

Abstract
Immunolocalization studies in epithelial cells revealed myo6 was associated with peripherally located vesicles that contained the transferrin receptor. Pulse-chase experiments after transferrin uptake showed that these vesicles were newly uncoated endocytic vesicles and that myo6 was recruited to these vesicles immediately after uncoating. GIPC, a putative myo6 tail binding protein, was also present. Myo6 was not present on early endosomes, suggesting that myo6 has a transient association with endocytic vesicles and is released upon early endosome fusion. Green fluorescent protein (GFP) fused to myo6 as well as the cargo-binding tail (M6tail) alone targeted to the nascent endocytic vesicles. Overexpression of GFP-M6tail had no effect on a variety of organelle markers; however, GFP-M6tail displaced the endogenous myo6 from nascent vesicles and resulted in a significant delay in transferrin uptake. Pulse-chase experiments revealed that transferrin accumulated in uncoated vesicles within the peripheries of transfected cells and that Rab5 was recruited to the surface of these vesicles. Given sufficient time, the transferrin did traffic to the perinuclear sorting endosome. These data suggest that myo6 is an accessory protein required for the efficient transportation of nascent endocytic vesicles from the actin-rich peripheries of epithelial cells, allowing for timely fusion of endocytic vesicles with the early endosome.
INTRODUCTION
Myosins are a large family of structurally diverse molecular motors. Myosins bind to F-actin and hydrolyze ATP to produce unidirectional movement along the filament. Actin filaments are polarized, and myosins traditionally travel toward the barbed or plus end of the actin filament. An exception to this rule is the unconventional myosin, myosin-VI (myo6), which travels backwards toward the pointed or minus end of actin filaments (Wells et al., 1999).
Myo6 has four structural domains. The N terminus encodes a conserved motor domain similar to that seen for other myosins, as well as a single IQ motif that serves as a light chain binding site for the calcium-binding protein calmodulin (Hasson and Mooseker, 1994). The myo6 motor domain is regulated by calcium (Yoshimura et al., 2001) and is sufficient for the pointed-end directed movement (Wells et al., 1999; Homma et al., 2001). After the motor is the tail domain, which is made up a 200 amino acid coiled-coil region and a C-terminal globular domain (Hasson and Mooseker, 1994). The coiled-coil region mediates dimerization of myo6 (De La Cruz et al., 2001). The globular tail contains no recognizable motifs but is highly conserved across all class VI myosins and probably associates with cargo.
Based on its direction of movement, myo6 has been implicated in multiple processes that involved the transport of components inwards into the cell (reviewed in Cramer, 2000). These functions stem from the observation that actin filaments within the cell tend to be oriented with their barbed ends at the peripheries of the cell and their pointed ends toward the center of the cell. Consistent with a role in endocytosis, in many epithelial cell types, myo6 is associated with endocytic domains. In sensory hair cells of the inner ear, myo6 is enriched in the pericuticular necklace, a region of the hair cell rich in membrane vesicles and clathrin, and the sole site of endocytosis for the hair cell (Hasson et al., 1997; Kachar et al., 1997). Myo6 is also enriched in endocytic regions in kidney. In the kidney proximal tubule, myo6 is present at the base of the brush-border microvilli where it overlaps with the clathrin adapter protein AP-2 (Buss et al., 2001; Biemesderfer et al., 2002).
The first direct evidence for an involvement of myo6 in endocytosis was presented in studies in normal rat kidney fibroblasts by using constructs that expressed differently spliced versions of chicken myo6 (Buss et al., 2001). A longer splice form of myo6 that contained an insert between the coiled-coil region and the globular tail was identified. This long myo6 splice form targeted to clathrin-coated pits (CCPs), whereas any form lacking this insert did not (Buss et al., 2001). This longer splice form of myo6 was implicated in endocytosis of transferrin; overexpression of a construct that contained the tail and the splice insert, but lacking the motor domain, caused a 50% reduction in transferrin uptake by normal rat kidney fibroblasts (Buss et al., 2001). In contrast, overexpression of tail constructs lacking the splice insert had no effect on transferrin endocytosis. This study suggested that myo6 might have a role in endocytosis but only in tissues such as intestinal brush border that express a myo6 isoform containing a tail splice insert (Buss et al., 2001).
Myo6 has been reported to be enriched in clathrin-coated vesicles (CCVs) isolated from brain; however this tissue does not express the myo6 isoform with the splice insert (Buss et al., 2001). Therefore, we hypothesized that all splice forms of myo6, including forms that lack the splice insert, may be involved in the process of endocytosis.
In this study, we evaluated myo6's role in endocytosis by using a retinal epithelial cell model system. We found that in this system myo6 is not present on CCPs, and instead, is recruited to newly uncoated endocytic vesicles that contain the transferrin receptor. GIPC, an adapter protein, is also present and may be involved in the recruitment myo6 to these transport intermediates. Overexpression of the cargo-binding tail domain of myo6 led to an accumulation of transferrin-containing uncoated vesicles within cell peripheries. These vesicles contained Rab5, suggesting that they were transport vesicles delayed in trafficking to the early endosome. These data suggest that myo6 is an accessory protein required for the efficient and timely traffic of nascent endocytic vesicles.
MATERIALS AND METHODS
Cell Culture
The retinal pigmented epithelial cell line ARPE-19 (Dunn et al., 1996) was grown at 37°C with 5% CO2 in DMEM-F12 with 10% fetal bovine serum fungizone (Invitrogen, Carlsbad, CA) and glutamine (Invitrogen). NIH3T3, COS-7, and A431 cells (American Type Culture Collection, Manassas, VA) were maintained at 37°C with 5% CO2 in DMEM supplemented as described above. UB/UE-1 utricular epithelium cells were maintained in culture and differentiated as described previously (Lawlor et al., 1999).
Antibody Production
A HindIII fragment encoding porcine myo6 amino acids 42–726 (Hasson and Mooseker, 1994) was cloned into HindIII cut pQE-31 (QIAGEN, Valencia, CA) to create a histidine-tagged myo6 fusion protein, HIS-myo6motor. HIS-myo6motor protein was isolated from isopropyl β-d-thiogalactoside-induced XL1-blue bacteria (Stratagene, La Jolla, CA) as described by QIAGEN. The protein was concentrated to 1 mg/ml and dialyzed into phosphate-buffered saline (PBS) before injection into rabbits as described previously (Hasson and Mooseker, 1994). To create a Myo6motor-specific affinity column, the HIS-myo6motor protein was dialyzed into 0.1 M carbonate buffer with 0.05% SDS and then coupled to cyanogen bromide-activated Sepharose (Amersham Biosciences, Piscataway, NJ). Anti-Myo6motor antibody was affinity purified using the fusion protein column as described previously (Hasson and Mooseker, 1994) and used at 2 μg/ml for immunoblot and 20 μg/ml for immunocytochemistry.
Other Antibodies
Antibodies used in this study were from the following sources: BD Transduction Laboratories (San Diego, CA): antibodies to clathrin heavy chain, early endosome antigen 1 (EEA1), Rab11, Rab4, Rab8, p96-Dab2, Rab5 (mouse clone 1), and caveolin. Sigma-Aldrich: human transferrin receptor (CD71) and tubulin (Tub2.1) antibodies. The Developmental Studies Hybridoma Bank (University of Iowa, Iowa City, IA): anti-Lamp2 (H4B4). Jackson Immunoresearch (West Grove, PA): rabbit nonimmune IgG and fluorescein isothiocyanate- and rhodamine-conjugated donkey anti-rabbit and donkey anti-mouse secondary antibodies. All purchased antibodies were used within the concentration range recommended by the manufacturer. Antibody to Sec31 (used at 1:100) was from W. E. Balch (The Scripps Research Institute, La Jolla, CA). Rough endoplasmic reticulum antibody was from Daniel Louvard (Institut Pasteur, Paris, France). Antibody AP.6 to the AP-2 alpha chain was from Sandy Schmid (The Scripps Research Institute) and used at 1:200. Grasp65 antibodies were from Vivek Malhotra (University of California, San Diego, La Jolla, CA) and used at 1:500. Antibody to the mannose-6-phosphate receptor (10C6) was from Bernard Hoflack (Institute dePasteur, deLille, France) and used at 1:50. GIPC antiserum (DeVries et al., 1998b) was from Marilyn Farquhar (University of California, San Diego, Medical School, La Jolla, CA) and used at 1:200. Monoclonal antibody to SAP97 (77.4) was provided by Craig Garner (University of Alabama at Birmingham, Birmingham, AL). Affinity-purified rabbit and mouse anti-myo6 tail domain antibodies were used as described previously (Hasson and Mooseker, 1994).
Immunofluorescence Microscopy
Coverslip-grown cells were processed for immunofluorescence in six-well plates essentially as described previously (Hasson and Mooseker, 1994). Cells were washed in PBS with 100 μM CaCl2 and 1 mM MgCl2 (PBS-CM) before fixation at room temperature for 20–30 min in 4% electron microscopy grade paraformaldehyde (EM Scientific, Gibbstown, NJ) in PBS-CM. In some cases, cells were treated with 50 μM cytochalasin D (ICN Pharmaceuticals, Costa Mesa, CA) or 5 μM cholchicine (Sigma-Aldrich, St. Louis, MO) for 30 min before fixation. Fixed cells were permeabilized by treatment for 5 min at room temperature in 4% paraformaldehyde in PBS-CM with 0.1% Triton X-100. Alternatively, cells that were stained with clathrin antibody were fixed and permeabilized with –20°C methanol for 10 min. After fixation and permeabilization, cells were blocked for 30 min at room temperature with 3% bovine serum albumin (BSA) and 1% normal donkey serum (NDS) in PBS. Primary antibodies were diluted in PBS + 0.1% BSA + 1% NDS and incubated with coverslips for 2 h at 37°C. Coverslips were washed three times for 15 min in 0.5% BSA-PBS before incubation with secondary antibodies. Secondary antibodies were diluted 1:100 in PBS + 0.1% BSA + 1% NDS and incubated with coverslips for 2 h at 37°C. Rhodamine-phalloidin or Oregon Green phalloidin (Molecular Probes, Eugene, OR) was used at 80 nM and added with the secondary antibodies. Coverslips were washed three times for 15 min in 0.5% BSA-PBS before being mounted in Vectashield immunofluorescence mounting media (Vector Laboratories, Burlingame, CA) and sealed with nail polish to minimize photobleaching.
Samples were observed with a Bio-Rad MRC1024 confocal microscope or with a Leica DMR light microscope fitted with an ORCA digital camera. Bleed-through during conventional light microscopy was minimized with the use of both excitation and emission filters and confirmed in control stainings lacking primary antibodies. All digital images were analyzed using Adobe Photoshop.
Overlap between myo6 or myo6-GFP and organelle markers was quantified by identifying myo6-positive vesicles and scoring whether the second marker (e.g., R-Tsfn, Rab5, AP-2, EEA1, and clathrin) was also present on the vesicle. Percentage of overlap was calculated by dividing the number of vesicles that stained positive for both markers by the total number of myo6-positive vesicles counted (n ≥100/experiment). Groups of 100 or more vesicles from at least three different cells were quantified and standard deviations were calculated in Microsoft Excel.
Immunoprecipitation
Rabbit anti-M6tail antibody, rabbit anti-GIPC antibody, or rabbit nonimmune IgG (35 μg) was coupled to protein A-Sepharose beads using methods described in the ImmunoPure protein A IgG orientation kit (Pierce Chemical, Rockford, IL). Initial studies with RIPA (50 mM Tris, pH 7.6, 50 mM NaCl, 1 mM EDTA, 1% NP-40, 0.5% deoxycholate, 0.1% SDS), a buffer traditionally used in immunoprecipitation, revealed little or no coimmunoprecipitation of GIPC with myo6 antibodies (our unpublished data), similar to what had been shown for the recently identified myo6 binding partner Dab2 (Morris et al., 2002). Analysis of the supernatant and pellet fractions after solubilization revealed that RIPA buffer did not solubilize the entire population of myo6 or GIPC, suggesting that the vesicle- and cytoskeleton-associated pool might never be accessible using this method of extraction. Therefore, we used the more stringent immunoprecipitation methods described by Hinck et al. (1994) that allowed for immunoprecipitation of both soluble and insoluble (cytoskeletal) fractions of GIPC and myo6 after reversible chemical cross-linking.
Cell extracts were prepared essentially as described by Hinck et al. (1994) with the following modifications. Extracts were prepared from ARPE-19 cells plated at ∼80% confluence. Cells were solubilized in extraction buffer (RIPA buffer with 300 mM sucrose, 5 mM ATP, containing 2 μg/ml aprotinin, 10 μg/ml leupeptin, 20 μg/ml chymostatin, 10 μg/ml pepstatin A, 0.2 mM pefabloc, and 1× phosphatase inhibitor cocktails I and II (Sigma-Aldrich)). After spinning at 20,800 × g, the soluble extracts were precleared by incubation with protein A-Sepharose beads (Amersham Biosciences). The clarified extract was incubated with the coupled Sepharose beads described above for at least 3 h at 4°C on a rotator. The immunoprecipitates were washed five times with ice-cold extraction buffer and analyzed by SDS-PAGE and immunoblotting as described below.
Immunoblot Analysis
Proteins isolated from cultured cells or immunoprecipitations were prepared for gel analysis as described previously (Hasson and Mooseker, 1994). Separated proteins were transferred to nitrocellulose and the filter was blocked with 5% nonfat dry milk in Tris-buffered saline (TBS) before incubation with antibodies diluted in TBS + 0.05% Tween 20 for 2 h at room temperature. Filters were washed extensively with TBS-Tween 20 before incubation for 30 min with 40 mU/ml horseradish peroxidase-conjugated anti-rabbit IgG, anti-mouse IgG, or anti-goat IgG (Jackson Immunoresearch Laboratories). The signal was visualized using a chemiluminescence detection kit (Pierce Chemical) and X-OMAT RP film (Eastman Kodak, Rochester, NY).
Myo6 GFP Constructs
Porcine myo6 clone 23, encoding the N terminus, and clone 79, encoding the C terminus (Hasson and Mooseker, 1994), were digested with KasI and BamHI. The resulting 4.3-kb fragment from the clone 23 digest and the 2.6-kb fragment from the clone 79 digest were ligated into pBluescript SK(–) (Stratagene) to create the full-length myo6 cDNA pBS-M6. pBS-M6 was further modified to remove N-terminal stop codons. The N-terminal 1.7 kb of clone 23 was amplified using the primers 5′-TTGAATTCACTCACTTCCAAAATGGAGGATGG-3′ and 5′-TGTTCCTTTGGCGCCCCC-3′. The polymerase chain reaction fragment was cut with EcoRI and AgeI and reinserted into EcoRI/AgeI cut pBS-M6, replacing the N-terminal sequence.
The modified pBS-M6 was cut with EcoRI and BamHI and ligated into pEGFP-C1 (BD Biosciences Clontech, Palo Alto, CA) to create the GFP-M6full construct. Then 0.7-kb KpnI/BamHI fragment from clone 79 (amino acids 1050–1254) was ligated into KpnI/BamHI cut pEGFP-C1 to create GFP-M6tail. All constructs were confirmed by sequencing and protein size confirmed in immunoblot analysis with antibody to green fluorescent protein (GFP) or the tail domain of myo6.
Transfections
ARPE-19 cells grown in six-well dishes were transfected with 2 μg of purified plasmid DNA/well by using the Trans-IT transfection reagent (Mirus, Madison, WI) as per manufacturer's instructions. Cells were incubated with transfection reagent for 16–20 h to obtain adequate expression levels and then were either fixed for immunofluorescence as described above or used for rhodamine-conjugated transferrin (R-Tsfn) uptake assays.
Pulse-Chase Uptake of R-Tsfn
Cells were incubated in serum-free DMEM-F12 media for 2 h at 37°C before initiation of uptake assays. To surface label the transferrin receptors, cells were chilled on ice for 30 min and then incubated for 30 min on ice with 10 μg/ml R-Tsfn (Molecular Probes) in DMEM-F12. Cells were washed with ice-cold DMEM-F12 to remove unbound R-Tsfn and then chased at 37°C in prewarmed media for the indicated time periods (1 min-30 min) before fixation.
Steady-State Uptake and Quantification of Trafficking
Twenty-four hours after transfection, coverslip-grown ARPE-19 cells were serum starved as described above. The medium was then replaced with DMEM-F12 containing 10 μg/ml R-Tsfn and uptake was allowed to proceed at 37°C for 15 min or 30 min. In parallel experiments to assess effects on fluid phase uptake, transfected ARPE-19 cells were incubated with 0.5 mg/ml rhodamine-conjugated dextran (Molecular Probes) for 10 or 30 min at 37°C. Uptake was stopped by fixation.
To quantify transferrin uptake, transfected and untransfected cells were photographed using both fluorescence and phase microscopy. Cells (100–400) were counted in three separate experiments, and the presence or absence of an accumulation of R-Tsfn in the perinuclear area was counted. Percentage of cells that had a perinuclear accumulation of R-Tsfn was calculated by dividing the number of cells that had a perinuclear accumulation of R-Tsfn by the total number of cells counted. The error bars represent the SD from three separate experiments (n ≥ 100 for each experiment).
RESULTS
Myo6 Is Enriched in a Peripheral Punctate Compartment in ARPE-19 Cells
Immunolocalization using antibodies to myo6 on cultured cells revealed a diffuse cytoplasmic stain, with some perinuclear enrichment. The intensity of the perinuclear staining varied from cell to cell, suggesting that it is due to an increase in cell thickness in this region but it also may be due to some myo6 associated with Golgi (Buss et al., 1998). Whereas the diffuse cytoplasmic stain for myo6 was seen in all cell lines tested, in cells of epithelial origin there was also an apparent enrichment for myo6 at the peripheries. As shown in Figure 1, a myo6 enrichment at cell peripheries was seen in epithelial cells derived from retina and inner ear (ARPE-19 and UB/UE-1, respectively), two cell types that express abundant myo6 in vivo (Avraham et al., 1995; Breckler et al., 2000). In the fibroblast cell lines COS-7 and NIH3T3, and the epidermoid line A431 (Figure 1; our unpublished data), the peripheral enrichment was not evident and, instead, myo6 exhibited a much more uniform punctate staining
Figure 1.

Localization of myo6 in cultured cell lines. Indirect immunofluorescence staining of cells by using affinity-purified rabbit antibodies directed to the tail domain of myo6 (Myo6), the motor domain of myo6 (Myo6 motor Ab) or affinity-purified mouse antibodies directed to the tail domain (Mouse Myo6 Ab). Cells were counterstained for F-actin by using rhodamine-conjugated phalloidin. The cortical actin is demarcated by filled arrows. Myo6 is enriched in a peripheral punctate compartment (open arrows) that overlaps with the cortical actin in both ARPE-19 and UB/UE-1 cell lines. Bar, 10 μm.
Because myo6 had been implicated in endocytosis, we hypothesized that this peripheral myo6 staining could represent the association of myo6 with an endocytic compartment. The peripheral enrichment of myo6 was detected in epithelial cells by using three distinct affinity-purified antibodies to myo6, directed to both the tail and the motor domains (Figure 1; Supplementary Figure 1). In all cases, the peripheral punctate staining was enriched in areas with dense F-actin (Figure 1). The peripheral staining was most evident in the cultured retinal pigmented epithelial cell line ARPE-19, so we focused our studies on myo6 function in these cells.
Myo6-associated Punctae Are Endocytic Vesicles Containing the Transferrin Receptor
Previous studies suggested that myo6 was associated with CCPs in intestinal epithelial cells (Buss et al., 2001). However, the myo6-containing punctae at the periphery of ARPE-19 cells did not significantly overlap with clathrin or AP-2, a clathrin adapter protein (Figure 2, A and B). The myo6-associated punctae were also distinct from caveolae, because there was no colocalization with either caveolin or GPI-linked GFP (Nichols et al., 2001), a fusion protein that targets to caveolae (our unpublished data).
Figure 2.

Myo6 overlaps with GIPC, transferrin receptor, and Rab5 on vesicles distinct from clathrin-coated pits and EEA1-positive early endosomes. (A–F) ARPE-19 cells were double labeled with antibodies directed to the tail domain of myo6 (M6; detected with a fluorescein isothiocyanate-conjugated secondary antibody) and to components of the endocytic pathway (detected with a rhodamine-conjugated secondary antibody). The top panels show an example of the whole-cell staining pattern seen for myo6 and the markers. The boxed area in the top panels is enlarged in the bottom panels. In the overlay, myo6 is shown in green, the markers are shown in red, and overlap between the two markers is yellow. Staining was for clathrin heavy chain (A) the clathrin adaptor AP-2 (B), Rab5 (C), EEA1 (D), TsfnR (E), and GIPC (F). Bars (A–F), 10 μm.
We next compared the location of myo6 to markers for many stages of the endocytic pathway found in cell peripheries (Figure 2; our unpublished data). Remarkably, the myo6-containing structures in the periphery were distinct from EEA1-containing early endosomes (Figure 2D), Rab4- and Rab11-staining recycling endosomes, Sec31-containing ER exit sites, Rab8-containing vesicles en route between the trans-Golgi and the plasma membrane, and mannose-6-phosphate receptor-positive late endosomes, trans-Golgi network, and lysosomes (our unpublished data). The only tested endosomal marker that overlapped to some extent with myo6 in cell peripheries was the GTPase Rab5 (Figure 2C). Rab5 is present on both EEA1-containing early endosomes found in the perinuclear region (our unpublished data) and small endocytic vesicles found in the periphery. Rab5 and myo6 colocation on perinuclear early endosomes was not evident, although in low-magnification views some diffuse myo6 staining in the perinuclear region was common. In contrast, in the cell peripheries some small vesicles that stained for myo6 also stained for Rab5 (Figure 2C, arrows). Quantitation showed that 7.2 ± 0.5% of the myo6-staining peripheral vesicles also contained Rab5. Although not a strong level of overlap, this overlap is significantly higher than that seen with AP-2 (2.0 ± 1.0%) or clathrin (3.6 ± 0.4%) and suggested that myo6 could be recruited to recently uncoated endocytic vesicles en route to the early endosome.
Because myo6 had been implicated in the endocytosis of transferrin (Buss et al., 2001), we investigated whether the transferrin receptor (TsfnR) was a component of the myo6-associated endocytic vesicles found at the peripheries of ARPE-19 cells. Confocal imaging revealed consistent colocation between myo6 and the TsfnR on vesicles in the cell periphery (Figure 2E, open arrows). This represents a small fraction of the total TsfnR, because the majority of the TsfnR was present in the pericentriolar recycling endosomes (Figure 2E, top). These results suggested that myo6 could be involved in the transport of transferrin-containing endocytic vesicles.
Myo6 Is Recruited to Transferrin-containing Endocytic Vesicles after Clathrin-coated Vesicle Uncoating but before Early Endosome Fusion
Testing our hypothesis that the myo6-associated vesicles seen in ARPE-19 cells were endocytic vesicles, we evaluated the kinetics of myo6 recruitment in pulse-chase experiments after transferrin uptake through the endocytic pathway. In these experiments, we prebound surface TsfnRs with R-Tsfn at 4°C and then incubated the cells for various times at 37°C before fixation for immunofluorescence microscopy. At 4°C, R-Tsfn bound the surface of ARPE-19 cells and this staining pattern overlapped in toto with AP-2, a marker of CCPs (Figure 3b). Overall 79.2 ± 12.5% of CCPs contained labeled transferrin. Therefore, TsfnRs are preclustered in clathrin-coated pits in ARPE-19 cells.
Figure 3.

Myo6 is recruited to uncoated endocytic vesicles. ARPE-19 cells were incubated with R-Tsfn at 4°C for 30 min. After surface labeling, the cells were either formaldehyde fixed (a and b) or warmed to 37°C for a 1-min chase (c–f) or a 10-min chase (g and h) before fixation. Cells were stained using antibodies to the tail domain of myo6 (a, c, d, and g), the clathrin-adapter AP-2 (b and e), or the early endosome protein EEA1 (f and h). Antibodies were visualized with a fluorescein-conjugated secondary antibody (green) and compared with the endocytosed rhodamine-conjugated transferrin (red) by immunofluorescence microscopy. Overlap between the R-Tsfn and either Myo6, AP-2, or EEA1 is shown in yellow. Cell peripheries of flat cells were chosen for analysis to allow clear visualization of the R-Tsfn as it moved through the endocytic pathway. Boxed areas in each top panel are enlarged in the bottom panel. The perinuclear early endosome is shown with arrows in f and h. Bars and brackets found in enlarged bottom panels are 10 μm.
The surface bound R-Tsfn did not overlap in location with myo6 (Figure 3a). Only 3.3 ± 1.0% of myo6-associated vesicles found in peripheries overlapped with the surface-associated R-Tsfn. This result agreed with the immunolocation studies showing that myo6 was not associated with CCPs on the plasma membrane (Figure 1), and suggested that the myo6-associated vesicles were not clathrin coated.
After 1 min of chase at 37°C, R-Tsfn was evident as distinct punctae dispersed throughout cytoplasm. The R-Tsfn no longer overlapped with AP-2 (Figure 3e), and quantitation revealed only 3.0 ± 1.0% of CCPs now contained R-Tsfn. Therefore, clathrin-coated vesicle formation and uncoating had occurred during the 1-min chase period.
Remarkably, after 1-min chase, the peripheral population of R-Tsfn now overlapped in many cases with myo6 (Figure 3, c and d). Myo6-associated vesicles (53.7 ± 7.0%) now contained labeled transferrin, suggesting that myo6 had been recruited to the vesicles immediately after uncoating. Not all transferrin-containing vesicles contained myo6; however, many of the R-Tsfn vesicles overlapped with EEA1 after the 1-min chase (Figure 3f), suggesting that even in this short time frame, a fraction of vesicles can be transported to and fuse with the early endosome.
Of the population of R-Tsfn vesicles that did stain with antibodies to myo6, this population was found primarily at the cell periphery, within 10 μm of the cell edge (see brackets in Figure 3, c and d). The EEA1-positive transferrin-containing endosomes, however, were more centrally located than these myo6-associated vesicles and were instead found >10 μm from the cell edge (Figure 3, brackets). Based on the positioning of the myo6-associated vesicles on the extreme peripheries of cells and the lack of overlap between these vesicles and EEA1-containing endosomes at the 1-min time point, we hypothesized that myo6 was being recruited to the endocytic vesicle immediately after uncoating, but before fusion with the early endosome. This positioning further suggested that myo6 could be involved in the translocation of these nascent endocytic vesicles from the far peripheries toward the more centrally located early endosome.
To test this hypothesis, we extended our pulse-chase experiment to include a chase of 10 min at 37°C. At the 10-min time point, the R-Tsfn had largely exited the peripheral areas of the cell (Figure 3g) and only 11.6 ± 1.1% of myo6-associated peripheral vesicles now contained R-Tsfn. Instead, R-Tsfn was now present in either EEA1-positive endosomes or the EEA1-positive pericentriolar region (Figure 3h, arrows). This location is in contrast to that seen after the 1-min time point when the R-Tsfn had not yet reached the EEA1-positive pericentriolar early endosome (Figure 3f, arrow). Because myo6 was no longer associated with the R-Tsfn–containing compartments after fusion of the endocytic vesicle with the early endosome, we theorized that myo6 departed or was released from the vesicle either concomitant with or just before fusion of the endocytic vesicle with the EEA1-positive early endosome.
GIPC Is Present on Myo6-associated Endocytic Vesicles
Our pulse-chase experiments suggested that myo6 had a transient association with uncoated endocytic vesicles. Myo6 is not known to bind to lipids directly, so we investigated whether three recently identified adapter proteins, GIPC, Sap97, and Dab2, identified in yeast-two-hybrid studies as putative myo6-tail binding proteins (Bunn et al., 1999; Morris et al., 2002; Wu et al., 2002), could be serving as the bridge to link myo6 to the uncoated vesicles. Double staining with antibodies to Sap97 or Dab2 revealed no overlap between these proteins and myo6 in peripheral regions of ARPE-19 cells (our unpublished data). Sap97 was found primarily at cell-cell contacts, where myo6 was not evident, and Dab2 was found exclusively in clathrin-coated pits, which, as shown in Figure 2, do not contain myo6 (our unpublished data). Therefore, our studies focused on GIPC as a potential linker between the uncoated vesicle and myo6.
GIPC is a PSD-95/Dlg/ZO-1 (PDZ)-domain containing adapter protein found in both CCPs and endocytic domains (DeVries et al., 1998a,b) that associates via its PDZ-domain with a variety of transmembrane proteins and receptors (Lou et al., 2002). Double-staining experiments in ARPE-19 cells revealed that GIPC colocated with myo6 in peripheral regions (Figure 2F). Essentially 100% of the myo6-associated vesicles stained positive for GIPC, suggesting that GIPC may be involved in the recruitment of myo6 to the uncoated vesicles or that GIPC was recruited along with myo6 to the vesicle surface.
Because myo6 and GIPC colocated on vesicles in cell peripheries, we predicted that the binding of myo6 and GIPC on the surface of these vesicles would be detectible using coimmunoprecipitation methods. As shown in Figure 4, using a method that fully solubilized myo6 and GIPC after reversible cross-linking, we successfully coimmunoprecipitated GIPC by using antibodies directed to the tail-domain of myo6. Antibodies to GIPC also coimmunoprecipitated this complex, although less effectively. The myo6–GIPC complex was not brought down with nonimmune IgG. Although these studies suggest that a fraction of the myo6 and GIPC present in the cell are bound in a complex, further studies will be required to determine whether the immunoprecipitated GIPC–myo6 complex reflects the small amount of GIPC and myo6 found on the surface of uncoated vesicles. Because myo6 was identified as a direct binding partner of GIPC by using yeast two-hybrid and glutathione S-transferase pull-down methods, the coimmunoprecipitation of the two proteins suggests a direct GIPC–myo6 interaction is occurring within the cell.
Figure 4.
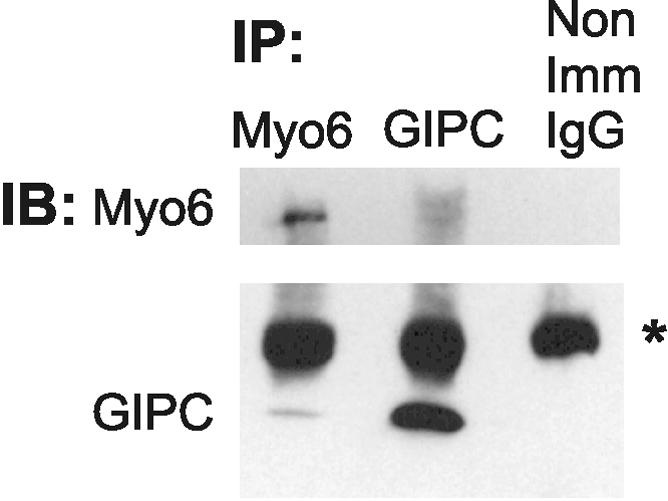
Coimmunoprecipiation of myo6 with GIPC. Extracts from ARPE-19 cells were immunoprecipitated (IP) with antibodies to Myo6, GIPC, or nonimmune IgG. Immunoprecipitated proteins were detected by immunoblot (IB) analysis with antibodies to myo6 (top) and GIPC (bottom). The position of the IgG is marked with an asterisk.
Positioning of Uncoated Vesicles Is Actin Dependent
Our localization and pulse-chase data supported a model in which myo6 associates with nascent endocytic vesicles and acts as an actin-based motor to allow these vesicles to associate with the cortical actin meshwork. To test whether the positioning of the myo6-associated endocytic vesicles was indeed actin dependent, we treated ARPE-19 cells with drugs that depolymerized the actin cytoskeleton. Because microtubules are important for endosome to lysosome trafficking (Jin and Snider, 1993), we also tested the effects of microtubule-depolymerizing drugs.
A 30-min treatment with cytochalasin D caused a reorganization and net disassembly of the F-actin (Figure 5A). Cytochalasin D treatment also had a dramatic effect on myo6 location because the protein was no longer evident in peripheral areas. In cases where a few remaining F-actin clusters were present in the peripheries after cytochalasin D treatment, the myo6 redistributed to overlap with this residual actin, suggesting an avid association between actin and myo6 within the cell. In contrast treatment with the microtubule-depolymerizing drug, colchicine (Figure 5A) had no effect on the myo6-staining pattern. Therefore, F-actin plays an important role in the positioning of myo6-associated vesicles at the cell periphery.
Figure 5.

Myo6 localization is dependent on F-actin and its C-terminal tail domain. (A) ARPE-19 cells were treated for 30 min with cytochalasin D (CytD) or colchicine (Colch) before staining with antibodies to myo6 and rhodamine-conjugated phalloidin or antitubulin antibody. Cytochalasin D treatment resulted in a collapse of the myo6-associated endocytic vesicles into peripheral clumps (arrowheads), whereas colchicine treatment had no effect on the location of the myo6-associated vesicles. (B) Fluorescence microscopy of ARPE-19 cells expressing GFP, GFP fused to full-length myo6 (GFP-M6full), or GFP fused to the C-terminal globular tail domain of myo6 (GFP-M6tail). Cells were costained with rhodamine-conjugated phalloidin to stain F-actin. Arrowheads point out actin-rich regions. Bars, 10 μm.
Myo6 Assembles on Endocytic Vesicles via Its C-Terminal Cargo-binding Domain
Because our pulse-chase experiments suggested that myo6 was recruited to the nascent vesicles after uncoating, we predicted that exogenously expressed myo6 should assemble onto the nascent vesicles. To test this theory, we created a fusion between GFP and myo6, GFP-M6full (see MATERIALS AND METHODS), and tested its ability to target to the peripheral vesicles in ARPE-19 cells. This fusion protein does not contain the insert implicated by Buss et al. (2001) as important for both CCP localization and endocytosis and represents the typical myo6 that has been cloned from kidney, retina, and cultured cell lines (Hasson and Mooseker, 1994; Avraham et al., 1995, 1997; Breckler et al., 2000).
When expressed at low levels, GFP-M6full targeted to small peripheral punctae along actin cables (Figure 5B) that did not overlap extensively with clathrin (Figure 6C). Confocal microscopy confirmed that these punctae overlapped with GIPC (Figure 6A) and the TsfnR (Figure 6B), confirming their identity as nascent endocytic vesicles and suggesting that GFP-M6full was fully functional.
Figure 6.

Myo6-GFP fusion proteins colocate with GIPC and Ts-fnR on an endocytic compartment in peripheries of ARPE-19 cells. Cells expressing GFP-M6full or GFP-M6tail were double stained with antibodies to GIPC (A) or the transferrin receptor (TsfnR) (B) and viewed by confocal microscopy. Both the full-length myo6 and the tail domain of myo6 targeted to the GIPC-associated, TsfnR-containing endocytic vesicles (arrowheads). (C) Cells expressing GFP-M6full (green) were stained with antibodies to clathrin (red). (D) Cells expressing GFP-M6tail (green) were incubated for 10 min with Rhod-dextran (red) to label endocytic compartments before fixation and visualization the confocal microscope. Myo6-tail-associated endocytic vesicles contain Rhod-dextran (open arrows; yellow color denotes overlap). Bars, 10 μm.
Myosins are targeted to distinct subcellular domains via their unique C-terminal tail domains. Therefore, we predicted that the tail domain of myo6, alone, should be capable of targeting to the vesicles. To test this hypothesis, we fused to GFP the C-terminal globular tail domain of myo6, which contains the cargo-binding region, and expressed this construct, GFP-M6tail, in ARPE-19 cells. Again, this construct lacked the insert implicated by Buss et al. (2001) as essential for endocytic function. Fluorescence microscopy revealed that when expressed at low levels, the GFP-M6tail fusion protein targeted to punctae on actin cables at the cell peripheries (Figure 5B). The GFP-M6tail punctae overlapped with GIPC (Figure 6A) and the TsfnR (Figure 6B), as judged by confocal microscopy. Therefore, the tail domain is sufficient to target myo6 the nascent endocytic vesicles.
If the myo6-associated vesicles were indeed an endocytic intermediate, we predicted that they would be accessible to the fluid phase uptake marker, rhodamine-conjugated dextran (Rhod-dextran). As shown in Figure 6D, after a 10-min incubation with Rhod-dextran there was significant overlap with the GFP-M6tail-decorated endocytic vesicles. Therefore, the tail of myo6 is targeting the motor protein to endocytic vesicles.
Removal of Myo6 from the Nascent Endocytic Vesicles Disrupts Trafficking of Transferrin
The immunolocalization, GFP-fusion, and uptake data presented thus far support a model whereby myo6 serves as the bridge between the TsfnR-containing nascent endocytic vesicle and the actin cytoskeleton and that myo6 is linked to the endocytic vesicle solely by its tail domain. Therefore, we predicted that overexpression of the tail domain would displace the endogenous myo6 from the vesicle. As shown in Figure 7, A and B, overexpression of GFP alone had no effect on the association of endogenous myo6 with peripheral vesicles, as judged by an antibody directed to the motor domain of myo6. In contrast, overexpression of GFP-M6tail resulted in a selective removal of myo6 stain from the peripheries of transfected cells, as judged by the motor-specific antibody (Figure 7, C–E). GFP-M6tail staining of peripheral vesicles was evident in these transfected cells, supporting the hypothesis that the endogenous myo6 is competed off the endocytic vesicles when the myo6 tail domain is overexpressed.
Figure 7.

GFP-M6tail displaces myo6 from peripheral endocytic vesicles. Immunofluorescence microscopy of ARPE-19 cells transiently transfected with GFP (A and B) or GFP-M6tail (C–E) and stained with antibodies to the motor domain of myo6. GFP fluorescence is shown in B, D, and E (green), and myo6-motor antibodies staining is shown in A, C, and E (red). The region of the cell where peripheral vesicles are found is demarcated by filled arrows in transfected cells and by open arrows in untransfected cells. Bar, 10 μm.
Presumably, myo6 was being recruited to endocytic vesicles to allow transportation through the actin meshwork. We predicted that, after overexpression of the M6tail domain, because no myo6 motor was present, the nascent endocytic vesicles would no longer be able to negotiate the actin-meshwork of the cell peripheries, precluding efficient fusion with the more centrally located early endosome. To test this theory, we evaluated whether overexpression of the GFP-m6tail construct would cause a block in transferrin uptake.
ARPE-19 cells were transfected with GFP, GFP-M6full, and GFP-M6tail and evaluated for the effects of transfection on the steady-state uptake of R-Tsfn (Figure 8). After a 15-min incubation, 84.7 ± 2.4% of untransfected cells and 82.3 ± 3.2% of GFP transfected cells exhibited a prominent accumulation of R-Tsfn in the perinuclear early endosome and recycling endosome compartments. Transfection with GFP-M6full decreased uptake slightly, with 58.1 ± 5.0% exhibiting perinuclear R-Tsfn accumulation. This decrease in uptake was statistically significant based on the Student's t test and may be due to a disruptive effect of GFP on the function of the nearby myo6 motor domain.
Figure 8.

Overexpression of the tail domain of myo6 delays endocytosis of transferrin. GFP, GFP-M6full, and GFP-M6tail–transfected cultures were incubated with R-Tsfn for either 15 or 30 min. (A) Overview of GFP-M6full and GFP-M6tail–transfected ARPE-19 cells after 15-min incubation with R-Tsfn. GFP-M6tail–expressing cells (demarcated by closed arrows) exhibited a decrease in net transferrin uptake, as is evidenced by a lack of perinuclear staining. In contrast, the majority of GFP-M6full–expressing cells accumulate transferrin normally (open arrows) compared with untransfected cells in the field. (B) Percentage of transfected and untransfected cells showing labeling of perinuclear endosomes after 15 min (gray bars) or 30 min (white bars) of R-Tsfn uptake. The total number of cells counted for each experiment is denoted as n. (C) Higher magnification view of GFP- (a and b), GFP-M6full–(c and d), and GFP-M6tail (e and f)–transfected cells after 15 min of incubation with R-Tsfn. Transferrin is shown in the top panels (a, c, and e); GFP in the bottom panels (b, d, and f). GFP-M6tail–transfected cells exhibit an increase in peripheral R-Tsfn staining, not seen in other cells. Bars, 10 μm.
Overexpression of GFP-m6tail, in contrast, caused a drastic decrease in the transferrin uptake, with only 14.6 ± 1.8% of cells exhibiting a perinuclear accumulation. In the GFP-M6tail–expressing cells, the transferrin was endocytosed, but instead of accumulating in perinuclear regions it was present in peripheral structures (Figures 8A, closed arrows; and C). These results suggested that transferrin endocytosis was occurring in the transfected cells but that the process of trafficking to the pericentriolar region was delayed.
GFP-M6tail Overexpression Delays Transport of Transferrin-Containing Endocytic Vesicles Out of Cell Peripheries
To evaluate the stage at which endocytosis was blocked after overexpression of the tail domain of myo6, we initiated a pulse-chase experiment similar to that shown in Figure 3. ARPE-19 cells were transfected with GFP-M6tail and low expressing cells were chosen for further study, because in these cells, we had found that the GFP-M6tail fusion targets to the nascent vesicles (Figure 6). These cells were incubated with R-Tsfn at 4°C and then warmed to 37°C to follow the transferrin as it trafficked through the endocytic pathway.
As seen for untransfected cells, in GFP-M6tail–transfected cells incubation with R-Tsfn at 4°C labeled the CCPs on the cell surface (Figure 3; our unpublished data). These CCPs were distinct from the GFP-M6tail–associated vesicles (Figure 9A, a). Quantitation revealed that only 4.6 ± 1.7% of GFP-M6tail–associated vesicles overlapped with the surface R-Tsfn label and only 2.3 ± 1.4% of the vesicles overlapped with AP-2 (Figure 9C). Therefore, these data confirm that the tail domain of myo6 is not recruited to CCPs.
Figure 9.

GFP-M6tail is recruited to transferrin-containing uncoated endocytic vesicles, slowing traffic of transferrin to the early endosome. A portion of a transfected cell is shown in each panel, oriented with the nucleus positioned to the left and the cell periphery to the right. (A) Pulse-chase experiments after R-Tsfn in GFP-M6tail–transfected ARPE-19 cells. Transfected cells were incubated with R-Tsfn at 4°C for 30 min (a) and then warmed to 37°C for 2 min (b), 10 min (c), or 30 min (d). Open arrows point to overlap between the R-Tsfn (red) and the GFP-M6tail (green) staining at the periphery of each cell. The boxed region is presented at the same scale as separate M6tail and R-Tsfn images below each panel. The position of GFP-M6tail–associated vesicles is denoted with arrows in both lower panels. PN in d demarcates the position of the perinuclear early endosome. (B) Localization of rab5 in GFP-M6tail–transfected ARPE-19 cells. Open arrows mark the position of GFP-M6tail–associated endocytic vesicles that also contain rab5. (C) Localization of AP-2 (red) in GFP-M6tail (green)–transfected ARPE-19 cells reveals no overlap in CCPs. (D) Localization of EEA1 (red) in GFP-M6tail (green)–transfected ARPE-19 cells reveals that a subset of the GFP-M6tail is associated with peripherally located endosomes. Overlap in location is seen as yellow and is demarcated with an arrow. Bars, 10 μm.
After 2 min of chase, similar to our observations in pulse-chase experiments in untransfected cells, a population of the R-Tsfn now colocated with the GFP-M6tail (Figure 9A, b). 56.8 ± 4.6% of the GFP-M6tail–labeled vesicles contained endocytosed transferrin, suggesting that the tail domain of myo6, like full-length myo6, is recruited to endocytic vesicles immediately after uncoating.
Unlike what we had seen previously for untransfected cells, in GFP-M6tail–expressing cells, the R-Tsfn was not observed to exit the cell periphery within 10 min (Figure 9A, c). Instead, R-Tsfn remained in the peripheral domain within the GFP-M6tail–associated endocytic vesicle. GFP-M6tail–associated peripheral vesicles (58.5 ± 16.1%) contained R-Tsfn after a 10-min chase. The magnitude of this trapping is more apparent when the transferrin-labeling pattern seen in GFP-M6tail–transfected cells after 15 min of uptake is compared with that seen in untransfected or transfected control cells (Figure 8C). In control cells, transferrin has accumulated significantly in the perinuclear region and there is little transferrin remaining in the peripheral cytoplasm. In contrast, in the GFP-M6tail–transfected cells (Figure 8C), the transferrin remains in peripheral zones and little enrichment in the perinuclear region is evident. These experiments suggest that the defect in trafficking seen upon overexpression of the tail domain of myo6 is due to a dominant effect of the tail domain binding to the nascent endocytic vesicles, and, because the tail domain lacks an actin-binding site or motor domain, its traffic along the endocytic pathway is delayed.
Even after 30 min of chase at 37°C, a significant population of the R-Tsfn remained within the GFP-M6tail–associated vesicles (28.3 ± 8.5%; Figure 9A, d); however, the percentage had diminished, suggesting that given time the endocytic vesicle could find and fuse with the early endosome leading to release of the GFP-M6tail. To test this hypothesis, we repeated our steady-state uptake experiments looking at the effects of transfection with GFP-M6tail on transferrin endocytosis but increased the uptake time to 30 min. As shown in Figure 8B, given sufficient time transferrin-containing endosomal vesicles could indeed reach the early endosome with 73.7 ± 3.7% of GFP-M6tail–transfected cells now seen to accumulate R-Tsfn in the perinuclear region. This percentage approaches that seen for control cells (85 ± 2.7%).
Our pulse-chase experiments suggested that the delay in endocytosis of transferrin was due to a block at the uncoated-vesicle transport step of endocytosis. However, the delay observed after GFP-M6tail overexpression could be attributed to blocks at one of many other steps in the endocytic pathway. To confirm that only vesicle trafficking was affected by GFP-M6tail overexpression, ARPE-19 cells were transfected and stained to look at the effects of overexpression on CCPs, endosome populations, organelles, and other cell structures.
Overexpression of the tail of myo6 had no effect on the distribution or number of CCPs on the surface of ARPE-19 cells (Figure 9C; our unpublished data), suggesting that the earliest stages of endocytosis, the formation of CCPs and vesicles, was unaffected by overexpression of the M6tail domain. We next evaluated the effect of GFP-M6tail expression on the early endosomes and other endocytic organelles. The location of mannose-6-phosphate receptor, which is present on trans-Golgi network, late endosome, and lysosomal membranes, was unaltered (our unpublished data) as well as markers for vesicle trafficking (Rab4, Rab11, and Rab8) lysosome (Lamp2), Golgi (Grasp65), ER markers, and early endosomes were not affected by GFP-M6tail domain overexpression (Figure 9; our unpublished data). Uptake of the fluid-phase marker Rhod-dextran was also unaffected by GFP-M6tail transfection (Supplementary Figure 2), suggesting that the delay in R-Tsfn uptake was not due to a general defect in endocytosis.
The one component that did exhibit a dramatic change after GFP-M6tail overexpression was the small GTPase Rab5. After overexpression of the tail domain, 86.4 ± 5.8% of the GFP-M6tail–associated endocytic vesicles now contained Rab5, a marked difference from that seen with the native myo6 where only ∼7% of the myo6-associated vesicles contained Rab5 (Figure 8B; our unpublished data) and GFP-M6full where only 6.4 ± 2.1% of the labeled vesicles contained Rab5 (our unpublished data). Other than this increase in association with the myo6-vesicles, the overall pattern of Rab5 location did not change after GFP-M6tail overexpression, suggesting that the increased Rab5 association with the GFP-M6tail vesicles reflected recruitment of Rab5 from the cytoplasmic pool.
Recruitment of Rab5 to endocytic vesicles is required for fusion with the early endosome. The fact that Rab5 is recruited to the GFP-M6tail–associated vesicles suggests that the delay in trafficking of transferrin is not due to a simple fusion defect and instead implicated myo6 as an accessory protein for movement of vesicles during endocytosis. In keeping with this hypothesis, although the tail domain was not wholly recruited to the early endosome, 9.0 ± 1.6% of the GFP-M6tail–associated vesicles did overlap with EEA1 (Figure 9D). This subset of GFP-M6tail–associated vesicles was found primarily >10 μm away from the cell peripheries (Figure 8D, arrows). This overlap with EEA1 was not seen with GFP-M6full constructs (our unpublished data), and because it is limited to a peripheral ring of early endosomes, we interpret it to be evidence of the fusion of the GFP-M6tail–associated vesicles with the peripheral-most early endosomes.
DISCUSSION
We find that myo6 is recruited to newly uncoated endocytic vesicles. Association of myo6 with the vesicles is transient, because myo6 departs upon fusion with the EEA1-positive early endosome. GIPC, an adapter protein, is also present on these endocytic vesicles. We found that disruption of myo6 function led to an accumulation of transferrin-containing and Rab5-containing uncoated vesicles within cell peripheries due to delayed trafficking to the early endosomes. These data suggest that myo6 is an accessory protein required for the efficient and timely traffic of nascent endocytic vesicles, even in the absence of any of the unique splice inserts previously implicated as essential for endocytic function. We interpret our findings to reflect an epithelial cell-specific need for translocation of endocytic vesicles through dense actin networks found at cell peripheries.
Myo6 Is Recruited to Uncoated Endocytic Vesicles before Early Endosome Fusion
Using both transferrin uptake and dominant negative expression studies, we have found that myo6 is recruited to nascent uncoated vesicles that accumulate 1 min after endocytosis is initiated (Figures 3 and 9). Transferrin uptake has been studied extensively and the endocytic pathway we have characterized in ARPE-19 cells mirrors the time course of endocytosis seen in baby hamster kidney (Eskelinen et al., 1991), A431 (Hopkins, 1983), KB (Hanover et al., 1984) and Chinese hamster ovary cells (Trischler et al., 1999). The observation that myo6 is recruited to the transferrin-containing vesicles within 1 min of initiation of endocytosis suggests that these vesicles are not involved in the recycling of receptors back to the cell surface because pulse-chase experiments in whole cells as well as cytoplasts have shown that traffic through recycling endosomes occurs at least 7 min and often 25 min after uptake (Hopkins, 1983; Hanover et al., 1984; Eskelinen et al., 1991; Mayor et al., 1993; Ghosh et al., 1994; Trischler et al., 1999; Sheff et al., 2002)
The myo6-associated uncoated vesicles also have components of the endosomal fusion machinery. Recruitment of the small GTPase Rab5 is required for subsequent early endosome fusion (Rubino et al., 2000). Although Rab5 was not found to be wholly present on the myo6-associated vesicles present in untransfected ARPE-19 cells, Rab5 was dramatically enriched on the GFP-M6tail–associated vesicles (Figure 9). We hypothesize that this enrichment of Rab5 on the GFP-M6tail–associated vesicles is due to the delay in trafficking. Normally the uncoated vesicles would be transported out of the cell peripheries (presumably by the myo6 motor) within 1 min of formation, precluding detection of Rab5 recruitment. Because the GFP-M6tail–labeled vesicles remain in the peripheries for tens of minutes, there is abundant time for Rab5 recruitment in advance of endosome fusion.
Our data suggest that myo6 associates with uncoated transferrin-containing vesicles transiently and before fusion with the EEA1-positive endosomes. It is unknown whether myo6 facilitates the fusion of the uncoated vesicle with the early endosome but given the fact that the observed defect in endocytosis is only a delay and not a strict block, suggests that fusion can occur in the absence of myo6 function. Interestingly, a peripheral subpopulation of small EEA1-positive endosomes found ∼10 μm from the cell edge was labeled with the GFP-M6tail construct (Figure 9). The presence of this endosome population supports a model whereby myo6 is present during fusion but is released after fusion is complete. Possibly the motor domain facilitates release of myo6, which may explain why we found some association of GFP-M6tail, but not GFP-M6full or the endogenous myo6, with these early endosomes.
Recruitment of Myo6 to Nascent Endocytic Vesicles
Our GFP-expression data suggests that it is the tail domain of myo6 that is binding to a component on uncoated vesicles. Our colocalization and coimmunoprecipitation data suggest that the component that may be involved in the recruitment of myo6 to vesicles is the adapter protein GIPC. The tail of myo6 is sufficient to interact with GIPC in a yeast-two hybrid assay and also in a glutathione S-transferase pull-down assay (Bunn et al., 1999), supporting a direct binding event between GIPC and myo6.
Presumably the association of GIPC with the vesicle reflects a binding event between its PDZ-domain and PDZ-binding motifs found at the C termini of transmembrane proteins associated with the vesicle. Therefore, GIPC likely serves as a bridge to link myo6 to the vesicle surface. Because only a small fraction of GIPC and myo6 coimmunoprecipitated (Figure 4), the two proteins are likely not found together as a stable complex in the cytoplasm. Ultimately, based on our pulse-chase experiments we cannot at this time determine whether GIPC recruits myo6 to the surface of the uncoated vesicle or whether, instead, the GIPC–myo6 complex together is recruited to the vesicle surface.
The mechanism that regulates GIPC's and myo6's association with the uncoated endocytic vesicles cells remains to be determined. We have found in immunolocalization studies that GIPC is not a component of CCPs in ARPE-19 cells (Supplementary Figure 3), whereas in other systems GIPC has been detected in CCPs (DeVries et al., 1998a,b; Lou et al., 2002). Therefore, GIPC may be recruited to endocytic vesicles earlier than myo6, or it may be recruited onto uncoated vesicles with myo6. Regardless of the timing of recruitment, GIPC, like myo6, departs once the vesicle fuses with the early endosome. This hypothesis is supported by the observation that although GIPC colocates extensively with myo6 in cell peripheries, it exhibits no colocation with EEA1-positive endosomes in untransfected cells (Supplementary Figure 4).
GIPC is not the only adapter protein shown to associate with the tail of myo6 using yeast-two hybrid methods. However, we did not find the other two identified adapters, Dab2 and Sap97 (Morris et al., 2002; Wu et al., 2002), associated with myo6 on nascent endocytic vesicles in ARPE-19 cells (unpublished data) suggesting that these two are not involved in myo6-dependent endocytosis in this system. Given the abundance of newly identified PDZ-domain containing adapters, it is conceivable that another adapter may facilitate myo6 binding to the uncoated vesicle, or that adapters may vary in a cell-type specific manner. Further studies will be required to determine the mechanism that regulates myo6's association with the uncoated vesicle.
Myo6's Role as a Motor for Endocytosis
Previous studies had suggested that only a splice variant of myo6 was involved in endocytosis and that the shorter version of myo6 used in these studies had no role in this process (Buss et al., 2001). In agreement with Buss et al., (2001), we found that myo6 constructs lacking the spliced insert did not target to CCPs (Figure 6). However, in direct contrast to their work, we found that the shorter form of myo6 protein was involved in a later step in endocytosis after CCV formation. We hypothesize that the effects of GFP-m6tail overexpression on transferrin uptake were missed by Buss et al. (2001) because they focused on the formation of CCVs rather than transport of components to the early endosome. Alternatively, myo6 without the splice insert may not have a role in vesicle trafficking in fibroblasts, compared with the epithelial cells used in our study.
We found that disruption of myo6 function selectively slowed trafficking of transferrin at the uncoated vesicle stage. Normally, the process of vesicle transport through the cell peripheries to the early endosome takes 1–2 min. After expression of GFP-M6tail, this transport took 10–20 min or more. We hypothesize that endocytosis was not totally blocked after GFP-M6tail overexpression because given sufficient time, even in the absence of a functional myosin motor, vesicles would encounter an endosome by diffusion and initiate fusion. Presumably, after fusion the endosome-associated motors would facilitate further internalization of the endocytosed receptors to the perinuclear sorting endosome.
Another explanation for the lack of a total endocytic block is that a secondary method for endocytic vesicle movement may be activated after disruption of myo6 function. This movement could be microtubule based or involve a different myosin motor, thereby allowing endocytosis to proceed. Of these two potential motor types, we believe that the induction of a microtubule-based motor is more likely. Based on our studies, myo6 is the only actin-binding protein present on the nascent endocytic vesicles, because overexpression of the tail domain alone was sufficient to release the endocytic vesicle from its actin tether and to cause trapping in peripheral regions.
Is Actin Acting in Endocytosis?
In polarized epithelial cells, apical endocytosis occurs in a clathrin-coated region found at the base of the microvilli (reviewed in Apodaca, 2001). Immediately below this clathrin-coated plasma membrane domain is the actin meshwork of the terminal web. We hypothesize that within actin-rich cell types such as epithelial cells, nascent endocytic vesicles must first traverse this dense actin network to reach the early endosome, implicating a myosin in transport. Myo6 is an excellent candidate for such a motor as in all epithelial cells studied to date, myo6 is found predominantly in the apical actin network at the base of microvilli, where it overlaps with the machinery for clathrin-mediated endocytosis (Hasson and Mooseker, 1994; Heintzelman et al., 1994; Buss et al., 2001; Biemesderfer et al., 2002). Therefore, our model is that myo6 is involved as an vesicle motor in systems where traversal of an actin-rich network is essential for fusion of nascent vesicles with the more centrally located early endosome. The cells used in our study are epithelial in origin, but they were not fully polarized for our study and did not have an apical microvillar domain. The peripheral regions contained a dense actin meshwork, however, akin to the terminal web, and it was traversal of vesicles through this region that was disrupted upon myo6tail domain overexpression.
Actin has not been shown unequivocably to have a role in endocytosis, because there is evidence both for and against its importance in the process (reviewed in Qualmann et al., 2000). Recent studies have implicated actin in the fission of clathrin-coated vesicles (Merrifield et al., 2002); however, studies that focus on this step have shown that drugs that depolymerize actin have little effect on endocytosis assayed in vitro in permeabilized fibroblasts or nonpolarized cells (Lamaze et al., 1997; Fujimoto et al., 2000).
We have implicated myo6 in a later stage in endocytosis, the movement of uncoated vesicles to the early endosome. In studies focusing on uptake to the early endosome, apical uptake by polarized epithelial cells was shown to be sensitive to actin-depolymerizing drugs. However, these results were not been seen in all polarized cell types (Gottlieb et al., 1993; Jackman et al., 1994; Shurety et al., 1996; Geckle et al., 1997). Based on our myo6 studies, we would argue that actin is a barrier to endocytosis and that myo6 is recruited to overcome that barrier. Ultimately, we hypothesize that myo6 would only play an accessory role in endocytic events that require traversal of uncoated vesicles from cell peripheries through actin meshworks. Further studies will be required to determine whether myo6 plays a role in basal-lateral uptake or uptake seen in nonpolarized cells.
Supplementary Material
Acknowledgments
We thank the many researchers that provided antibodies used in our study. We especially thank Sandy Schmid and Vivek Malhotra for critical reading of the manuscript and general support of our endeavors. This work was supported by Research Grant no. 6-FY02-150 and a Basil O'Connor Starter Scholar Research Grant from the March of Dimes Birth Defects Foundation.
Article published online ahead of print. Mol. Biol. Cell 10.1091/mbc.E02–11–0767. Article and publication date are available at www.molbiolcell.org/cgi/doi/10.1091/mbc.E02-11-0767.
Abbreviations used: CCP, clathrin-coated pit; CCV, clathrin-coated vesicle; EEA1, early endosome antigen 1; GFP, green fluorescent protein; PDZ, PSD-95/Dlg/ZO-1; R-Tsfn, rhodamine-conjugated transferrin; TsfnR, transferrin receptor.
References
- Apodaca, G. (2001). Endocytic traffic in polarized epithelial cells: role of the actin and microtubule cytoskeleton. Traffic 2, 149–159. [DOI] [PubMed] [Google Scholar]
- Avraham, K.B., Hasson, T., Sobe, T., Balsara, B., Testa, J., Copeland, N.G., and Jenkins, N.A. (1997). Characterization of human unconventional myosin-VI, a gene responsible for deafness in Snell's waltzer mice. Hum. Mol. Genet. 6, 1225–1231. [DOI] [PubMed] [Google Scholar]
- Avraham, K.B., Hasson, T., Steel, K.P., Kingsley, D.M., Russell, L.B., Mooseker, M.S., Copeland, N.G., and Jenkins, N.A. (1995). The mouse Snell's waltzer deafness gene encodes an unconventional myosin required for structural integrity of inner hair cells. Nat. Genet. 11, 369–374. [DOI] [PubMed] [Google Scholar]
- Biemesderfer, D., Mentone, S.A., Mooseker, M., and Hasson, T. (2002). Expression of myosin-VI within the endocytic pathway in the adult and developing proximal tubule. Am. J. Physiol. 282, F785–F794. [DOI] [PubMed] [Google Scholar]
- Breckler, J., Au, K., Cheng, J., Hasson, T., and Burnside, B. (2000). Novel myosin VI isoform is abundantly expressed in retina. Exp. Eye Res. 70, 121–134. [DOI] [PubMed] [Google Scholar]
- Bunn, R.C., Jensen, M.A., and Reed, B.C. (1999). Protein interactions with the glucose transporter binding protein GLUT1CBP that provide a link between GLUT1 and the cytoskeleton. Mol. Biol. Cell 10, 919–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buss, F., Arden, S.D., Lindsay, M., Luzio, J.P., and Kendrick-Jones, J. (2001). Myosin VI isoform localized to clathrin-coated vesicles with a role in clathrin-mediated endocytosis. EMBO J. 20, 3676–3684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buss, F., Kendrick-Jones, J., Lionne, C., Knight, A.E., Cote, G.P., and Luzio, J.P. (1998). The localization of myosin VI at the Golgi complex and leading edge of fibroblasts and its phosphorylation and recruitment into membrane ruffles of A431 cells after growth factor stimulation. J. Cell Biol. 143, 1535–1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cramer, L.P. (2000). Myosin VI: roles for a minus end-directed actin motor in cells. J. Cell Biol. 150, F121–F126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De La Cruz, E.M., Ostap, E.M., and Sweeney, H.L. (2001). Kinetic mechanism and regulation of myosin VI. J. Biol. Chem. 276, 32373–32381. [DOI] [PubMed] [Google Scholar]
- De Vries, L., Elenko, E., McCaffery, M., Fischer, T., Hubler, L., McQuistan, T., Watson, N., and Farquhar, M.G. (1998a). RGS-GAIP, a GTPase-activating protein for Gαi3 heterotrimeric G proteins, is located on clathrin-coated vesicles. Mol. Biol. Cell 9, 1123–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vries, L., Lou, X., Zhao, G., Zheng, B., and Farquhar, M.G. (1998b). GIPC, a PDZ domain containing protein, interacts specifically with the C terminus of RGS-GAIP. Proc. Natl. Acad. Sci. USA 95, 12340–12345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn, K.C., Aotaki-Keen, A.E., Putkey, F.R., and Hjelmeland, L.M. (1996). ARPE-19, a human retinal pigment epithelial cell line with differentiated properties. Exp. Eye Res. 62, 155–169. [DOI] [PubMed] [Google Scholar]
- Eskelinen, S., Kok, J.W., Sormunen, R., and Hoekstra, D. (1991). Coated endosomal vesicles: sorting and recycling compartment for transferrin in BHK cells. Eur. J. Cell Biol. 56, 210–222. [PubMed] [Google Scholar]
- Fujimoto, L.M., Roth, R., Heuser, J.E., and Schmid, S.L. (2000). Actin assembly plays a variable but not obligatory role in receptor mediated endocytosis in mammalian cells. Traffic 1, 161–171. [DOI] [PubMed] [Google Scholar]
- Geckle, M., Mildenberger, S., Freudinger, R., Schwerdt, G., and Silbernagl, S. (1997). Albumin endocytosis in OK cells: dependence on actin and microtubules and regulation by protein kinases. Am. J. Physiol. 272, F668–F677. [DOI] [PubMed] [Google Scholar]
- Ghosh, R.N., Gelman, D.L., and Maxfield, F.R. (1994). Quantification of low density lipoprotein and transferrin endocytic sorting in HEp2 cells using confocal microscopy. J. Cell Sci. 107, 2177–2189. [DOI] [PubMed] [Google Scholar]
- Gottlieb, T.A., Ivanov, I.E., Adesnik, M., and Sabatini, D.D. (1993). Actin microfilaments play a critical role in endocytosis at the apical but not the basolateral surface of polarized epithelial cell. J. Cell Biol. 120, 695–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanover, J.A., Willingham, M.C., and Pastan, I. (1984). Kinetics of transit of transferrin and epidermal growth factor through clathrin-coated membranes. Cell. 39, 283–293. [DOI] [PubMed] [Google Scholar]
- Hasson, T., Gillespie, P.G., Garcia, J.A., MacDonald, R.B., Zhao, Y.-D., Yee, A.G., Mooseker, M.S., and Corey, D.P. (1997). Unconventional myosins in inner-ear sensory epithelia. J. Cell Biol. 137, 1287–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasson, T., and Mooseker, M.S. (1994). Porcine myosin-VI: characterization of a new mammalian unconventional myosin. J. Cell Biol. 127, 425–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heintzelman, M.B., Hasson, T., and Mooseker, M.S. (1994). Multiple unconventional myosin domains in the intestinal brush border cytoskeleton. J. Cell Sci. 107, 3535–3543. [DOI] [PubMed] [Google Scholar]
- Hinck, L., Nathke, I.S., Papkoff, J., and Nelson, W.J. (1994). Dynamics of cadherin/catenin complex formation. J. Cell Biol. 125, 1327–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homma, K., Yoshimura, M., Saito, J., Ikebe, R., and Ikebe, M. (2001). The core of the motor domain determines the direction of myosin movement. Nature 412, 831–834. [DOI] [PubMed] [Google Scholar]
- Hopkins, C.R. (1983). Intracellular routing of transferrin and transferrin receptors in epidermoid carcinoma A431 cells. Cell 35, 321–330. [DOI] [PubMed] [Google Scholar]
- Jackman, M.R., Shurety, W., Ellis, J.A., and Luzio, J.P. (1994). Inhibition of apical but not basolateral endocytosis of ricin and folate in Caco-2 cells by cytochalasin D. J. Cell Sci. 107, 2547–2556. [DOI] [PubMed] [Google Scholar]
- Jin, M., and Snider, M.D. (1993). Role of microtubules in transferrin receptor transport from the cell surface to endosomes and the Golgi complex. J. Biol. Chem. 268, 18390–18397. [PubMed] [Google Scholar]
- Kachar, B., Battaglia, A., and Fex, J. (1997). Compartmentalized vesicular traffic around the hair cell cuticular plate. Hearing Res. 107, 102–112. [DOI] [PubMed] [Google Scholar]
- Lamaze, C., Fujimoto, L.M., Yin, H.L., and Schmid, S.L. (1997). The actin cytoskeleton is required for receptor mediated endocytosis in mammalian cells. J. Biol. Chem. 272, 20332–20335. [DOI] [PubMed] [Google Scholar]
- Lawlor, P., Marcotti, W., Rivolta, M.N., Kros, C.J., and Holley, M.C. (1999). Differentiation of mammalian vestibular hair cells from conditionally immortal, postnatal supporting cells. J. Neurosci. 19, 9445–9458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou, X., McQuistan, T., Orlando, R.A., and Farquhar, M.G. (2002). GAIP, GIPC and Gαi3 are concentrated in endocytic compartments of proximal tubule cells: putative role in regulating megalin's function. J. Am. Soc. Nephrol. 13, 918–927. [DOI] [PubMed] [Google Scholar]
- Mayor, S., Presley, J.F., and Maxfield, F.R. (1993). Sorting of membrane components from endosomes and subsequent recycling to the cell surface occurs by a bulk flow process. J. Cell Biol. 121, 1257–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrifield, C.J., Feldman, M.E., Wan, L., and Almers, W. (2002). Imaging actin and dynamin recruitment during invagination of single clathrin-coated pits. Nat. Cell Biol. 4, 691–698. [DOI] [PubMed] [Google Scholar]
- Morris, S.M., Arden, S.D., Roberts, R.C., Kendrick-Jones, J., Cooper, J.A., Luzio, J.P., and Buss, F. (2002). Myosin VI binds to and localizes with Dab2, potentially linking receptor-mediated endocytosis and the actin cytoskeleton. Traffic 3, 331–341. [DOI] [PubMed] [Google Scholar]
- Nichols, B.J., Kenworthy, A.K., Polishchuk, R.S., Lodge, R., Roberts, T.H., Hirschberg, K., Phair, R.D., and Lippincott-Schwartz, J. (2001). Rapid cycling of lipid raft markers between the cell surface and Golgi complex. J. Cell Biol. 153, 529–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qualmann, B., Kessels, M.M., and Kelly, R.B. (2000). Molecular links between endocytosis and the actin cytoskeleton. J. Cell Biol. 150, F111–F116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubino, M., Miaczynska, M., Lippe, R., and Zerial, M. (2000). Selective membrane recruitment of EEA1 suggests a role in directional transport of clathrin-coated vesicles to early endosomes. J. Biol. Chem. 275, 3745–3748. [DOI] [PubMed] [Google Scholar]
- Sheff, D., Pelletier, L., O'Connell, C.B., Warren, G., and Mellman, I. (2002). Transferrin receptor recycling in the absence of perinuclear recycling endosomes. J. Cell Biol. 156, 797–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shurety, W., Bright, N.A., and Luzio, J.P. (1996). The effects of cytochalasin D and phorbol myristate acetate on the apical endocytosis of ricin in polarised Caco-2 cells. J. Cell Sci. 109, 2927–2935. [DOI] [PubMed] [Google Scholar]
- Trischler, M., Stoorvogel, W., and Ullrich, O. (1999). Biochemical analysis of distinct rab5- and rab11-positive endosomes along the transferrin pathway. J. Cell Sci. 112, 4773–4783. [DOI] [PubMed] [Google Scholar]
- Wells, A.L., Lin, A.W., Chen, L.-Q., Safer, D., Cain, S.M., Hasson, T., Carragher, B.O., Milligan, R.A., and Sweeney, H.L. (1999). Myosin VI is a myosin that moves backwards. Nature 401, 505–508. [DOI] [PubMed] [Google Scholar]
- Wu, H., Nash, J.E., Zamorano, P., and Garner, C.C. (2002). Interaction of SAP97 with minus-end-directed actin motor myosin VI. Implications for AMPA receptor trafficking. J. Biol. Chem. 277, 30928–30934. [DOI] [PubMed] [Google Scholar]
- Yoshimura, M., Homma, K., Saito, J., Inoue, A., Ikebe, R., and Ikebe, M. (2001). Dual regulation of mammalian myosin VI motor function. J. Biol. Chem. 276, 39600–39607. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}