

Abstract
Kallikreins cleave plasma kininogens to release the bioactive peptides bradykinin (BK) or kallidin (Lys-BK). These peptides then activate widely disseminated B2 receptors with consequences that may be either noxious or beneficial. We used cultured cells to show that kallikrein can bypass kinin release to activate BK B2 receptors directly. To exclude intermediate kinin release or kininogen uptake from the culture medium, we cultured and maintained cells in medium entirely free of animal proteins. We compared the responses of stably transfected CHO cells that express human B2 receptors (CHO-B2) and cells that co-express angiotensin I-converting enzyme (ACE) as well (CHO AB). We found that BK (1 nM or more) and tissue kallikrein (1–10 nM) both significantly increased release of arachidonic acid beyond unstimulated or baseline levels. An enzyme-linked immunoassay for kinin established that kallikrein did not release a kinin from CHO cells. We confirmed the absence of kininogen mRNA with RT-PCR to rule out kininogen synthesis by CHO cells. Next, we tested an ACE inhibitor for enhanced BK receptor activation in the absence of kinin release and synthesized an ACE-resistant BK analogue as a control for these experiments. Enalaprilat (1μM) potentiated kallikrein (100 nM) in CHO AB cells but was ineffective in CHO B2 cells that do not bear ACE. We concluded that kallikrein activated B2 receptors without releasing a kinin. Furthermore, inhibition of ACE enhanced the receptor activation by kallikrein, an action that may contribute to the manifold therapeutic effects of ACE inhibitors.
Keywords: arachidonic acid, ACE inhibitor, kallidin, immuno-assay, ACE
INTRODUCTION
The functions of the kallikrein-kinin system have been widely studied since discovery of its components (1, 14, 19, 46). For example, decrease in urinary excretion of kallikrein is a characteristic feature of clinical or experimentally induced hypertension (36). Kinins release potent vasodilators, such as prostaglandins (PG), nitric oxide (NO), and endothelial-derived hyperpolarizing factor (EDHF) (3–5), which influence blood pressure and vessel tone, but they can cause pain and enhance capillary permeability in inflammation as well (3). The beneficial effects of angiotensin I-converting enzyme (ACE) (18, 40), or kininase II are attributed at least in part to prolongation of the half-life of bradykinin (BK) (6–8, 20–22) and potentiation of its effects on the B2 receptor (15, 33–35). Although plasma kininogen is the primary substrate of kallikreins, these serine proteases can cleave other proteins as well, such as Factor XII, the activator of prokallikrein (12, 41, 42). Generation of BK or kallidin (Lys-BK) by kallikrein results from a complex, multi-step enzymatic cascade starting with prokallikrein activation and ending with kinin release from plasma kininogen (12, 29, 32, 41, 42). The peptides then must evade cleavage by the kininases we characterized [such as carboxypeptidase N or M, as kininase I, and angiotensin I-converting enzyme (ACE) and neprilysin, as kininase II] in order to activate the B2 receptors (3, 16, 26, 43, 47). When carboxypeptidases N or M cleave the C-terminal arginine from either BK or Lys-BK (14, 44), the resulting des-Arg kinins become ligands for the second kinin receptor, B1 (3).
Because activation of B2 receptors has such important consequences in many diverse tissues, we considered that another pathway, a shunt, may activate B2 receptors independent of the complex enzymatic cascade required for kinin release (23–25). Similar dual back-up systems occur in numerous other biological reactions. Indeed, we found that B2 receptors can be activated directly by kallikreins and certain other serine proteases. The B2 BK receptors, similar to the PAR 1–4 receptors of thrombin or trypsin (38), belong to a G protein-linked, hepta-helical transmembrane receptor group that is activated by serine proteases. However, our experiments with cultured cells of various origins indicate that activation of the B2 BK receptor proceeds through a different mechanism than for PARs by thrombin or TRAP peptide ligand (24).
Culture of specialized mammalian cells usually requires a rich medium that contains 5-10% fetal bovine serum. Previously, we used cells washed free of added proteins (23, 25) or washed and serum-starved. Nevertheless, we wanted to exclude a possibility that proteases could activate B2 receptors by cleaving traces of adherent kininogen. Consequently, we have grown and maintained cells transfected with human BK B2 receptors, or B2 and ACE, in media entirely free of serum and animal proteins.
We measured release of [3H] arachidonic acid (AA) from Chinese hamster ovary (CHO) and found that kallikreins activated transfected human B2 BK receptors in the absence of any cell-bound kininogen. We also investigated the effects of ACE inhibitors, because these agents appear to potentiate kinin effects through crosstalk between ACE and B2 receptors (15, 33–35). The results from these experiments enabled us to exclude kinin release as a mechanism for kallikrein activation of B2 receptors. We showed that ACE inhibitors potentiated the actions of kallikrein by a mechanism independent of kinin inactivation also by employing an ACE resistant BK analogue in CHO cells expressing human ACE and B2 receptors.
Materials and Methods
Materials
CHO cells were purchased from American Type Culture Collection (Rockville, MD). The cDNA of human B2 receptor was obtained from Dr. K. Jarnigan, (Syntex Co., Palo Alto, CA) and cDNA of human ACE was from Prof. P. Corvol (College de France, Paris). Superfect was from Qiagen (Valencia, CA), geneticin (G418) was from Invitrogen (Carlsbad, CA), and [3H] bradykinin was from Amersham (Piscataway, NJ). CHO Protein-free, animal-component-free medium for attached cells (CHO PF-AF) was purchased from Sigma Aldrich (St. Louis, MO). [3H] arachidonic acid was purchased from American Radiolabeled Chemicals (St. Louis, MO). BK, porcine pancreatic kallikrein, HOE-140, fatty acid free BSA, protease inhibitor cocktail and other reagents were from Sigma Aldrich, and human plasma kallikrein was purchased from Enzyme Research Labs (South Bend, IN). Crystalline BSA and crystalline aprotinin were from Calbiochem (San Diego, CA). The enzyme-linked immunoassay (ELISA) high sensitivity kit for bradykinin was purchased from Bachem (King of Prussia, PA). RNA STAT-60 kit for total RNA isolation was from TEL-TEST (Friendswood, TX). Hamster liver was from PEL-FREEZ (Rogers, AR), the rat liver mRNA sample was a gift from D. Riley, Dept. of Anesthesiology, University of Illinois at Chicago. SuperScript™ one-step RT-PCR with Platinum Taq kit was purchased from Invitrogen (Carlsbad, CA). We synthesized a BK analogue by increasing its size at the N-terminus (11, 39), dansylating both the α- and ɛ-NH2 of Lys1 [didansyl-Lys-BK (didnasyl-kallidin; DidnsKD)] that rendered it resistant to ACE (11).
Cell culture
Initially CHO cells were grown in 100 mm Petri dishes in Ham’s F-12 culture medium (Cellgro) supplemented with antibiotic and 10% fetal bovine serum under 5% CO2 and a water-saturated environment. Cells were routinely subcultured using trypsin-EDTA for detachment and transfer. CHO cells stably transfected with either bradykinin B2 receptor or both ACE and B2 receptor were adapted to culture in protein-free, animal component-free medium for attached cells (Sigma). The medium was supplemented with 200 mM L-glutamine, and the cultures were maintained at 37°C under 5% CO2 in a water-saturated environment. The cells were subcultured by rinsing with the same serum-free medium, then they were scraped and transferred to 100 mm Petri dishes in fresh medium. For measurements of AA release and other experiments, cells were cultured in CHO PF-AF medium for at least 8–10 passages to exclude artifacts from prior exposure to fetal calf serum before transfer into 12-well plates for experiments.
Transfection with human B2 receptor cDNA and human ACE cDNA
pcDNA3 plasmid containing human BK B2 receptor was used to transfect CHO cells (24, 25). To obtain CHO cells co-expressing both human ACE and bradykinin B2 receptor, the cells were first transfected with human ACE pcDNA6 plasmid, and individual clones were screened for ACE activity (11, 34). Clones with high ACE activity were then transfected with human BK B2 receptor cDNA as described above (CHO AB).
[3H]AA release
Experiments were performed essentially as previously described (11, 24 ). Briefly, the cells were grown to confluence in a 12-well dish. The medium was replaced with 1 ml of culture medium containing 0.5 mCi/ml of [3H]AA, and cells were loaded for 16 h at 37°C. After three washings with incubation medium (Ham’s F-12 medium containing 0.1% albumin) the cells were incubated for 10 min at 37°C in the presence of enalaprilat (1 μM). Then kallikrein, DidnsKD or other agonist was added, and cells were incubated for additional 20 min at 37°C. In parallel samples we added a B2 receptor blocker, HOE 140, prior to addition of the test reagent. After completion of the incubation, the medium was removed and the [3H]AA content was measured in a scintillation counter. To trap released [3H] AA and block its re-uptake, we added albumin that adsorbed the [3H] AA to be counted. We have used in some experiments defatted bovine serum albumin, crystallized albumin in others, but the results were the same. Neither preparation had contaminating kininogen.
ELISA
Enzyme immunoassays used a high sensitivity kit for BK (Bachem) according to the manufacturer’s protocol. Briefly, 50 μl of standard solutions or test samples were added to immunoplate multiwells, then 25 μl of each primary antisera and biotinylated peptide solution were added, and the plates were incubated for 2 h at room temperature with mild agitation. Then the plates were washed 5 times, and 100 μl of diluted streptavidin-conjugated horse radish peroxidase solution was added to each well. After 60 min incubation at room temperature the immunoplates were washed 5 times, and 100 μl of 3, 3’,5, 5’-tetramethyl benzidine.diHCl (TMB) solution was added to each well. After further 20 min incubation at room temperature, the reaction was stopped with 100 μl of 2N HCl. Absorbance was read at 450 nm, using 100 μl of TMB solution and 100 μl 2N HCl as a blank control.
Radioligand binding
Cells were grown in 24-well plates. In [3H]BK saturation binding experiments cells were washed with serum free Ham’s F-12 medium and increasing concentrations of [3H]BK were added to wells (24). After 1 h incubation at 4°C cells were washed 3 times with serum-free Ham’s F-12 medium, solubilized in 0.5 ml of a solution containing 0.1 M NaOH, 0.1 M NaHCO3 and 1% SDS, and counted. Non-specific binding was determined in the presence of 10 μM unlabelled BK, and specific binding was calculated as the difference between total and non-specific binding. CHO AB/PFAF cells were pre-incubated with ACE inhibitor enalaprilat (1 μM) for 20 min before adding BK.
In radioligand displacement experiments approximately 1 nM [3H]BK was competing for the binding with increasing concentrations of Di-dns-Kd (ranging from 10−12 to 10−4 M). As control homologous displacement using unlabelled BK was performed.
Total RNA/mRNA isolation
We used RNA STAT-60 isolation reagent according to the manufacturer’s protocol. The cells were lysed using RNA STAT-60 and homogenized, then chloroform was added, and the samples were vortexed and held at room temperature for 2–3 min. All samples were centrifuged at 12,000 g for 15 min at 4°C, and the aqueous phase was transferred to a fresh tube and mixed with isopropanol. After 10 minutes incubation at room temperature, the samples were centrifuged at 12,000 g for 10 min at 4°C. The resulting RNA pellet was washed with 75% ethanol by vortexing and centrifugation at 7,500 g for 5 min at 4°C. The samples were then air-dried briefly and dissolved in RNase-free water.
Reverse transcription polymerase chain reaction (RT-PCR)
We used SuperScript™ one-step RT-PCR with Platinum Taq kit (Invitrogen) according to the manufacturer’s protocol. PCR primers for kininogen were kng3 (5’gcccagagctgaaggagg) and kng4 (5’catgtacacgttagcattgcag), and GAPDH1 (5’cgaccccttcattgacctc) and GAPDH2 (5’ctccacgacatactcagcacc) for GAPDH.
Statistical analysis
Means and standard errors were calculated for the experiments and statistical significance of differences between means was tested by a paired t-test (Microsoft Excel).
Results
CHO cells in serum-free medium
CHO cells stably transfected with human BK B2 (CHO-B2) receptors were grown and maintained in medium entirely free of animal proteins for 8 or more passages. Activation of the B2 receptor on these cells released [3H]AA, presumably by a mechanism involving a Gαi protein coupled to the receptor, which in turn activates phospholipase A2 (4). Taking basal release as 1.0, (Figure 1) tissue kallikrein (1 and 10 nM; Panel A) increased the amount of released [3H]AA by 1.9 ± 0.3 or 2.9 ± 0.4 fold (n ≥ 4). All experiments were done in triplicate, and in a pilot experiment, human plasma kallikrein was as active as tissue kallikrein. BK (0.1–1 nM; Panel B) also significantly enhanced the release of [3H]AA, increasing it 1.8 ± 0.2 and 3.3 ± 0.4 fold beyond the basal level. The B2 receptor blocker HOE-140 (0.5 μM) almost completely abolished the activation of the receptor by either BK or kallikrein.
Fig. 1.

Release of [3H] arachidonic acid (AA) from CHO B2/PF-AF cells by tissue kallikrein (KLK) or bradykinin (BK). The cells were stably transfected with human BK B2 receptors, cultured and maintained in entirely serum-free medium. Incubation with tissue kallikrein (1 and 10 nM; Panel A) and BK (0.1 and 1 nM; Panel B) for 30 min stimulated the release of [3H] AA from previously labeled cells. Pretreatment of cells with HOE 140 (0.5 μM) inhibited both kallikrein and BK-induced release of [3H] AA. Data are relative amount of [3H]AA released, with basal (spontaneous release) = 1. Data are mean ± SEM from 4 or more separate experiments done in triplicates. Significant differences (p<0.001) indicated by***.
ELISA
To rule out any possible release of BK by kallikrein from kininogen expressed by or adhering to CHO-B2 cells in protein-free medium we used an ELISA for BK. The linear portion of the assay standard curve ranged from 2 x 10−9 to 1 x 10−10 M BK in >4 experiments. Tissue kallikrein (10 nM) was tested first by adding it to a medium containing 10% fetal bovine serum. It released bradykinin in excess of 10−8 M in 30 min. Serum-free culture medium as well as conditioned medium from kallikrein-treated cells were assayed along with appropriate reagent controls. We measured only a basal value 0.2 ng/ml from protein-free medium sampled both in presence or absence of CHO-B2 cells (data not shown); these findings confirm the absence of kininogen on cell membranes or in the culture medium. Because immunoreactivity of the medium collected from kallikrein-treated (10 nM) cells did not exceed the reagent baseline, we concluded that kallikrein failed to release kinin from either from cell-free medium or CHO-B2 cells and that the enzyme activated the BK B2 receptor by another mechanism.
Lack of kininogen expression
We considered the possibility that CHO cells constitutively express kininogen, even when grown in protein-free medium, so we used RT-PCR to test for kininogen expression. Since the exact sequence of hamster kininogen was not available, primers were synthesized from identical regions found after comparison of sequences from rat (Rattus norvegicus) kininogen 1 and mouse (Mus musculus) kininogen 2 mRNA (obtained from GenBank). We also synthesized primers based on identical regions of rat and mouse glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA sequences for RNA quality control. The total RNA isolated from CHO B2 cells cultured in serum-free medium was compared to total RNA from rat and hamster liver (Fig. 2). RNA was isolated with RNA STAT-60 and quantified by measuring UV-absorbance at 260 nm. As a control, a kininogen cDNA fragment of the expected size (approx. 247 bp) was amplified from 50 ng rat liver RNA after 35 cycles (not shown). Samples not subjected to RT-PCR had no detectable signal. Similarly, with hamster liver RNA samples, strong bands of kininogen cDNA appeared with RT-PCR after 35 cycles using 50 or 500 ng RNA (Fig. 2A). In contrast, the same amount of CHO B2 cell RNA, either with or without reverse transcription, yielded no detectable bands (Fig. 2A). The 200 bp cDNA fragments of GAPDH were amplified by RT-PCR from either the CHO B2 cell RNA preparation or the rat liver RNA preparation (Fig. 2B).
Fig. 2.
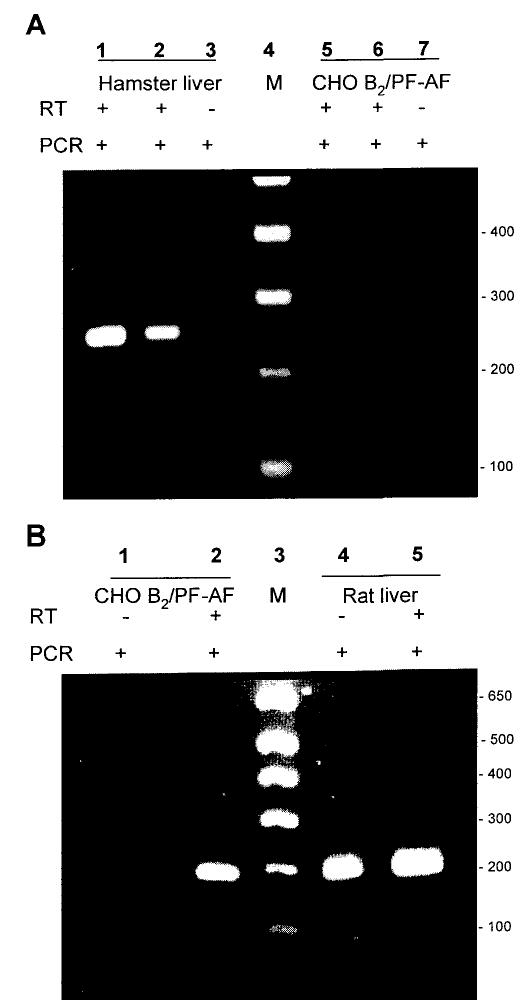
Kininogen mRNA is detected in hamster liver, but not in CHO B2/PF-AF cells. Total RNA was reverse-transcribed (RT), and the resulting cDNA was amplified by PCR and separated by agarose gel electrophoresis.
A: PCR products of total RNA samples from hamster liver and CHO B2/PF-AF cells amplified using kininogen primers. Lane 1 = hamster liver (500 ng); lane 2 = hamster liver (50 ng); lane 3 = hamster liver (500 ng); lane 4 = marker; lane 5 = CHO B2/PF-AF cells (500 ng); lane 6 = CHO B2/PF-AF cells (50 ng); lane 7 = CHO B2/PF-AF cells (500 ng).
B: PCR products of total RNA samples from rat liver and CHO B2/PF-AF cells amplified using GAPDH primers. Lanes 1 and 2 = CHO B2/PF-AF cells; lane 3 = marker; lanes 4 and 5 = rat liver.
Kininogens originate mainly from the liver in mammalian species (3). Thus, the rat and hamster livers were strongly positive, showing appreciable amounts of mRNA, but cultured CHO cells lacked kininogen mRNA, confirming the results of our experiments (23, 25) that kallikrein indeed activates the B2 BK receptors without releasing kinin.
Effect of ACE inhibitor
We previously reported that inhibitors of ACE potentiated the effects of BK B2 receptor agonists in cultured cells and organs ex vivo (15, 33–35) by a mechanism independent of blocking BK inactivation. To determine how ACE inhibitors potentiate kinin receptor agonists, we stably transfected CHO cells bearing human B2 BK receptors with human ACE and adapted them to grow in serum-free medium (CHO AB/PFAF cells). These co-transfected CHO cells (CHO AB/PAF) were less sensitive to B2 receptor agonists than CHO B2 cells. This in spite of both cell lines had very similar Bmax and Kd values. These were for CHO B2 /PFAF cells: Bmax 214 ± 62 fmol/106 cells, Kd 17.7 ± 5.3 nM and for CHO AB/PFAF cells Bmax 174 ± 70 fmol/106 cells, Kd 11.3 ± 5.7 nM (n = 3). Figure 3 shows that kallikrein (100 nM) increased release of [3H] AA 1.73 ± 0.14 fold over the basal level and the ACE inhibitor enalaprilat (1 μM) significantly enhanced it, augmenting release of [3H] AA 5.6 ± 0.47 fold over basal level. This potentiation was blocked when B2 receptor antagonist HOE 140 (0.5 μM) was added to the reaction mixture (Fig 3). We also used the ACE-resistant peptide, DidnsKD, as a positive control. This B2 receptor agonist 97% resists degradation by ACE (11) because dansylation of both α and ɛ NH2 groups of Lys1 blocks hydrolysis by ACE, which cleaves peptides only of restricted size (13, 16, 43). In displacing labeled BK, this B2 agonist had a Ki = 1.96 ± 0.2 μM at 4°C. DidnsKD (1 μM) stimulated [3H]AA release 3.07 ± 0.2 over the basal level. Addition of enalaprilat enhanced [3H]AA release by DidnsKD even further to 6.3 ± 0.9 fold (Fig. 3) and it was inhibited by HOE 140 (0.5 μM). Enalaprilat potentiates only active kallikrein, when inhibited by crystalline aprotinin (1 μM) (Fig 4A), it was inactive, and enalaprilat had no effect.
Fig. 3.

Effect of enalaprilat on [3H]AA release by either kallikrein or ACE resistant BK analogue in CHO AB/PF-AF cells. Cells expressing both BK B2 receptor and ACE were grown in serum-free medium as described in Methods. Pretreatment of cells with enalaprilat (1 μM) for 10 min significantly enhanced release of [3H]AA in response to either kallikrein (100 mM) or DidnsKD (1 μM). HOE 140 (0.5 μM) blocked activation of B2 receptor by either agonist. Data are relative release of [3H] AA above basal (spontaneous) release and are means ± SEM of 3–6 separate experiments done in triplicates. Significant differences (p<0.001) indicated by***.
Fig. 4.

Effects of aprotinin and lack of ACE. Conditions and ordinate as in Fig. 1. A: release of [3H]AA in CHO AB/PF-AF cells after pretreatment of cells with enalaprilat (EPT 1 μM), prior to addition kallikrein (KLK 100 nM) treated with crystalline aprotinin (1 μM) for 20 min. Data are means ± SEM of 4–5 separate experiments. B: release of [3H]AA in CHO B2/PF-AF cells that lack ACE but express BK B2 receptors. Cells were pretreated with EPT (1 μM) for 10 min before to addition of either KLK (10 nM) or BK (1 nM) and incubated for 20 min. Data are means ± SEM from 4 separate experiments done in triplicates. EPT did not potentiate [3H] AA release when kallikrein was inhibited or ACE was not expressed in the cells.
Lack of ACE expression
To show that the ACE inhibitor is effective only when both B2 receptors and ACE are expressed, we tested CHO B2 cells which have no ACE. Also, CHO B2 cells lack B1 receptors that might be activated by an ACE inhibitor (28). Figure 4B shows that, as previously noted (15), enalaprilat was ineffective in the absence of ACE; it failed to enhance [3H]AA release by either BK or kallikrein (n ≥ 3). We concluded that kallikrein must be active to act as a B2 receptor agonist (25) and to be potentiated by ACE inhibitor requires the expression of ACE.
Finally, we used an ELISA to see if enalaprilat might increase the putative presence of BK locally by blocking its inactivation by CHO AB/PFAF cells. However, cells pretreated with enalaprilat (1 μM) prior to stimulation by kallikrein did not release BK (results not shown). BK was absent in the samples with or without added enalaprilat, allowing us to rule out BK release or protection from degradation as a cause of enhanced AA release.
DISCUSSION
We found that cells grown and maintained in protein-free medium can be activated by kallikrein, despite the fact that they were never been exposed to plasma kininogen, the precursor of kinins. Human plasma has two kininogens. These proteins, high and low molecular weight kininogen, are products of the same gene; both are synthesized in the liver and released into circulation (3, 14). Plasma kallikrein cleaves the high molecular weight substrate, while tissue kallikrein hydrolyzes both proteins. When blood coagulates, much of the plasma kininogen is metabolized, which is likely what happens to the fetal bovine serum commonly used to supplement media for cultured cells.
Nevertheless, we eliminated the possibilities that in our experiments kallikrein activated the B2 receptors of cultured cells, because of the presence of kininogen. Initially, we used serum-starved CHO cells to exclude the possibility of an uptake of kininogen from the medium. Next, although Zn2+ ions facilitate adsorption of kininogen from the medium to the cell membrane (29, 41, 42), we demonstrated previously that activation of B2 receptors by kallikrein was unaffected in the absence of zinc (25). Here, we report that cells were cultured in entirely serum-free medium, when we applied an ELISA to measure putative kinin release from these cells, the results were negative. Finally, to eliminate the possibility that the CHO cells could synthesize kininogen, we showed that the cells we used lack kininogen mRNA. Taken together, these findings effectively eliminate participation of kinin release by kallikrein as a mechanism of B2 BK receptor activation in our studies.
We measured [3H] AA release from CHO cells by BK or kallikrein as an indication of receptor activation and showed that HOE 140, a B2 receptor blocker, inhibited release by either agonist.
ACE inhibitors are used successfully in therapy of a variety of cardiovascular diseases (18, 20, 22, 40). At least part of the effects of an ACE inhibitor may depend on actions beyond blocking the inactivation of BK by ACE, specifically the direct potentiation of BK at its B2 receptors, (33–35). Although the first clinically tested ACE inhibitor was a peptide derived from the so-called bradykinin potentiating factor in snake venom (22), bradykinin potentiation and kininase, ACE inhibition did not go parallel (15). Indeed, they differed a great deal in a series of synthetic peptide congeners (37); peptides potentiated also in absence of ACE expression. Synthetic ACE inhibitors can enhance the actions of BK on its B2 receptors by a crosstalk between the receptor and ACE (15). For this reason, we wondered if ACE inhibitors would potentiate kallikrein effects on the B2 BK receptors in a similar manner. To confirm that potentiation occurred in the absence of kinin generation and independent of BK inactivation, we synthesized a BK analogue (DidnsKD) with a modified N-terminus. This structural change rendered the peptide almost entirely (97%) resistant to ACE (11). ACE inhibitor can enhance ACE and cyclooxygenase 2 expression in endothelial cells. This involves the phosphorylation of Ser 1270 of the C-terminal cytosolic sequence of ACE (30, 31). Our acute experiments would unlikely involve protein synthesis, which may take 24–36 h (30, 31). ACE inhibitor potentiated bradykinin and resensitized B2 receptors to the peptide even when in a mutated, truncated ACE, the C-terminal 19 amino acids of the cytosolic portions of human ACE were deleted (33), thus very likely in absence of phosphorylation.
Enalaprilat had no effect on release of [3H] AA from labeled CHO-B2 cells in serum-free medium. These cells were transfected to express B2 receptors but lack ACE. In contrast, adding the ACE inhibitor to CHO cells that stably express both human ACE and B2 receptors potentiated the effect of kallikrein and an ACE-resistant BK analogue. These findings suggest that an additional contribution to the multiple beneficial effects of ACE inhibitors could include the enhanced activation of B2 receptors by kallikrein.
We reported that kallikreins (tissue or plasma) activate B2 receptors differently than BK. For example, kallikreins and BK both desensitized B2 receptors, but there was no cross-desensitization (25). Experiments with a carboxypeptidase BK inactivator and site directed mutagenesis underscored differences between the protease and the peptide B2 agonists. Our investigations confirm that ACE inhibitors act beyond blocking kinin inactivation by ACE. Enalaprilat enhanced both the ACE-resistant BK analogue and kallikrein activity in an apparently kinin-free system. Because ACE and B2 can form heterodimers (33) and can colocalize closely on cell membranes (Z. Chen, F. Tan, E.G. Erdös, P.A. Deddish, to be published), our findings suggest that ACE inhibitors may induce conformational changes via ACE, acting as indirect allosteric enhancers (9) of B2 BK receptor agonists. Once the receptor is activated, the mechanism presumably follows a common path, namely the coupling Gαi protein then to phospholipase A2 activation.
Over the long history of kallikrein research, some experimental results could not be explained by kallikreins acting exclusively on plasma kininogen to release a kinin (45). For example, rat urinary kallikrein or trypsin applied at short intervals stimulated the isolated estrogen-primed rat uterus repeatedly (2, 10, 17). This repetitive action on the isolated organ cannot be due to a continuous replenishment of kininogen.
Furthermore, kallikreins release one molecule of kinin from each kininogen molecule, and the liberated kinins are rapidly cleaved by kininases. Kallidin (Lys-BK) is converted to BK by the removal of the N-terminal Lys (16). The half-life of injected BK is only 15-20 sec, indicating that kinin release and action would necessarily occur within a discrete, local environment. In this regard, kinins function as paracrine agents (8). Kinin generation requires a very rapid reaction where possibly only a fraction of the kininogen substrate is cleaved. If only 10% of the plasma protein kininogen substrate were hydrolyzed, high or low molecular weight kininogen with a molecular mass of approximately 50 or 100 kDa would need to be in 500 to 1000-fold excess to release a single molecule of BK (1 kDa) near to a receptor. This assumption then raises the question as to whether this weight ratio would always be obtained at the outer surface of cellular plasma membranes.
The direct activation of the human B2 peptide receptor (25, 27) offers an alternate pathway of activation for this multi-step enzymatic processes. We have identified such a pathway in cultured cells, and we believe that this mechanism may explain some of the phenomena associated with the therapeutic actions of ACE inhibitors in vivo.
Acknowledgments
This work was supported by National Institutes of Health (NIH) Grant HL36473 and HL68580. The authors would like to thank Dr. Peter A. Deddish of UIC College of Medicine for his help and suggestions.
References
- 1.Beraldo WT, Andrade SP. Discovery of bradykinin and the kallikrein-kinin system. In: Farmer SG, editor. The Kinin System. San Diego, CA: Academic Press Ltd; 1997. pp. 1–8. [Google Scholar]
- 2.Beraldo WT, Araujo RL, Mares-Guia M. Oxytocic esterase in rat urine. Am J Physiol. 1966;211:975–980. doi: 10.1152/ajplegacy.1966.211.4.975. [DOI] [PubMed] [Google Scholar]
- 3.Bhoola KD, Figueroa CD, Worthy K. Bioregulation of kinins: kallikreins, kininogens, and kininases. Pharmacol Rev. 1992;44:1–80. [PubMed] [Google Scholar]
- 4.Campbell WB, Halushka PV. Lipid-derived autacoids: eicosanoids and platelet-activating factor. In: Hardman JC, Limberd LE, Molinoff PB, Ruddon RW, Gilman AG, editors. Goodman & Gilman's The Pharmacological Basis of Therapeutics. 9th ed. New York: McGraw-Hill; 1996. pp. 602–616. [Google Scholar]
- 5.Campbell WB, Harder DR. Endothelium-derived hyperpolarizing factors and vascular cytochrome P450 metabolites of arachidonic acid in the regulation of tone. Circ Res. 1999;84:484–488. doi: 10.1161/01.res.84.4.484. [DOI] [PubMed] [Google Scholar]
- 6.Carretero OA, Miyazaki S, Scicli AG. Role of kinins in the acute antihypertensive effect of the converting enzyme inhibitor, captopril. Hypertension. 1981;3:18–22. doi: 10.1161/01.hyp.3.1.18. [DOI] [PubMed] [Google Scholar]
- 7.Carretero OA, Scicli AG. The kallikrein-kinin system as a regulator of cardiovascular and renal function. In: Laragh JH, Brenner BM, editors. Hypertension: Pathophysiology, Diagnosis, and Management. 2nd ed. New York, NY: Raven Press Publishers; 1995. pp. 983–999. [Google Scholar]
- 8.Carretero OA, Scicli AG, Maitra SR. Role of kinins in the pharmacological effects of converting enzyme inhibitors. In: Horovitz Z, editor. Angiotensin Converting Enzyme Inhibitors. Mechanisms of Action and Clinical Implications. Baltimore: Urban & Schwarzenberg; 1981. pp. 105–121. [Google Scholar]
- 9.Changeux JP, Edelstein SJ. Allosteric mechanisms of signal transduction. Science. 2005;308:1424–1428. doi: 10.1126/science.1108595. [DOI] [PubMed] [Google Scholar]
- 10.Chao J, Buse J, Shimamoto K, Margolius HS. Kallikrein-induced uterine contraction independent of kinin formation. Proc Natl Acad Sci U S A. 1981;78:6154–6157. doi: 10.1073/pnas.78.10.6154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen Z, Tan F, Erdös EG, Deddish P. Hypertension. 2005. Hydrolysis of angiotensin peptides by human ACE and the resensitization of B2 kinin receptors. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Colman RW. Handbook of Proteolytic Enzymes. 2nd ed. San Diego, CA: Academic Press; 2004. Plasma prekallikrein and kallikrein; pp. 1644–1651. [Google Scholar]
- 13.Corvol P, Eyries M, Soubrier F. Peptidyl-dipeptidase A/angiotensin I-converting enzyme. In: Barrett AJ, Rawlings ND, Woessner JF, editors. Handbook of Proteolytic Enzymes. 2nd ed. San Diego, CA: Academic Press; 2004. pp. 337–346. [Google Scholar]
- 14.Erdös EG, editor. Bradykinin, Kallidin and Kallikrein. Handbook of Experimental Pharmacology. suppl to XXV. Berlin: Springer-Verlag; 1979. pp. 1–817. [Google Scholar]
- 15.Erdös EG, Deddish PA, Marcic BM. Potentiation of bradykinin actions by ACE inhibitors. Trends Endocrinol Metab. 1999;10:223–229. doi: 10.1016/s1043-2760(99)00156-3. [DOI] [PubMed] [Google Scholar]
- 16.Erdös EG, Skidgel RA. Metabolism of bradykinin by peptidases in health and disease. In: Farmer SG, editor. The Kinin System. London: Academic Press; 1997. pp. 111–141. [Google Scholar]
- 17.Erdös EG, Tague LL, Miwa I. Kallikrein in granules of the submaxillary gland. Biochem Pharmacol. 1968;17:667–674. doi: 10.1016/0006-2952(68)90003-8. [DOI] [PubMed] [Google Scholar]
- 18.Frohlich ED. Left ventricular enlargement in hypertension: it's not only LVH. In: Giles TD, editor. Angiotensin-Converting Enzyme (ACE): Clinical and Experimental Insights. Fort Lee, NJ: Health Care Communications, Inc.; 2001. pp. 189–195. [Google Scholar]
- 19.Gaddum JH. Polypeptides Which Stimulate Plain Muscle. London: E. & S. Livingston Ltd; 1955. pp. 1–140. [Google Scholar]
- 20.Gainer JV, Morrow JD, Loveland A, King DJ, Brown NJ. Effect of bradykinin-receptor blockade on the response to angiotensin-converting-enzyme inhibitor in normotensive and hypertensive subjects. N Engl J Med. 1998;339:1285–1292. doi: 10.1056/NEJM199810293391804. [DOI] [PubMed] [Google Scholar]
- 21.Gavras I, Gavras H. Hypertension, vasoactive peptides and coagulation factors. J Hypertens. 2004;22:1091–1092. doi: 10.1097/00004872-200406000-00007. [DOI] [PubMed] [Google Scholar]
- 22.Gavras I, Gavras H. The use of ACE inhibitors in hypertension. In: Kostis JB, DeFelice EA, editors. Angiotensin Converting Enzyme Inhibitors. New York: Alan R. Liss, Inc.; 1987. pp. 93–122. [Google Scholar]
- 23.Hecquet C, Becker RP, Tan F, Erdös EG. Kallikreins when activating bradykinin B2 receptor induce its redistribution on plasma membrane. Int Immunopharmacol. 2002;2:1795–1806. doi: 10.1016/s1567-5769(02)00176-5. [DOI] [PubMed] [Google Scholar]
- 24.Hecquet C, Biyashev D, Tan F, Erdös EG. Am J Physiol - Heart & Circ. 2005. Positive cooperativity between the thrombin and bradykinin B2 receptors enhances arachidonic acid release. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hecquet C, Tan F, Marcic BM, Erdös EG. Human bradykinin B2 receptor is activated by kallikrein and other serine proteases. Mol Pharmacol. 2000;58:828–836. doi: 10.1124/mol.58.4.828. [DOI] [PubMed] [Google Scholar]
- 26.Hess JF. Molecular pharmacology of kinin receptors. In: Farmer SG, editor. The Kinin System. San Diego: Academic Press, Ltd.; 1997. pp. 45–55. [Google Scholar]
- 27.Hess JF, Borkowski JA, Macneil T, Stonesifer GY, Fraher J, Strader CD, Ransom RW. Differential pharmacology of cloned human and mouse B2 bradykinin receptors. Mol Pharmacol. 1994;45:1–8. [PubMed] [Google Scholar]
- 28.Ignjatovic T, Tan F, Brovkovych V, Skidgel RA, Erdös EG. Novel mode of action of angiotensin I converting enzyme inhibitors. Direct activation of bradykinin B1 receptor. J Biol Chem. 2002;277:16847–16852. doi: 10.1074/jbc.M200355200. [DOI] [PubMed] [Google Scholar]
- 29.Kaplan AP, Joseph K, Silverberg M. Pathways for bradykinin formation and inflammatory disease. J Allergy Clin Immunol. 2002;109:195–209. doi: 10.1067/mai.2002.121316. [DOI] [PubMed] [Google Scholar]
- 30.Kohlstedt K, Brandes RP, Muller-Esterl W, Busse R, Fleming I. Angiotensin-converting enzyme is involved in outside-in signaling in endothelial cells. Circ Res. 2004;94:60–67. doi: 10.1161/01.RES.0000107195.13573.E4. [DOI] [PubMed] [Google Scholar]
- 31.Kohlstedt K, Busse R, Fleming I. Signaling via the angiotensin-converting enzyme enhances the expression of cyclooxygenase-2 in endothelial cells. Hypertension. 2005;45:126–132. doi: 10.1161/01.HYP.0000150159.48992.11. [DOI] [PubMed] [Google Scholar]
- 32.Mahdi F, Shariat-Madar Z, Schmaier AH. The relative priority of prekallikrein and factors XI/XIa assembly on cultured endothelial cells. J Biol Chem. 2003;278:43983–43990. doi: 10.1074/jbc.M304239200. [DOI] [PubMed] [Google Scholar]
- 33.Marcic B, Deddish PA, Skidgel RA, Erdös EG, Minshall RD, Tan F. Replacement of the transmembrane anchor in angiotensin I-converting enzyme (ACE) with a glycosylphosphatidylinositol tail affects activation of the B2 bradykinin receptor by ACE inhibitors. J Biol Chem. 2000;275:16110–16118. doi: 10.1074/jbc.M909490199. [DOI] [PubMed] [Google Scholar]
- 34.Marcic BM, Deddish PA, Jackman HL, Erdös EG, Tan F. Effects of the N-terminal sequence in the N-domain of ACE on the properties of its C-domain. Hypertension. 2000;36:116–121. doi: 10.1161/01.hyp.36.1.116-a. [DOI] [PubMed] [Google Scholar]
- 35.Marcic BM, Erdös EG. Protein kinase C and phosphatase inhibitors block the ability of angiotensin I-converting enzyme inhibitors to resensitize the receptor to bradykinin without altering the primary effects of bradykinin. J Pharmacol Exp Ther. 2000;294:605–612. [PubMed] [Google Scholar]
- 36.Margolius HS. The kallikrein-kinin system and the kidney. Ann Rev Physiol. 1984;46:309–326. doi: 10.1146/annurev.ph.46.030184.001521. [DOI] [PubMed] [Google Scholar]
- 37.Mueller S, Gothe R, Siems WD, Vietinghoff G, Paegelow I, Reissmann S. Potentiation of bradykinin actions by analogues of the bradykinin potentiating nonapeptide BPP9alpha. Peptides. 2005;26:1235–1247. doi: 10.1016/j.peptides.2005.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.O'Brien PJ, Molino M, Kahn M, Brass LF. Protease activated receptors: theme and variations. Oncogene. 2001;20:1570–1581. doi: 10.1038/sj.onc.1204194. [DOI] [PubMed] [Google Scholar]
- 39.Odya CE, Levin Y, Erdös EG, Robinson CJG. Soluble dextran complexes of kallikrein, bradykinin, and enzyme inhibitors. Biochem Pharmacol. 1978;27:173–179. doi: 10.1016/0006-2952(78)90297-6. [DOI] [PubMed] [Google Scholar]
- 40.Pfeffer M. Angiotensin-converting enzyme (ACE) inhibitors in the prevention of cardiovascular disease: clinical evidence. In: Giles TD, editor. Angiotensin-Converting Enzyme (ACE): Clinical and Experimental Insights. Fort Lee, NJ: Health Care Communications, Inc.; 2001. pp. 219–225. [Google Scholar]
- 41.Schmaier AH. The kallikrein-kinin and the renin-angiotensin systems have a multilayered interaction. Am J Physiol Regul Integr Comp Physiol. 2003;285:R1–13. doi: 10.1152/ajpregu.00535.2002. [DOI] [PubMed] [Google Scholar]
- 42.Schmaier AH, Rokjaer R, Shariat-Madar Z. Activation of the plasma kallikrein/kinin system on cells: a revised hypothesis. Thrombosis and Haemostasis. 1999;82:226–233. [PubMed] [Google Scholar]
- 43.Skidgel RA, Erdös EG. Angiotensin converting enzyme (ACE) and neprilysin hydrolyze neuropeptides: a brief history, the beginning and follow-ups to early studies. Peptides. 2004;25:521–525. doi: 10.1016/j.peptides.2003.12.010. [DOI] [PubMed] [Google Scholar]
- 44.Skidgel RA, Erdös EG. Lysine carboxypeptidase. In: Barret AJ, Rawlings ND, W JF, editors. Handbook of Proteolytic Enzymes. 2nd ed. San Diego, CA: Academic Press; 2004. pp. 837–839. [Google Scholar]
- 45.Trabold F, Pons S, Hagege AA, Bloch-Faure M, Alhenc-Gelas F, Giudicelli JF, Richer-Giudicelli C, Meneton P. Cardiovascular phenotypes of kinin B2 receptor- and tissue kallikrein- deficient mice. Hypertension. 2002;40:90–95. doi: 10.1161/01.hyp.0000021747.43346.95. [DOI] [PubMed] [Google Scholar]
- 46.Werle E. Discovery of the most important kallikreins and kallikrein inhibitors. In: Erdös EG, editor. Bradykinin, Kallidin and Kallikrein. Handbook of Experimental Pharmacology. Vol XXV. Berlin: Springer Verlag; 1970. pp. 1–6. [Google Scholar]
- 47.Yang HYT, Erdös EG, Levin Y. Characterization of a dipeptide hydrolase (kininase II; angiotensin I converting enzyme) J Pharmacol Exper Therap. 1971;177:291–300. [PubMed] [Google Scholar]