

Abstract
Theaflavin, a major constituent of black tea, possesses biological functions such as the antioxidative, antiviral, and anti-inflammatory ones. The purpose of this study was to verify whether theaflavin reduces focal cerebral ischemia injury in a rat model of middle cerebral artery occlusion (MCAO). Male Sprague-Dawley rats were anesthetized and subjected to 2 hours of MCAO followed 24 hours reperfusion. Theaflavin administration (5, 10, and 20 mg/kg, IV) ameliorated infarct and edema volume. Theaflavin inhibited leukocyte infiltration and expression of ICAM-1, COX-2, and iNOS in injured brain. Phosphorylation of STAT-1, a protein which mediates intracellular signaling to the nucleus, was enhanced 2-fold over that of sham group and was inhibited by theaflavin. Our study demonstrated that theaflavin significantly protected neurons from cerebral ischemia-reperfusion injury by limiting leukocyte infiltration and expression of ICAM-1, and suppressing upregulation of inflammatory-related prooxidative enzymes (iNOS and COX-2) in ischemic brain via, at least in part, reducing the phosphorylation of STAT-1.
INTRODUCTION
Acute ischemic stroke is the leading cause of adult disability and it is also an important cause of death in industrialized countries with a high incidence affecting up to 0.2% of the population every year [1]. Although pathologic mechanisms leading to cerebral ischemic injury remained unclear, it has been emphasized that inflammatory process had fundamental roles in both the etiology of ischemic cerebrovascular disease and the pathophysiology of cerebral ischemia [2, 3]. Neutrophils are critically involved in the early stage of inflammatory reaction after ischemia, initiating scavenger functions which are later subsumed by macrophages [4, 5]. Endothelial cells actively participate in inflammatory events by regulating leukocyte recruitment via the expression of inflammation-related genes such as ICAM-1, VCAM-1, E-selectin, IL-6, IL-8, and cyclooxygenase-2 [6, 7].
Cyclooxygenase (COX), a rate-limiting enzyme in the metabolism of arachidonic acid into prostanoids, produces PGH2 which in subsequent steps gives rise to PGs with various physiological functions [8–10]. It has been demonstrated in previous reports that cerebral ischemia upregulated the inducible form of COX (COX-2) in neurons, glial cells and infiltrating leukocytes in injured brain [11–13]. Inhibition of COX-2 activity during or after ischemia and genetic deletion of COX-2 reduce infarct volume [14]. In addition, neuronal overexpression of COX-2 increases cerebral infarction [15, 16]. These observations suggest that COX-2 plays a deleterious role in cerebral ischemia. Interestingly, nitric oxide produced by inducible form of nitric oxide synthase (iNOS) has been found to positively regulate COX-2 activity in focal cerebral ischemia [17]. Cerebral ischemia enhanced iNOS expression in neurons, endothelial cells, and microglia [18, 19]. iNOS clearly plays a role in stroke outcome, as evidenced by its selective inhibition in the rat model of MCAO or its genetic deletion [20, 21].
STAT-1 is a member of the signal transducers and activators of transcription proteins family (STATs), which mediate intracellular signaling initiated at cytokine cell surface receptors and transmit to the nucleus. The C terminal domains of STAT proteins contain a transcriptional transactivation domain which is essential for maximal STAT function. Recent study has shown that myocardial ischemia activates JAKs, followed by recruitment of STAT-1, resulting in transcriptional upregulation of inducible nitric oxide synthase (iNOS) and cyclooxygenase-2 (COX-2) [22–24]. It has been demonstrated that focal cerebral ischemia induced STAT-1 activation [25] and STAT-1 knockout mice developed smaller lesions and less pronounced neurological deficit following transient focal cerebral ischemia [26].
Catechins and theaflavins are two groups of natural polyphenols found in green tea and black tea, respectively [27]. These tea polyphenols possess a broad spectrum of biological functions such as antioxidative, antibacteria, antitumor, antiviral, anti-inflammatory, and cardiovascular protection activities [28–31]. It has been reported that epigallocatechin-3-gallate (EGCG), a major constituent of catechins, may attenuate cerebral ischemia-reperfusion injury in rats [32] and it was also a potent inhibitor of STAT-1 phosphorylation [33]. Therefore, the present study was undertaken to evaluate the neuronal protective potential of theaflavin (TF1), a major constituent of theaflavins, in middle cerebral artery occlusion (MCAO) induced focal cerebral ischemia-reperfusion model in rats.
MATERIALS AND METHODS
Experimental protocol and drug
Male Sprague-Dawley rats weighing 220–260 g were housed at room temperature under a controlled 12 h light/dark cycle and allowed access to food and water ad libitum. All experiments were performed as approved by the institutional animal care and use committee. Rats were divided into six groups and each group had ten animals. The first was vehicle-treated group, that is, ischemia was induced for 2 h of MCAO followed by reperfusion for 24 h. The second was sham group. The theaflavin (a pure natural product collection provided by MicroSource Discovery System Inc, Gaylordsville, Conn)-treated groups were separated into a low dosage group (TF1, 5 mg/kg), a middle dosage group (TF1, 10 mg/kg), and a high dosage group (TF1, 20 mg/kg). The intravenous injection of theaflavin was conducted directly before the reperfusion. The sixth was nimodipine-treated group (1 mg/kg, IP). All other chemicals and reagents were of the highest analytical grades available locally.
Middle cerebral artery occlusion induced focal cerebral ischemia
Focal cerebral ischemia was produced by occluding the left middle cerebral artery according to the methods by Longa et al [34]. Briefly, the rats were anaesthetized with Chloral hydrate (400 mg/kg, IP). Through a midline neck incision, the left common and external carotid artery were isolated from muscles and coagulated. A 3-0 nylon suture with a blunted tip was inserted into the internal carotid through the external carotid artery stump and advanced up to 21 mm or till resistance was left. After 2 h of MCAO, the suture was removed to restore blood flow. In sham group, the same surgical procedure was performed except that the suture was introduced into the external carotid artery but not advanced. After surgery, the incision was sutured and the rats were returned to their cage with free access to water and food. Twenty-four hours after reperfusion, rats were sacrificed by rapid decapitation under deep anesthesia and the brains were taken out for biochemical estimations.
Infarct and edema volume
Twenty-four hours after reperfusion, whole brains were rapidly removed. Immediately after being weighed, the brains were sliced into 2-mm-thick coronal sections and stained with 2%2,3,5-triphenyltetrazoliumchloride (TTC, Sigma-Aldrich) at 37°C for 30 minutes in the dark, followed by fixation with 10% formalin at room temperature overnight. The sections were photographed with a digital camera (Kodak DC240, USA) connected to a computer. The unstained areas, defined as infarct tissue, were calculated by using an image analysis program (Adobe Photoshop 5.0CS). The infarct volume was calculated by measuring the unstained area in each slice. Edema correction of infarct volume was done using the equation, volume correction = (infarct volume × contralateral volume)/ipsilateral volume. The volumes of both the hemispheres were calculated from which edema volume was calculated by subtracting the contralateral volume from the ipsilateral volume.
Measurement of lipid peroxidation
The estimate of lipid peroxidation of the cerebral cortex was determined by measuring the formed malondialdehyde (MDA). Briefly, brain tissues were homogenized (10%, w/v) with cold 1.5% KCl. The homogenate was mixed with a 1% phosphoric acid and 6% TBA (Sigma-Aldrich) aqueous solution. The mixture was heated for 45 minutes in a boiling water bath. After cooling, n-butanol was added and mixed vigorously. The absorbance of the butanol phase was measured at 525 nm. A serially diluted MDA (Sigma-Aldrich) solution was prepared and used as a standard. The data (MDA) was expressed as nmol/mg protein.
Myeloperoxidase assay
The activity of myeloperoxidase (MPO) was determined as an indicator of PMNs migration, as previously described [35]. The method of assaying MPO activity was according to the guide of the assay kit (Nanjing Jiancheng Bioengineering Co Ltd, China).
Immunohistochemistry detection
The procedures were processed according to the protocols recommended for ICAM-1, iNOS, and COX-2 immunohistochemistry kit. Following deparaffinization and rehydration, the cortices sections were exposed to 3% hydrogen peroxide for 10 minutes to bleach endogenous peroxidases. Then microwave oven-based antigen retrieval was performed. Slides were probed with either anti-ICAM-1 (1 : 100, rat monoclonal, Santa Cruz Biotechnology), anti-iNOS (1 : 100, rat polyclonal, Santa Cruz Biotechnology), or anti-COX-2 (1 : 50, rat monoclonal, Santa Cruz Biotechnology) for 1 hour at 37°C, washed 3 times in PBS, incubated with biotin-labeled anti-rat IgG for 1 hour at 37°C, respectively. Incubation with PBS instead of the primary antibody served as a negative control. After washing in PBS, tissues were visualized with 3, 3′-diaminobenzidine tetrahydrochloride (DAB) and counterstained with hematoxylin. Finally, the sections were dehydrated in graded ethanol, immersed in xylene and coverslipped. In specimens the positive cells were counted in cortex in ten randomly selected areas from each case and expressed as number of immunopositive/mm2. Results are presented as mean ± SEM.
RT-PCR
Total RNA was extracted from cortex using TRIzol reagent (Sigma Co). cDNA was synthesized according to the manufacturer's instruction of reverse transcription kit (GIBCO-BRL, USA), and then amplified with a multiplex PCR kit (GIBCO-BRL, USA). Conditions for amplification were as follows: initial denaturation for 2 minutes at 94°C, 35 cycles of 94°C for 30 seconds, 60°C for 45 seconds, 72°C for 60 seconds, and a final extension stop at 72°C for 7 minutes. The rat specific primers (sense and antisense primers) for iNOS, COX-2, and β-actin were 5′-CGGTGCTGTATTTCCTTACGAGGCGAAGAAGG-3′ and 5′-GGTGCTGTCTGTTAGGAGGTCAAGTAAAGGGC-3′ (iNOS,259 bp); 5′-CCATGTCAAAA-CCGTGGTGAATG-3′ and 5′-A-TGGGAGTTGGGCAGTCATCAG-3′ (COX-2,374 bp); 5-ATGGATGACGATATCGCTG-3 and 5-ATGAGGTAGTCTGTCAGGT-3 (β-actin, 568 bp), respectively. Reaction products were then separated on a 1.5% agarose gel, stained with ethidium-bromide, and visualized by UV transillumination. HPIAS-1000 software analysis system was used to determine the relative absorbance of mRNA expression.
Western blot analysis
The cortices of brains were removed and used for Western analysis. Protein concentrations were determined using the Bio-Rad protein assay kit (Bio-Rad, Hercules, Calif) and all samples were adjusted to an equal protein content before analysis. Samples (30 μg of total protein) were separated on 8% denaturing polyacrylamide gel. Following electrophoresis, proteins were transferred to a nitrocellulose membrane (80 V, 90 minutes; transfer buffer 25 mM Tris, 190 mM glycine, 20% methanol, 0.5% sodium dodecyl sulfate) by an electroblotter (Bio-rad). After being blocked for two hours at room temperature in blocking buffer (5% nonfat milk in 20 mM Tris/HCl, pH 7.6, 140 mM NaCl, 0.5% Tween 20), membranes were incubated over night at 4°C with primary antibodies against antiphospho-STAT-1Tyr-701 (Zymed, South San Francisco, Calif), or anti-STAT-1 (Santa Cruz Biotechnology, Santa Cruz, Calif). Membranes were then washed (in 20 mM Tris/HCl, pH 7.6, 140 mM NaCl, 0.1% Tween 20) and incubated with a peroxidase-conjugated secondary antibody at room temperature for 50 minutes. The immunoblots were visualized using Western blotting luminal reagent (Cell Signal Corp). The density of protein band was scanned and analyzed with an image analyzer.
Statistical analysis
Unless otherwise stated, all the results were finally presented as means ± SEM. Statistical differences between different groups were assessed by a one-way analysis of variance and Student-Newman-Keuls test. P value less than .05 was considered statistically significant.
RESULTS
Effect of theaflavin on cerebral infarction and edema
Infarct volume was measured in the coronal brain sections which were stained with TTC. Two hours of MCAO and 24 hours of reperfusion showed an infarct volume of 220.87 ± 27.42 mm3. The infarct volume was decreased to 183.49 ± 19.33 mm3, 139.06 ± 11.28 mm3, and 118.25 ± 10.36 mm3 in 5, 10, and 20 mg/kg theaflavin-treated rats, respectively, (Figure 1). Theaflavin at the doses of 10 and 20 mg/kg produced 40.79 ± 8.71% and 52.30 ± 9.79% reduction in infarct volume, respectively, as compared to vehicle-treated group (P < .01, Figure 1). Two hours of MCAO and 24 hours of reperfusion resulted in 133.63 ± 11.07 mm3 increase in the ipsilateral volume due to edema. Theaflavin at the doses of 5, 10, and 20 mg/kg resulted in reduction of edema volume to 98.61 ± 25.34 mm3, 61.37 ± 14.13 mm3, and 51.25 ± 9.97 mm3 of ipsilateral hemisphere, respectively (Figure 1). Theaflavin at the doses of 10 and 20 mg/kg showed 48.63±7.84% and 55.04±8.01% reduction in edema volume, respectively as compared to vehicle-treated group (P < .01, Figure 1). Nimodipine-treatment also reduced the infarct and edema volumes. The infarct and edema volumes of Nimodipine-treated group were 110.39 ± 10.17 mm3 and 55.26 ± 10.65 mm3, respectively (P < .01, Figure 1).
Figure 1.

(a) Representative coronal brain sections stained with TTC after 2 hours of MCAO and 24 hours of reperfusion showing infarction. Dark-colored region in the TTC stained sections indicated nonischemic portion of brain and pale-colored region indicated ischemic portion of brain. Theaflavin and nimodipine-treatment reduced infarct volume. (b) Volume of infarction after 2 hours of MCAO and 24 hours of reperfusion in vehicle, theaflavin (5, 10, and 20 mg/kg) and nimodipine (1 mg/kg)-treated rats. (c) Volume of edema after 2 hours of MCAO and 24 hours of reperfusion in vehicle, theaflavin (5, 10, and 20 mg/kg) and nimodipine-treated (1 mg/kg) rats, **P < .01 as compared to the vehicle-treated group.
Effect of theaflavin on MDA
The level of MDA significantly increased in the vehicle-treated group more than in the sham group. As compared to the vehicle-treated group, the levels of MDA significantly decreased in the theaflavin and nimodipine-treated groups (P < .01, Table 1). The theaflavin-treated group (20 mg/kg) had the same effect as compared to the nimodipine-treated group (P > .05). However, the MDA levels of theaflavin-treated groups were still higher than that of sham group.
Table 1.
Effect of theaflavin on MDA and MPO activities ( ± s n = 10).
| Parameters | Dose | MDA | MPO |
| (mg · kg−1) | (nmol/mg protein) | (U · g−1) | |
| Vehicle | — | 4.13 ± 1.56 | 2.13 ± 0.69 |
| Sham | — | 1.45 ± 0.09 | 0.34 ± 0.12 |
| TF1 | 5 | 3.12 ± 1.62** | 1.92 ± 0.51 |
| TF1 | 10 | 2.77 ± 1.09** | 1.43 ± 0.45** |
| TF1 | 20 | 2.34 ± 1.13** | 1.21 ± 0.39** |
| Nimodipine | 1 | 2.41 ± 1.10** | 1.24 ± 0.33** |
**denotes that P < .01 versus the vehicle-treated group.
Effect of theaflavin on inflammatory injury of cerebral ischemia
Infiltration of leukocytes to CI/R-injured tissue provides predominant sources for MPO, an important prooxidative enzyme responsible for oxidative stress in CI/R-injured brain. In this study, the MPO activity was relatively low in the sham group, and significantly increased in the vehicle-treated group. Treatment with 10 and 20 mg/kg theaflavin significantly reduced MPO activity in the CI/R-injured cerebral tissue. Nimodipine-treatment also reduced MPO activity (Table 1).
Effect of theaflavin on ICAM-1, iNOS, and COX-2 protein production
The protein expressions of ICAM-1, iNOS, and COX-2 in the ischemic cortex of the vehicle-treated group significantly increased compared with those of the sham group. The expression of ICAM-1 was obviously identified on the microvascular endothelial cells in the ischemic hemisphere (Figure 2). The positive cells of iNOS and COX-2 were found with brown cytoplasma and predominantly located within the neurons, glial cells, and infiltrating leukocytes (Figures 3, 4). The protein expressions of ICAM-1, iNOS, and COX-2 decreased dose dependently in theaflavin-treated groups (Table 2). Effect of 20 mg/kg theaflavin was similar to that of nimodipine (1 mg/kg).
Figure 2.

Immunohistochemical staining of ICAM-1 in brain tissues of (a) vehicle-treated rats and (b) theaflavin-treated rats (20 mg/kg), SP×400. ICAM-1 protein is mainly expressed on the microvascular endothelial cells. ICAM-1 expression decreases dramatically in theaflavin-treated groups. Scale bar = 10 μm.
Figure 3.

Immunohistochemical staining for iNOS protein expression in (a) vehicle-treated rats and (b) theaflavin-treated rats (20 mg/kg), SP × 400. The number of iNOS immunoreactive positive cells in theaflavin-treated groups is significantly less than that of vehicle-treated group. Scale bar = 10 μm.
Figure 4.

Immunohistochemical staining for COX-2 protein expression in (a) vehicle-treated rats and (b) theaflavin-treated rats (20 mg/kg), SP × 400. The number of COX-2 immunoreactive positive cells in theaflavin group is significantly less than that of vehicle-treated group. Scale bar = 10 μm.
Table 2.
ICAM-1, iNOS, and COX-2 protein production in vehicle and theaflavin-treated groups ( ± s n = 10).
| Parameters | Dose | ICAM-1 | iNOS | COX-2 |
| (mg · kg−1) | (number of immunopositive/mm2) | (number of immunopositive/mm2) | (number of immunopositive/mm2) | |
| Vehicle | — | 166.21 ± 34.26 | 61.21 ± 20.34 | 67.41 ± 22.29 |
| Sham | — | 12.36 ± 7.09 | 13.24 ± 6.98 | 10.36 ± 7.06 |
| TF1 | 5 | 97.28 ± 24.67** | 42.11 ± 21.06** | 39.14 ± 21.74** |
| TF1 | 10 | 63.12 ± 22.30** | 31.21 ± 16.57** | 28.57 ± 12.39** |
| TF1 | 20 | 32.17 ± 16.55** | 24.88 ± 14.89** | 21.36 ± 14.21** |
| Nimodipine | 1 | 38.66 ± 19.87** | 24.31 ± 15.33** | 23.61 ± 12.58** |
**denotes that P < .01 versus the vehicle-treated group.
Effect of theaflavin on COX-2 and iNOS mRNA expressions
The mRNA expressions of COX-2 and iNOS were analyzed by RT-PCR. The brain tissue obtained from the sham group showed low mRNA expression levels of COX-2 and iNOS. After 2 hours of MCAO and 24 h reperfusion, the expressions of COX-2 and iNOS remarkably increased in ischemic hemisphere in the vehicle-treated group as compared with the sham group. Theaflavin-treatment could reduce molecule mRNA expressions dose dependently and nimodipine also reduced the expressions of molecule mRNA (Figures 5, 6).
Figure 5.

(a) The mRNA expression of COX-2 was assessed by using RT-PCR as standardized by coamplifying the housekeeping gene β-actin. Lanes 1–7: Marker, Vehicle, Sham, TF1 (5 mg · kg−1), TF1 (10 mg · kg−1), TF1 (20 mg · kg−1), Nimodipine. (b) Statistical analysis revealed that theaflavin-treatment markedly decreased mRNA expression of COX-2 dose dependently, **P < .01.
Figure 6.

(a) The mRNA expression of iNOS was assessed by using RT-PCR as standardized by coamplifying the housekeeping gene β-actin. Lanes 1–7: Marker, Vehicle, Sham, TF1 (5 mg · kg−1), TF1 (10 mg · kg−1), TF1 (20 mg · kg−1), Nimodipine. (b) Statistical analysis revealed that theaflavin-treatment markedly decreased mRNA expression of iNOS dose dependently, **P < .01.
Effect of theaflavin on STAT-1 protein expression
The levels of STAT-1 phosphorylation on tyrosine 701 were markedly enhanced in brains subjected to 2 hours of MCAO followed 24 hours reperfusion. However, the brains treated with theaflavin and nimodipine reduced STAT-1 phosphorylation levels on tyrosine 701 (Figure 7). Theaflavin-treatment could reduce STAT-1 phosphorylation dose dependently. These results demonstrate that theaflavin could have the ability to inhibit STAT-1 701 phosphorylation as well as protect brain against I/R-induced inflammation.
Figure 7.
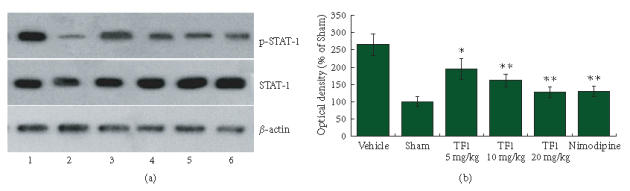
(a) Western blotting showed levels of STAT-1 in brain tissue of rats. Lanes 1–6: Vehicle, Sham, TF1 (5 mg · kg−1), TF1 (10 mg · kg−1), TF1 (20 mg · kg−1), Nimodipine. (b) Statistical analysis revealed that theaflavin-treatment markedly decreased STAT-1 phosphorylation dose dependently, *P < .05, **P < .01.
DISCUSSION
In the current study theaflavin-treatment showed protective effects on brain injuries induced by middle cerebral artery occlusion followed by reperfusion in rats by blocking inflammation-related events (MPO and ICAM-1) and expressions of prooxidative enzymes such as COX-2 and iNOS. Further, the protective effect of theaflavin was associated with downregulation of STAT-1 phosphorylation. The neuronal protective potential of theaflavin was dose dependently and the effect of 20 mg/kg theaflavin was similar to that of nimodipine.
Rats subjected to cerebral ischemia-reperfusion showed typical markers of cerebral inflammation and oxidative/nitrosative injury including leukocyte infiltration into the infarct area (enhanced MPO activity), upregulation of adhesion molecules (ICAM-1), and induction of prooxidative enzymes (COX-2 and iNOS) [36, 37]. Ischemia activates a cascade that leads to the induction and expression of genes in a variety of cell types throughout the central nervous system (CNS). COX-2, one product of such immediate early genes, has become the focus of attention because it is the rate-limiting enzyme involved in arachidonic acid metabolism, thereby generating prostaglandins and thromboxanes which play important roles in supporting and sustaining the inflammatory response [38]. In rodents as well as in humans, cerebral ischemia upregulated COX-2 expression in neurons, blood vessels, and inflammatory cells in the injured brain [13, 39, 40]. Moreover, administration of the selective COX-2 inhibitor NS398 attenuated the elevation of PGE2 and reduced the infarct in a model of MCAO [13]. COX-2 reaction products may also contribute to NMDA-induced neuronal injury and the pathogenesis of nitric oxide after ischemia [41, 42].
Nitric oxide (NO) is an important mediator in the cerebral ischemic injury [43]. Specifically, Nitric oxide derived from the inducible isoform (iNOS) expressed by many cells is very important in excitotoxic injury cascades [18, 19]. Pharmacologically selective inhibitors of iNOS attenuated infarct volume after focal cerebral ischemia [21, 44, 45]. Nitric oxide produced by iNOS has been shown to contribute to COX-2 activity (possibly without altering COX-2 expression) [17]. Inhibition of iNOS could also serve as neuroprotection through COX-2 inhibition just before the start of the delayed death of CA1 neurons [46]. We confirmed that cortex tissue obtained from rats with 2 hours of MCAO followed 24 hours reperfusion exhibited significantly more COX-2 and iNOS protein expressions than that of sham group, which supported the idea that inflammatory molecules participate in the occurrence and development of cerebral ischemia. At the same time, we found that theaflavin-treatment dose dependently inhibited COX-2 and iNOS protein expressions.
In order to elucidate the mechanism of theaflavin on inflammation-related events, we investigated the mRNA expression of COX-2 and iNOS in cerebral ischemic tissues of rats and determined the influence of theaflavin-treatment on mRNA production of COX-2 and iNOS. We found that the mRNA expressions of COX-2 and iNOS were in accordance with the results of immunohistochemistry detection. RT-PCR analysis revealed that the mRNA levels of COX-2 and iNOS increased in brain tissues of the vehicle-treated group. Similarly, theaflavin had a dose-dependent effect on decreasing mRNA expressions of COX-2 and iNOS. This prompted us to investigate the regulation of COX-2 and iNOS gene transcriptions in the process of inflammatory responses.
Many cytokines such as IL-6, IL-11, and inflammatory mediators produced by ischemic brain cells, play important roles contributing to ischemic pathophysiology [47, 48]. JAK-STAT is an important downstream signal pathway of these cytokines [49]. Binding of neurokines to the membrane receptor leads to dimerization of gp130, followed by activation of JAK, which in turn phosphorylates cytoplasmic STAT. Phosphorylated STAT forms homo- or heterodimers and translocates into the nucleus, stimulating gene transcription. Therefore, the JAK-STAT pathway provides cells with a vital mechanism for responding to various extracellular stimuli including ischemic stress. Accumulation in the nucleus of tyrosine phosphorylated STAT dimers is followed by DNA binding, activation of target gene transcription, dephosphorylation, and returns to the cytoplasm [50]. STAT-1 induces expression of the transcription factor IRF-1, which then itself binds to specific DNA elements of the iNOS promoter to further promote iNOS expression [51]. Pretreatment with the Janus tyrosine kinase (JAK) inhibitor AG-490 before the six occlusion-reperfusion cycles blocked both the tyrosine phosphorylation of STAT1/3 and the subsequent upregulation of COX-2 protein, demonstrating a necessary role of the JAK-STAT pathway in the induction of COX-2 [52]. We therefore investigated the effect of theaflavin on tyrosine phosphorylation of STAT-1. Our results have shown that the levels of STAT-1 phosphorylation on tyrosine 701 were markedly enhanced in brains subjected to 2 h of MCAO followed by 24 hours reperfusion. Theaflavin-treatment dose dependently inhibited phosphorylation of STAT-1 and mRNA expressions (COX-2 and iNOS) controlled by it.
In conclusion, our study demonstrated that theaflavin significantly protected neurons from cerebral ischemia-reperfusion injury by limiting lipid peroxidation, leukocyte infiltration and expression of ICAM-1. Theaflavin also suppressed upregulations of inflammatory-related prooxidative enzymes (iNOS and COX-2) in ischemic brain via, at least in part, reducing STAT-1 phosphorylation. As a potent antioxidative drug, theaflavin could be beneficial for the prevention and/or amelioration of cerebral ischemia-reperfusion injury. Thus, the protection of neurons by theaflavin may provide clinically beneficial outcomes alone or in combination with thrombolytic therapy.
ACKNOWLEDGMENT
We thank Doctor Yao Liu for checking the spelling of this manuscript.
References
- 1.Klijn CJM, Hankey GJ. Management of acute ischaemic stroke: new guidelines from the American Stroke Association and European Stroke Initiative. Lancet Neurology. 2003;2(11):698–701. doi: 10.1016/s1474-4422(03)00558-1. [DOI] [PubMed] [Google Scholar]
- 2.Lo EH, Dalkara T, Moskowitz MA. Mechanisms, challenges and opportunities in stroke. Nature Reviews Neuroscience. 2003;4(5):399–415. doi: 10.1038/nrn1106. [DOI] [PubMed] [Google Scholar]
- 3.Nurmi A, Lindsberg PJ, Koistinaho M, et al. Nuclear factor-κB contributes to infarction after permanent focal ischemia. Stroke. 2004;35(4):987–991. doi: 10.1161/01.STR.0000120732.45951.26. [DOI] [PubMed] [Google Scholar]
- 4.Kochanek PM, Hallenbeck JM. Polymorphonuclear leukocytes and monocytes/macrophages in the pathogenesis of cerebral ischemia and stroke. Stroke. 1992;23(9):1367–1379. doi: 10.1161/01.str.23.9.1367. [DOI] [PubMed] [Google Scholar]
- 5.Heinel LA, Rubin S, Rosenwasser RH, Vasthare US, Tuma RF. Leukocyte involvement in cerebral infarct generation after ischemia and reperfusion. Brain Research Bulletin. 1994;34(2):137–141. doi: 10.1016/0361-9230(94)90010-8. [DOI] [PubMed] [Google Scholar]
- 6.Kułdo JM, Westra J, Àsgeirsdóttir SA, et al. Differential effects of NF-κB and p38 MAPK inhibitors and combinations thereof on TNF-α- and IL-1β-induced proinflammatory status of endothelial cells in vitro. American Journal of Physiology - Cell Physiology. 2005;289(5):C1229–C1239. doi: 10.1152/ajpcell.00620.2004. [DOI] [PubMed] [Google Scholar]
- 7.Sugama Y, Tiruppathi C, Offakidevi K, Andersen TT, Fenton JW II, Malik AB. Thrombin-induced expression of endothelial P-selectin and intercellular adhesion molecule-1: a mechanism for stabilizing neutrophil adhesion. Journal of Cell Biology. 1992;119(4):935–944. doi: 10.1083/jcb.119.4.935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hayaishi O. Molecular mechanisms of sleep-wake regulation: roles of prostaglandins D2 and E2. FASEB Journal. 1991;5(11):2575–2581. [PubMed] [Google Scholar]
- 9.Kaufmann WE, Andreasson KI, Isakson PC, Worley PF. Cyclooxygenases and the central nervous system. Prostaglandins. 1997;54(3):601–624. doi: 10.1016/s0090-6980(97)00128-7. [DOI] [PubMed] [Google Scholar]
- 10.Matsuoka Y, Okazaki M, Zhao H, Asai S, Ishikawa K, Kitamura Y. Phosphorylation of c-Jun and its localization with heme oxygenase-1 and cyclooxygenase-2 in CA1 pyramidal neurons after transient forebrain ischemia. Journal of Cerebral Blood Flow and Metabolism. 1999;19(11):1247–1255. doi: 10.1097/00004647-199911000-00009. [DOI] [PubMed] [Google Scholar]
- 11.Iadecola C, Forster C, Nogawa S, Clark HB, Ross ME. Cyclooxygenase-2 immunoreactivity in the human brain following cerebral ischemia. Acta Neuropathologica. 1999;98(1):9–14. doi: 10.1007/s004010051045. [DOI] [PubMed] [Google Scholar]
- 12.Miettinen S, Fusco FR, Yrjänheikki J, et al. Spreading depression and focal brain ischemia induce cyclooxygenase-2 in cortical neurons through N-methyl-D-aspartic acid-receptors and phospholipase A2. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(12):6500–6505. doi: 10.1073/pnas.94.12.6500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nogawa S, Zhang F, Ross ME, Iadecola C. Cyclo-oxygenase-2 gene expression in neurons contributes to ischemic brain damage. Journal of Neuroscience. 1997;17(8):2746–2755. doi: 10.1523/JNEUROSCI.17-08-02746.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nakayama M, Uchimura K, Zhu RL, et al. Cyclooxygenase-2 inhibition prevents delayed death of CA1 hippocampal neurons following global ischemia. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(18):10954–10959. doi: 10.1073/pnas.95.18.10954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Govoni S, Masoero E, Favalli L, et al. The Cycloxygenase-2 inhibitor SC58236 is neuroprotective in an in vivo model of focal ischemia in the rat. Neuroscience Letters. 2001;303(2):91–94. doi: 10.1016/s0304-3940(01)01675-5. [DOI] [PubMed] [Google Scholar]
- 16.Doré S, Otsuka T, Mito T, et al. Neuronal overexpression of cyclooxygenase-2 increases cerebral infarction. Annals of Neurology. 2003;54(2):155–162. doi: 10.1002/ana.10612. [DOI] [PubMed] [Google Scholar]
- 17.Nogawa S, Forster C, Zhang F, Nagayama M, Ross ME, Iadecola C. Interaction between inducible nitric oxide synthase and cyclooxygenase-2 after cerebral ischemia. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(18):10966–10971. doi: 10.1073/pnas.95.18.10966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Samdani AF, Dawson TM, Dawson VL. Nitric oxide synthase in models of focal ischemia. Stroke. 1997;28(6):1283–1288. doi: 10.1161/01.str.28.6.1283. [DOI] [PubMed] [Google Scholar]
- 19.Iadecola C. Bright and dark sides of nitric oxide in ischemic brain injury. Trends in Neurosciences. 1997;20(3):132–139. doi: 10.1016/s0166-2236(96)10074-6. [DOI] [PubMed] [Google Scholar]
- 20.Goyagi T, Goto S, Bhardwaj A, Dawson VL, Hurn PD, Kirsch JR. Neuroprotective effect of σ1-receptor ligand 4-phenyl-1-(4-phenylbutyl) piperidine (PPBP) is linked to reduced neuronal nitric oxide production. Stroke. 2001;32(7):1613–1620. doi: 10.1161/01.str.32.7.1613. [DOI] [PubMed] [Google Scholar]
- 21.Iadecola C, Zhang F, Casey R, Nagayama M, Ross ME. Delayed reduction of ischemic brain injury and neurological deficits in mice lacking the inducible nitric oxide synthase gene. Journal of Neuroscience. 1997;17(23):9157–9164. doi: 10.1523/JNEUROSCI.17-23-09157.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bolli R, Manchikalapudi S, Tang X-L, et al. The protective effect of late preconditioning against myocardial stunning in conscious rabbits is mediated by nitric oxide synthase: evidence that nitric oxide acts both as a trigger and as a mediator of the late phase of ischemic preconditioning. Circulation Research. 1997;81(6):1094–1107. doi: 10.1161/01.res.81.6.1094. [DOI] [PubMed] [Google Scholar]
- 23.Guo Y, Jones WK, Xuan Y-T, et al. The late phase of ischemic preconditioning is abrogated by targeted disruption of the inducible NO synthase gene. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(20):11507–11512. doi: 10.1073/pnas.96.20.11507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shinmura K, Tang X-L, Wang Y, et al. Cyclooxygenase-2 mediates the cardioprotective effects of the late phase of ischemic preconditioning in conscious rabbits. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(18):10197–10202. doi: 10.1073/pnas.97.18.10197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Takagi Y, Harada J, Chiarugi A, Moskowitz MA. STAT1 is activated in neurons after ischemia and contributes to ischemic brain injury. Journal of Cerebral Blood Flow and Metabolism. 2002;22(11):1311–1318. doi: 10.1097/01.WCB.0000034148.72481.F4. [DOI] [PubMed] [Google Scholar]
- 26.West DA, Valentim LM, Lythgoe MF, et al. MR image-guided investigation of regional signal transducers and activators of transcription-1 activation in a rat model of focal cerebral ischemia. Neuroscience. 2004;127(2):333–339. doi: 10.1016/j.neuroscience.2004.05.022. [DOI] [PubMed] [Google Scholar]
- 27.Leung LK, Su Y, Chen R, Huang Y, Chen Z-Y. Theaflavins in black tea and catechins in green tea are equally effective antioxidants. Journal of Nutrition. 2001;131(9):2248–2251. doi: 10.1093/jn/131.9.2248. [DOI] [PubMed] [Google Scholar]
- 28.Lambert JD, Yang CS. Mechanisms of cancer prevention by tea constituents. Journal of Nutrition. 2003;133(10):3262S–3267S. doi: 10.1093/jn/133.10.3262S. [DOI] [PubMed] [Google Scholar]
- 29.Higdon JV, Frei B. Tea catechins and polyphenols: health effects, metabolism, and antioxidant functions. Critical Reviews in Food Science and Nutrition. 2003;43(1):89–143. doi: 10.1080/10408690390826464. [DOI] [PubMed] [Google Scholar]
- 30.Yang CS, Landau JM. Effects of tea consumption nutrition health. Journal of Nutrition. 2000;130(10):2409–2412. doi: 10.1093/jn/130.10.2409. [DOI] [PubMed] [Google Scholar]
- 31.Mukhtar H, Ahmad N. Tea polyphenols: prevention of cancer and optimizing health. American Journal of Clinical Nutrition. 2000;71(suppl 6):1698S–1704S. doi: 10.1093/ajcn/71.6.1698S. [DOI] [PubMed] [Google Scholar]
- 32.Choi YB, Kim YI, Lee KS, Kim BS, Kim DJ. Protective effect of epigallocatechin gallate on brain damage after transient middle cerebral artery occlusion in rats. Brain Research. 2004;1019(1-2):47–54. doi: 10.1016/j.brainres.2004.05.079. [DOI] [PubMed] [Google Scholar]
- 33.Menegazzi M, Tedeschi E, Dussin D, et al. Anti-interferon gamma action of epigallocatechin-3-gallate mediated by specific inhibition of STAT1 activation. FASEB journal. 2001;15(7):1309–1311. doi: 10.1096/fj.00-0519fje. [DOI] [PubMed] [Google Scholar]
- 34.Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989;20(1):84–91. doi: 10.1161/01.str.20.1.84. [DOI] [PubMed] [Google Scholar]
- 35.Matsuo Y, Onodera H, Shiga Y, et al. Correlation between myeloperoxidase-quantified neutrophil accumulation and ischemic brain injury in the rat: effects of neutrophil depletion. Stroke. 1994;25(7):1469–1475. doi: 10.1161/01.str.25.7.1469. [DOI] [PubMed] [Google Scholar]
- 36.Liu S-J, Zhou S-W, Xue C-S. Effect of tetrandrine on neutrophilic recruitment response to brain ischemia/reperfusion. Acta Pharmacologica Sinica. 2001;22(11):971–975. [PubMed] [Google Scholar]
- 37.Jander S, Schroeter M, Stoll G. Role of NMDA receptor signaling in the regulation of inflammatory gene expression after focal brain ischemia. Journal of Neuroimmunology. 2000;109(2):181–187. doi: 10.1016/s0165-5728(00)00317-9. [DOI] [PubMed] [Google Scholar]
- 38.Vane JR, Bakhle YS, Botting RM. Cyclooxygenases 1 and 2. Annual Review of Pharmacology and Toxicology. 1998;38(1):97–120. doi: 10.1146/annurev.pharmtox.38.1.97. [DOI] [PubMed] [Google Scholar]
- 39.Candelario-Jalil E, González-Falcón A, García-Cabrera M, et al. Assessment of the relative contribution of COX-1 and COX-2 isoforms to ischemia-induced oxidative damage and neurodegeneration following transient global cerebral ischemia. Journal of Neurochemistry. 2003;86(3):545–555. doi: 10.1046/j.1471-4159.2003.01812.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ohtsuki T, Kitagawa K, Yamagata K, et al. Induction of cyclooxygenase-2 mRNA in gerbil hippocampal neurons after transient forebrain ischemia. Brain Research. 1996;736(1-2):353–356. doi: 10.1016/0006-8993(96)00948-1. [DOI] [PubMed] [Google Scholar]
- 41.Hewett SJ, Uliasz TF, Vidwans AS, Hewett JA. Cyclooxygenase-2 contributes to N-methyl-D-aspartate-mediated neuronal cell death in primary cortical cell culture. Journal of Pharmacology and Experimental Therapeutics. 2000;293(2):417–425. [PubMed] [Google Scholar]
- 42.Kittaka M, Giannotta SL, Zelman V, et al. Attenuation of brain injury and reduction of neuron-specific enolase by nicardipine in systemic circulation following focal ischemia and reperfusion in a rat model. Journal of Neurosurgery. 1997;87(5):731–737. doi: 10.3171/jns.1997.87.5.0731. [DOI] [PubMed] [Google Scholar]
- 43.Dirnagl U, Iadecola C, Moskowitz MA. Neurobiology of ischemic stroke: an integrated view. Trends in Neurosciences. 1989;22(9):391–397. doi: 10.1016/s0166-2236(99)01401-0. [DOI] [PubMed] [Google Scholar]
- 44.Jiang MH, Kaku T, Hada J, Hayashi Y. 7-Nitroindazole reduces nitric oxide concentration in rat hippocampus after transient forebrain ischemia. European Journal of Pharmacology. 1999;380(2-3):117–121. doi: 10.1016/s0014-2999(99)00555-5. [DOI] [PubMed] [Google Scholar]
- 45.Nanri K, Montécot C, Springhetti V, Seylaz J, Pinard E. The selective inhibitor of neuronal nitric oxide synthase, 7- nitroindazole, reduces the delayed neuronal damage due to forebrain ischemia in rats. Stroke. 1998;29(6):1248–1254. doi: 10.1161/01.str.29.6.1248. [DOI] [PubMed] [Google Scholar]
- 46.Endoh M, Maiese K, Wagner J. Expression of the inducible form of nitric oxide synthase by reactive astrocytes after transient global ischemia. Brain Research. 1994;651(1-2):92–100. doi: 10.1016/0006-8993(94)90683-1. [DOI] [PubMed] [Google Scholar]
- 47.Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: an integrated view. Trends in Neurosciences. 1999;22(9):391–397. doi: 10.1016/s0166-2236(99)01401-0. [DOI] [PubMed] [Google Scholar]
- 48.Heinrich PC, Behrmann I, Müller-Newen G, Schaper F, Graeve L. Interleukin-6-type cytokine signalling through the gp130/Jak/STAT pathway. Biochemical Journal. 1998;334(pt 2):297–314. doi: 10.1042/bj3340297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cattaneo E, Conti L, De-Fraja C. Signalling through the JAK-STAT pathway in the developing brain. Trends in Neurosciences. 1999;22(8):365–369. doi: 10.1016/s0166-2236(98)01378-2. [DOI] [PubMed] [Google Scholar]
- 50.Shen Y, Schlessinger K, Zhu X, et al. Essential role of STAT3 in postnatal survival and growth revealed by mice lacking STAT3 serine 727 phosphorylation. Molecular and Cellular Biology. 2004;24(1):407–419. doi: 10.1128/MCB.24.1.407-419.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kamijo R, Harada H, Matsuyama T, et al. Requirement for transcription factor IRF-1 in NO synthase induction in macrophages. Science. 1994;263(5153):1612–1615. doi: 10.1126/science.7510419. [DOI] [PubMed] [Google Scholar]
- 52.Xuan Y-T, Guo Y, Zhu Y, et al. Mechanism of cyclooxygenase-2 upregulation in late preconditioning. Journal of Molecular and Cellular Cardiology. 2003;35(5):525–537. doi: 10.1016/s0022-2828(03)00076-2. [DOI] [PMC free article] [PubMed] [Google Scholar]