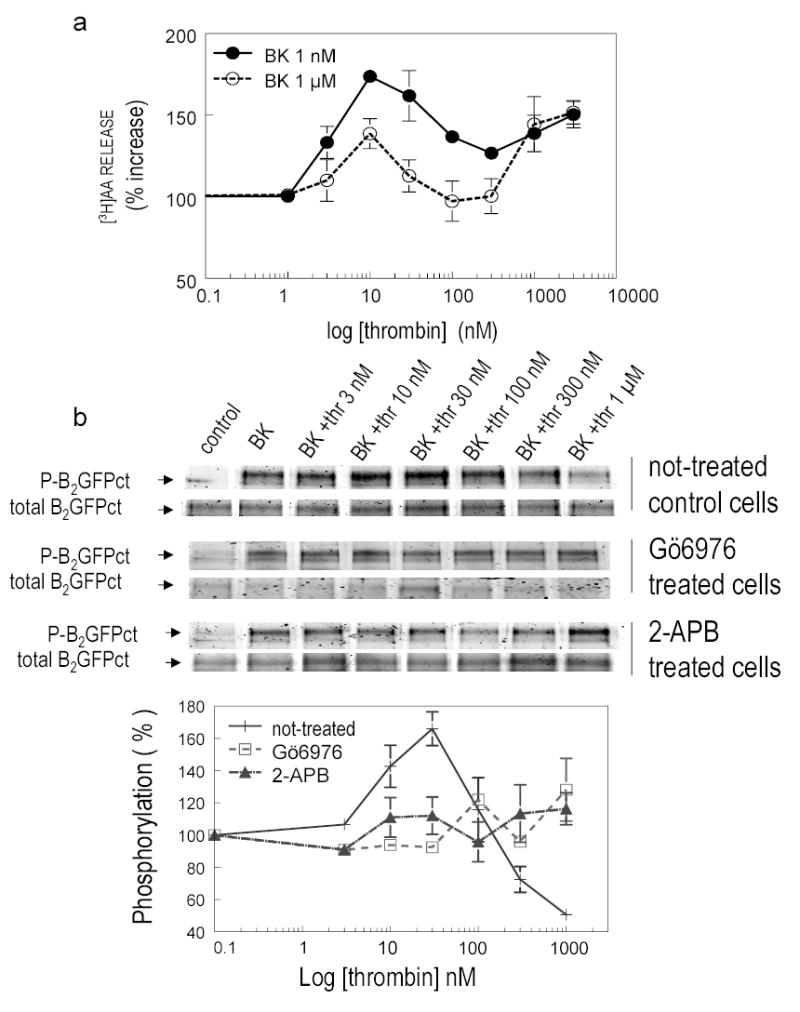
Panel a: CHO/B2 transfected cells were incubated with 1 nM BK ( -•- ) or 1 μM BK (-○-) in the presence or absence of increasing concentrations of thrombin (abscissa). The increase in [3H]AA is expressed as % on the ordinate. The effect of BK without thrombin is taken as 100 %; P < 0.05. Panel b: thrombin modified BK-induced B2R phosphorylation in a concentration dependent manner. CHO/B2-GFPct transfected cells untreated ( + ) or treated with 100 nM Gö6976 ( -□-) or 75 μM 2-APB(-▲-) were incubated with 1 μM BK (30 min) and increasing concentrations of thrombin (0.1 – 1 μM. BK). Data are from three separate determinations. BK increased the basal level of phosphorylation of the B2R by 4.3 ± 0.5 fold (n=3) taken as 100%. P < 0.05 vs not-treated cells without thrombin. Thrombin in low concentrations enhanced phosphorylation of B2-GFP by BK, higher concentrations blocked it; effects were abolished by IP3 R and [Ca2+]i release inhibitors.
Fig 6. Effect of aminopeptidase inhibitor on [3H]AA release and B2 R phosphorylation stimulated by TRAP.
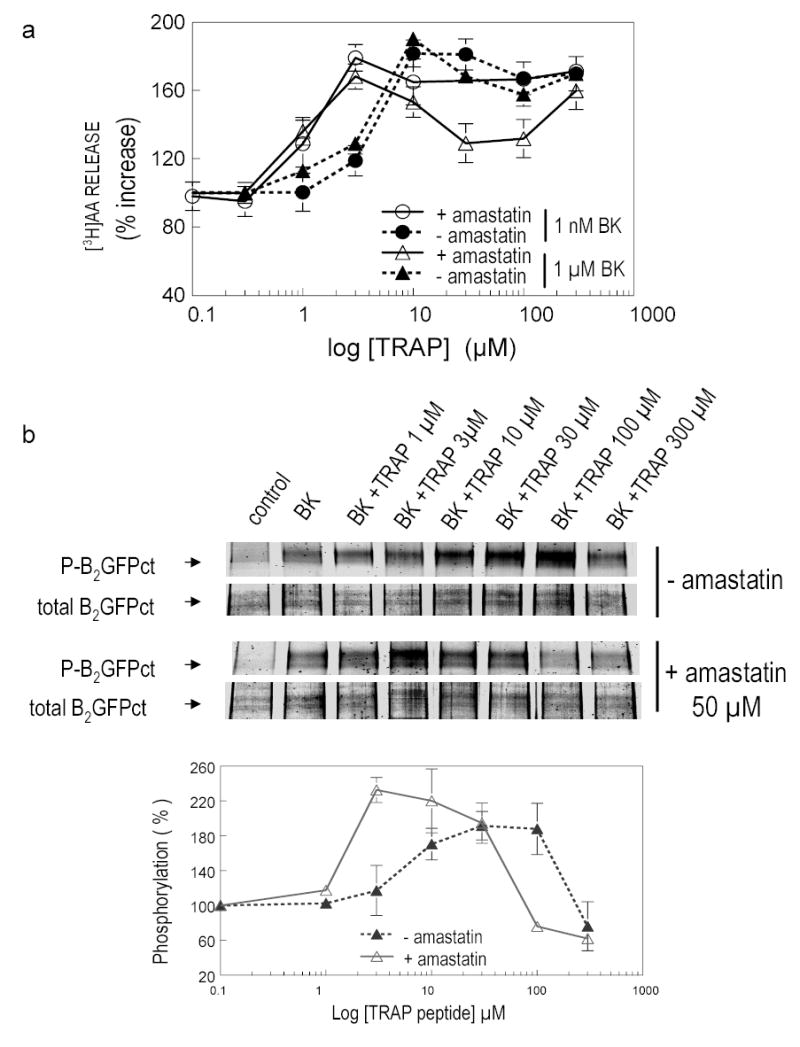
Panel a: TRAP potentiates stimulation of CHO/B2 transfected cells by 1 nM BK (-•-) or 1 μM BK (-▲-) to release AA in a concentration dependent manner. Effects of TRAP were increased by 50 μM amastatin (-○-for 1 nM BK and -⋄- for 1 μM BK). Ordinate and abscissa as in Fig 5. Panel b: TRAP modifies BK-induced B2R phosphorylation in a concentration-dependent manner. CHO/B2-GFPct transfected cells were incubated with 1 μM BK (30 min) and increasing concentration of TRAP ranging from 1 – 300 μM with or without 50 μM amastatin. Details as in Fig 5. Aminopeptidase inhibitor amastatin blocked cleavage of N-terminus of TRAP and enhanced its activity.
Similarly to thrombin, TRAP potentiated either 1 nM or 1 μM BK on CHO/B2 transfected cells. [3H]AA was released in a concentration-dependent manner with maximum at 10–30 nM of peptide. BK alone increased the basal level of phosphorylation; this increase was taken as 100 %, and data from additional treatments are expressed relative to this (Fig 6a). Because an aminopeptidase on the cell surface rapidly degrades TRAP, we repeated this experiment in the presence of amastatin (50 μM), an aminopeptidase inhibitor. When hydrolysis of the N-terminal Ser of TRAP was blocked by amastatin, as little as 3 μM TRAP potentiated the BK response. Increasing amounts of TRAP augmented B2R phosphorylation by BK (1 μM); maximum phosphorylation (192 % of control) was obtained with 30–100 μM TRAP in the absence of the aminopeptidase inhibitor. When 50 μM amastatin was added to the reaction mixture, the effect of TRAP resembled that of thrombin (Fig 5b, 6b). TRAP potentiation of BK-induced B2R phosphorylation reached a maximum (232 %) between 3 to 10 μM concentration of the peptide, and higher concentrations decreased it (Fig 6b).
DISCUSSION
We found that activation of PAR1 by either thrombin or the hexapeptide, TRAP, potentiates BK B2R agonists to enhance release of [3H]AA. AA is the precursor of prostaglandins, such as prostacyclin, a coronary vasodilator, and prostaglandin E2 that prevents platelet aggregation. It is also a precursor of thromboxane (8, 35). Although thrombin releases AA from many cell types, we used CHO cells in most of our experiments because they express native PAR1. Thrombin or TRAP released relatively small amounts of AA above baseline, but these agents both potentiated B2R agonists. In experiments repeated all together 35 – 40 times with human plasma kallikrein (10 nM) or BK (1 nM) we found that thrombin and TRAP consistently doubled AA release by these agonists. We obtained qualitatively similar results in experiments with human arterial endothelial cells that constitutively express both PAR1 and B2Rs.
Besides BK, we also tested human plasma kallikrein, because kallikreins affect B2R differently than BK. It is clear that kallikrein action at the B2R does not depend upon release of a kinin (19, 20). As with BK, stimulation of the B2R by kallikrein was blocked by HOE 140, and both prokallikrein and inhibited kallikrein were inactive. Kallikrein and BK both desensitized the R, but there was no cross-desensitization. [3H]AA release by BK was reduced 40% by addition of carboxypeptidase M, which removes Arg9. Carboxypeptidase M did not influence R activation by kallikreins and other proteases. Site-directed mutagenesis of the Arg169 residue of the B2R further emphasized differences in the mechanisms of activation by the proteases and BK. The mutation decreased R activation by BK but not by kallikrein. In addition, our study shows that B2R activation by BK depends, at least in part, on Ca2+-independent PLA2 activation while activation by kallikrein does not.
Potentiation at the B2R is not simply due to direct activation of PAR1 by the protease action of kallikrein, because we obtained similar results with the peptide agonist, BK. We further ruled out a proteolytic action of thrombin at the B2R because TRAP, a specific agonist of PAR1, potentiated both BK and kallikrein. DFP-inhibited thrombin (inactive on PAR1) failed to enhance B2R activation by agonists. A scrambled peptide sequence of TRAP was equally inactive, indicating that the potentiation mechanism is initiated and mediated through activation of PAR1. TRAP did not affect substrate hydrolysis by kallikrein (not shown).
Lipid rafts are believed to be essential for BK B2R and epidermal growth factor R crosstalk (23), and methyl-β-cyclodextrin blocks this interaction by depleting cholesterol which disrupts complex formation. We found that 5 mM methyl-β-cyclodextrin failed to modify thrombin-induced potentiation in CHO/B2 transfected cells. Thus, it seems unlikely that the potentiation phenomenon depends on PAR1/B2R dimer formation, even though B2 Rs can form heterodimers with angiotensin converting enzyme (ACE; Z. Chen and E.G. Erdös, unpublished observations and 28). In addition, because of the discrepancy in the ratio of PAR1 to B2 expression in CHO/B2 transfected cells, potentiation cannot be due simply to co-localization; it is presumably a consequence of enhanced signal transduction.
In a comparable system, thrombin first activates Gαq/PKCα and the Gα12/13/Rho pathways to mobilize [Ca2+]i in endothelial cells (21, 31). Upon stimulation, PAR1 coupled to Gαq protein generates DAG and IP3; the latter then binds to IP3 Rs on the endoplasmic reticulum to trigger [Ca2+]i release. The interaction of IP3 R with transient receptor potential channel 1 (TRPC1) activates Ca2+ entry by store depletion (21, 27, 31). Yet, beside Gαq, stimulated PAR1 couples to Gα12/13 proteins (13, 17, 27). The released α subunit of G12/13 induces P115 Rho guanine nucleotide exchange factor (p115RhoGEF) activation that, in turn, activates Rho. Upon activation of Gα12/13/Rho signaling cascade, activated Rho associates with IP3 R and TRPC1 by actin filament polymerization to provoke [Ca2+]i store depletion (31). Following emptying of stored Ca2+, the Rho/IP3 R/ TRPC1 complex translocates to the plasma membrane to trigger extracellular Ca2+ entry (21, 31). The phosphorylation of p115RhoGEF depends on PKCα (21), which is activated via Gαq signaling pathway stimulation. These experiments with endothelial cells show that increased [Ca2+]i upon PAR1 activation requires cooperative interaction of both Gα12/13 and Gαq /PKCα pathways.
Our results are consistent with those findings and indicate that [Ca2+]i release indeed triggers thrombin-mediated potentiation of BK. We found that enhancement of AA release by thrombin was attenuated after inhibition of either Gαq/PKCα or Gα12/13 /Rho pathways. The inactivation of the IP3 R-dependent [Ca2+]i mobilization in CHO/B2 cells by 2-APB, the inhibition of PKCα by Gö6976, and inhibition of Rho by a Rho kinase inhibitor Y27632 all blocked potentiation of BK. Together these findings suggest that PAR1 R stimulation causes formation of the Rho/TRPC1/IP3 R complex, and that the resulting [Ca2+]i release initiates signaling events leading to increased [3H]AA release.
However, interestingly thrombin potentiates the effects of plasma kallikrein and BK on the B2R differently. While an inhibitor of the IP3 R completely blocked the potentiation of kallikrein by thrombin to release [3H] AA, the selective inhibitor of [Ca2+]i release (TMB-8), or that of Rho kinase (Y27632) did not modify it. Thus, both the IP3 R activation and the resulting elevation of [Ca2+]i level are involved in potentiation of BK, only the IP3 R activation is necessary for potentiation of kallikrein by thrombin. Altogether, these data are showing that IP3 R is indeed the critical point of interaction between the pathways leading to positive cooperativity. Because PAR1 initiates enhancement of [3H]AA release by B2R agonists and Ca2+ stimulates cPLA2, cPLA2 is likely involved in potentiation. Indeed, a specific inhibitor of Ca2+-dependent PLA2 blocked the potentiation by thrombin, while an inhibitor of the Ca2+-independent isoform was ineffective, implying that [Ca2+]i mediates enhanced B2R activity by agonists. Both thrombin and TRAP modified B2R phosphorylation by agonists in a concentration-dependent manner. Once activated by agonist, B2R undergoes phosphorylation and desensitization. ACE inhibitors decreased the phosphorylation of B2R by BK and thus enhanced the activity of the peptide agonist (29). Now we find that low concentrations of thrombin (10–30 nM) or TRAP (30–100 μM without addition of the aminopeptidase inhibitor amastatin and 1–10 μM with amastatin) elevate BK B2R activity and increase phosphorylation of the R. Higher amounts of enzyme or peptide reversed the process and inhibited BK-initiated phosphorylation. Blocking of these effects by the IP3 R inhibitor, 2-APB, and by the PKCα inhibitor Gö6976 can be explained by co-activation of Gα12/13 and Gαq /PKCα pathways by thrombin and the subsequent [Ca2+]i mobilization that modulates B2R phosphorylation and activity. Increased phosphorylation is not likely due to increased expression of B2R, because we found no concomitant increase in B2-GFP total protein with the increase in R phosphorylation (Fig. 5). In addition, inhibitors would unlikely alter protein synthesis and receptor expression during the brief time course of our experiments, but only modify activity. Additional controls where we measured [3H]BK binding to the cell membrane indicated no modification by thrombin (data not shown).
Fig 7 provides a scheme that summarizes pathways potentially involved in the interaction of PAR1 and B2R agonists. We conclude that PAR1 R activation enhances the effects of BK B2R agonists in signal transduction which then results in augmented release of AA. Since AA is a precursor of several vasoactive mediators, regulation of these pathways influences many diverse physiological functions. The cooperativity between the two Rs that we described for signal transduction pathways involved in AA release may underline the cardioprotective nature of kinins in vivo.
Fig 7. Scheme for the enhanced AA release.
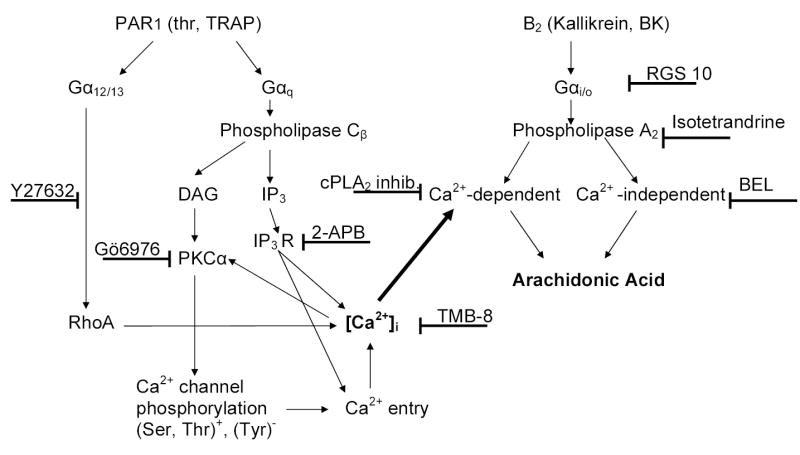
Upon thrombin activation, Gαq and Gα12/13 signaling pathways converge to empty Ca2+ stores and redirect subsequent PKCα dependent Ca2+ entry. The elevation of [Ca2+]i enhances Ca2+-dependent PLA2 activation initiated by B2 R agonists and increases release of AA.
Acknowledgments
We would like to thank Drs. C. Tiruppathi and D. Mehta of UIC College of Medicine for their assistance in the project, and Dr. P. Deddish for collaborating in the calculation of expression levels. This work was supported by National Institutes of Health (NIH) Grants HL36473 and HL68580.
Footnotes
Table 1. Ineffective inhibitors: [3H]AA-labeled cells were incubated for 30 min in the presence or absence of the indicated drugs and then treated for an additional 30 min with 1 nM BK in the presence or absence of 10 nM human thrombin. After treatments, [3H]AA release was measured as described in Methods. Results were calculated by taking basal release as 1.0. All data are means ± S.E.M. of 3 separate experiments using triplicate samples. Significance is indicated by p < 0.01 (paired t-test). The release of AA by BK was significantly (p < 0.01) potentiated in spite of the presence of an inhibitor.
References
- 1.Abe M, Nakamura F, Tan F, Deddish PA, Colley KJ, Becker RP, Skidgel RA, Erdös EG. Expression of rat kallikrein and epithelial polarity in transfected Madin-Darby canine kidney cells. Hypertension. 1995;26:891–898. doi: 10.1161/01.hyp.26.6.891. [DOI] [PubMed] [Google Scholar]
- 2.Ahmmed GU, Mehta D, Vogel S, Holinstat M, Paria BC, Tiruppathi C, Malik AB. Protein Kinase C{alpha} Phosphorylates the TRPC1 Channel and Regulates Store-operated Ca2+ Entry in Endothelial Cells. J Biol Chem. 2004;279:20941–20949. doi: 10.1074/jbc.M313975200. [DOI] [PubMed] [Google Scholar]
- 3.Bhoola KD, Figueroa CD, Worthy K. Bioregulation of kinins: kallikreins, kininogens, and kininases. Pharmacol Rev. 1992;44:1–80. [PubMed] [Google Scholar]
- 4.Blaukat A, Pizard A, Breit A, Wernstedt C, Alhenc-Gelas F, Muller-Esterl W, Dikic I. Determination of bradykinin B2 receptor in vivo phosphorylation sites and their role in receptor function. J Biol Chem. 2001;21:21. doi: 10.1074/jbc.M107024200. [DOI] [PubMed] [Google Scholar]
- 5.Burch RM. Diacylglycerol in the synergy of bradykinin and thrombin stimulation of prostaglandin synthesis. Eur J Pharmacol. 1989;168:39–42. doi: 10.1016/0014-2999(89)90630-4. [DOI] [PubMed] [Google Scholar]
- 6.Burch RM, Axelrod J. Dissociation of bradykinin-induced prostaglandins formation, from other phosphatidylinositol turnover in Swiss 3T3 fibroblasts: Evidence for G protein regulation of phospholipase A2. Proc Natl Acad Sci USA. 1987;84:6374–6378. doi: 10.1073/pnas.84.18.6374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burch RM, Ma AL, Axelrod J. Phorbol esters and diacylglycerols amplify bradykinin-stimulated prostaglandin synthesis in Swiss 3T3 fibroblasts. Possible independence from protein kinase C. J Biol Chem. 1988;263:4764–4767. [PubMed] [Google Scholar]
- 8.Campbell WB, Halushka PV. Lipid-derived autacoids: eicosanoids and platelet-activating factor. In: Hardman JC, Limberd LE, Molinoff PB, Ruddon RW, Gilman AG, editors. Goodman & Gilman's The Pharmacological Basis of Therapeutics. 9th ed. New York: McGraw-Hill; 1996. pp. 602–616. [Google Scholar]
- 9.Carretero O, Nasjletti A, Fasciolo JC. The kinin content of human blood at rest and during vasodilatation. Experientia. 1965;21:141–142. doi: 10.1007/BF02141980. [DOI] [PubMed] [Google Scholar]
- 10.Carretero OA, Carbini LA, Scicli AG. The molecular biology of the kallikrein-kinin system: I. General description, nomenclature and the mouse gene family. J Hypertens. 1993;11:693–697. doi: 10.1097/00004872-199307000-00002. [DOI] [PubMed] [Google Scholar]
- 11.Carretero OA, Scicli AG. Kinin as regulators of blood flow and blood pressure. In: Laragh JH, Brenner BM, editors. Hypertension: Pathophysiology, Diagnosis and Management. New York: Raven Press; 1990. pp. 805–817. [Google Scholar]
- 12.Coughlin SR. How the protease thrombin talks to cells. Proc Natl Acad Sci U S A. 1999;96:11023–11027. doi: 10.1073/pnas.96.20.11023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Coughlin SR, Vu TK, Hung DT, Wheaton VI. Characterization of a functional thrombin receptor. Issues and opportunities. J Clin Invest. 1992;89:351–355. doi: 10.1172/JCI115592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Crouch MF, Belford DA, Milburn PJ, Hendry IA. Pertussis toxin inhibits EGF-, phorbol ester- and insulin-stimulated DNA synthesis in BALB/c3T3 cells: evidence for post-receptor activation of Giα. Biochem Biophys Res Commun. 1990;167:1369–1376. doi: 10.1016/0006-291x(90)90674-c. [DOI] [PubMed] [Google Scholar]
- 15.Davey MG, Luscher EF. Actions of thrombin and other coagulant and proteolytic enzymes on blood platelets. Nature. 1967;216:857–858. doi: 10.1038/216857a0. [DOI] [PubMed] [Google Scholar]
- 16.Erdös EG, editor. Bradykinin, Kallidin and Kallikrein. Handbook of Experimental Pharmacology. Heidelberg: Springer-Verlag; 1979. pp. 1–817. [Google Scholar]
- 17.Gohla A, Offermanns S, Wilkie TM, Schultz G. Differential involvement of Gα12 and Gα13 in receptor-mediated stress fiber formation. J Biol Chem. 1999;274:17901–17907. doi: 10.1074/jbc.274.25.17901. [DOI] [PubMed] [Google Scholar]
- 18.Gutowski S, Smrcha A, Nowak L, Wu D, Simon M, Sternweis P. Antibodies to the αq subfamily of guanine nucleotide-binding regulatory protein alpha subunits attenuate activation of phosphatidylinositol 4,5-bisphosphate hydrolysis by hormones. J Biol Chem. 1991;266:20519–20524. [PubMed] [Google Scholar]
- 19.Hecquet C, Becker RP, Tan F, Erdös EG. Kallikreins when activating bradykinin B2 receptor induce its redistribution on plasma membrane. Int Immunopharmacol. 2002;2:1795–1806. doi: 10.1016/s1567-5769(02)00176-5. [DOI] [PubMed] [Google Scholar]
- 20.Hecquet C, Tan F, Marcic BM, Erdös EG. Human bradykinin B2 receptor is activated by kallikrein and other serine proteases. Mol Pharmacol. 2000;58:828–836. doi: 10.1124/mol.58.4.828. [DOI] [PubMed] [Google Scholar]
- 21.Holinstat M, Mehta D, Kozasa T, Minshall RD, Malik AB. Protein kinase Cα-induced p115RhoGEF phosphorylation signals endothelial cytoskeletal rearrangement. J Biol Chem. 2003;278:28793–28798. doi: 10.1074/jbc.M303900200. [DOI] [PubMed] [Google Scholar]
- 22.Huntington JA, Baglin TP. Targeting thrombin--rational drug design from natural mechanisms. Trends Pharmacol Sci. 2003;24:589–595. doi: 10.1016/j.tips.2003.09.002. [DOI] [PubMed] [Google Scholar]
- 23.Hur EM, Park YS, Lee BD, Jang IH, Kim HS, Kim TD, Suh PG, Ryu SH, Kim KT. Sensitization of epidermal growth factor-induced signaling by bradykinin is mediated by c-Src. Implications for a role of lipid microdomains. J Biol Chem. 2004;279:5852–5860. doi: 10.1074/jbc.M311687200. [DOI] [PubMed] [Google Scholar]
- 24.Kramer RM, Sharp JD. Structure, function and regulation of Ca2+-sensitive cytosolic phospholipase A2 (cPLA2) FEBS Lett. 1997;410:49–53. doi: 10.1016/s0014-5793(97)00322-0. [DOI] [PubMed] [Google Scholar]
- 25.LaMorte VJ, Harootunian AT, Spiegel AM, Tsien RY, Feramisco JR. Mediation of growth factor induced DNA synthesis and calcium mobilization by Gq and Gi2. J Cell Biol. 1993;121:91–99. doi: 10.1083/jcb.121.1.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liao JK, Homcy CJ. The G proteins of the Gαi and Gαq family couple the bradykinin receptor to the release of endothelium-derived relaxing factor. J Clin Invest. 1993;92:2168–2172. doi: 10.1172/JCI116818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mackie EJ, Pagel CN, Smith R, de Niese MR, Song SJ, Pike RN. Protease-activated receptors: a means of converting extracellular proteolysis into intracellular signals. IUBMB Life. 2002;53:277–281. doi: 10.1080/15216540213469. [DOI] [PubMed] [Google Scholar]
- 28.Marcic B, Deddish PA, Skidgel RA, Erdös EG, Minshall RD, Tan F. Replacement of the transmembrane anchor in angiotensin I-converting enzyme (ACE) with a glycosylphosphatidylinositol tail affects activation of the B2 bradykinin receptor by ACE inhibitors. J Biol Chem. 2000;275:16110–16118. doi: 10.1074/jbc.M909490199. [DOI] [PubMed] [Google Scholar]
- 29.Marcic BM, Erdös EG. Protein kinase C and phosphatase inhibitors block the ability of angiotensin I-converting enzyme inhibitors to resensitize the receptor to bradykinin without altering the primary effects of bradykinin. J Pharmacol Exp Ther. 2000;294:605–612. [PubMed] [Google Scholar]
- 30.Martiny-Baron G, Kazanietz MG, Mischak H, Blumberg PM, Kochs G, Hug H, Marme D, Schachtele C. Selective inhibition of protein kinase C isozymes by the indolocarbazole Gö 6976. J Biol Chem. 1993;268:9194–9197. [PubMed] [Google Scholar]
- 31.Mehta D, Ahmmed GU, Paria BC, Holinstat M, Voyno-Yasenetskaya T, Tiruppathi C, Minshall RD, Malik AB. RhoA interaction with inositol 1,4,5-trisphosphate receptor and transient receptor potential channel-1 regulates Ca2+ entry. Role in signaling increased endothelial permeability. J Biol Chem. 2003;278:33492–33500. doi: 10.1074/jbc.M302401200. [DOI] [PubMed] [Google Scholar]
- 32.Oldgren J, Wallentin L, Grip L, Linder R, Norgaard BL, Siegbahn A. Myocardial damage, inflammation and thrombin inhibition in unstable coronary artery disease. Eur Heart J. 2003;24:86–93. doi: 10.1016/s0195-668x(02)00312-3. [DOI] [PubMed] [Google Scholar]
- 33.Popov S, Yu K, Kozasa T, Wilkie TM. The regulators of G protein signaling (RGS) domains of RGS4, RGS10, and GAIP retain GTPase activating protein activity in vitro. Proc Natl Acad Sci U S A. 1997;94:7216–7220. doi: 10.1073/pnas.94.14.7216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schievella AR, Regier MK, Smith WL, Lin LL. Calcium-mediated translocation of cytosolic phospholipase A2 to the nuclear envelope and endoplasmic reticulum. J Biol Chem. 1995;270:30749–30754. doi: 10.1074/jbc.270.51.30749. [DOI] [PubMed] [Google Scholar]
- 35.Schmitz JM, Apprill PG, Buja LM, Willerson JT, Campbell WB. Vascular prostaglandin and thromboxane production in a canine model of myocardial ischemia. Circ Res. 1985;57:223–231. doi: 10.1161/01.res.57.2.223. [DOI] [PubMed] [Google Scholar]
- 36.Takada Y, Skidgel RA, Erdös EG. Purification of human prokallikrein. Identification of the site of activation by the metalloproteinase thermolysin. Biochem J. 1985;232:851–858. doi: 10.1042/bj2320851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vanhauwe JF, Thomas TO, Minshall RD, Tiruppathi C, Li A, Gilchrist A, Yoon EJ, Malik AB, Hamm HE. Thrombin receptors activate Go proteins in endothelial cells to regulate intracellular calcium and cell shape changes. J Biol Chem. 2002;277:34143–34149. doi: 10.1074/jbc.M204477200. [DOI] [PubMed] [Google Scholar]
- 38.Vu TK, Wheaton VI, Hung DT, Charo I, Coughlin SR. Domains specifying thrombin-receptor interaction. Nature. 1991;353:674–677. doi: 10.1038/353674a0. [DOI] [PubMed] [Google Scholar]