

Abstract
Erythrocyte invasion by Plasmodium requires molecules present both on the merozoite surface and within the specialized organelles of the apical complex. The Plasmodium erythrocyte binding protein family includes the Plasmodium falciparum sialic acid-binding protein, EBA-175 (erythrocyte binding antigen-175), which binds sialic acid present on glycophorin A of human erythrocytes. We address the role of the conserved 3′-cysteine rich region, the transmembrane, and cytoplasmic domains through targeted gene disruption. Truncation of EBA-175 had no measurable effect on either the level of EBA-175 protein expression or its subcellular localization. Similarly, there appears to be no impairment in the ability of soluble EBA-175 to be released into the culture supernatant after schizont rupture. Additionally, the 3′-cys rich region, transmembrane, and cytoplasmic domains of EBA-175 are apparently non-essential for merozoite invasion. In contrast, erythrocyte invasion via the EBA-175/glycophorin A route appears to have been disrupted to such a degree that the mutant lines have undergone a stable switch in invasion phenotype. As such, EBA-175 appears to have been functionally inactivated within the truncation mutants. The sialic acid-independent invasion pathway within the mutant parasites accounts for approximately 85% of invasion into normal erythrocytes. These data demonstrate the ability of P. falciparum to utilize alternate pathways for invasion of red blood cells, a property that most likely provides a substantial survival advantage in terms of overcoming host receptor heterogeneity and/or immune pressure.
The ability of Plasmodium merozoites to invade erythrocytes is contingent on a rapid cascade of specific interactions occurring between parasite molecules and host erythrocyte receptors. Despite a good understanding of the recognition and invasion processes at the ultrastructural level, the nature and timing of these events at the molecular level remain poorly defined. The recent development of techniques to transfect Plasmodium falciparum (1, 2) and target specific genetic loci through homologous recombination (3) provides the opportunity to address the role of molecules believed to play a role in invasion.
P. falciparum is known to use different receptors for invasion of erythrocytes and commonly invades via sialic acid residues present on glycophorin A or B (4). Mutant red blood cells (RBCs) with modifications or deficiencies in glycophorins are, in general, less susceptible to invasion by P. falciparum than normal erythrocytes (5, 6). Similarly, treatment of erythrocytes by enzymes that modify the structure of glycophorins renders them less susceptible to invasion (7, 8). Moreover, P. falciparum appears not to be restricted to invasion via a single class of erythrocyte receptor. Indeed, there is mounting evidence of receptor heterogeneity in both laboratory (4, 6, 9) and field isolates (10). The possibility of “switching” between, or up-regulating, novel invasion phenotypes introduces a further element of complexity into the invasion process and was demonstrated through the ability of Dd2 to be selected for invasion into neuraminidase (Nm)-treated erythrocytes (11).
Based on high degrees of structural conservation at both the genetic and protein levels, the Plasmodium erythrocyte binding ligands reported to date have been ascribed to an erythrocyte binding protein family (12) that includes the P. falciparum sialic acid-binding protein, EBA-175 (erythrocyte binding antigen 175). EBA-175 exclusively binds sialic acid residues present on glycophorin A of human erythrocytes (13, 14). This protein and other members of the erythrocyte binding protein family have extracellular N- and C-terminal cysteine-rich regions in addition to transmembrane and cytoplasmic domains. The two cysteine-rich regions (often referred to as regions II and VI) bear numerous conserved cysteine residues, with region II possessing the erythrocyte binding function (14–16).
This study presents an example of the targeted disruption of a member of the erythrocyte binding protein family in P. falciparum. The EBA-175 protein was truncated such that only regions I–V were expressed and no longer contains the 3′ cysteine-rich domain (region VI), the transmembrane domain, or cytoplasmic tail. Although the resultant molecule retains a reduced capacity to bind human erythrocytes in vitro, a switch in invasion phenotype has resulted such that the parasites are no longer reliant on EBA-175 binding to sialic acid residues present on glycophorin A for invasion.
Materials and Methods
Plasmid Construction.
Genomic DNA from D10 parasites was used to PCR a 1.5-kb DNA fragment from the central portion of the eba-175 coding sequence (exon I; regions III–V) using oligonucleotide primers EBA 1 (5′-ataagaaataatgaacaaac-3′) and EBA 2 (5′-atgtagattattcatggtatgg-3′). XhoI restriction sites were incorporated into each oligonucleotide to facilitate cloning into XhoI-restricted pHC1 (17). The pHC1-Δeba plasmid generated in this manner was subsequently restricted with XmnI/XhoI to release a 1,457-bp eba-175 fragment that was subcloned into HinCII/XhoI-restricted pHH1 to create pHH1-Δeba. The pHH1 series of plasmids (18) contain a human dhfr fragment mutated to encode resistance to WR99210 (19).
Parasite Materials and Transformation.
P. falciparum parasites were grown in human O+ erythrocytes as described (20). Because of the inability to Percoll purify schizont-infected erythrocytes derived from W2-mef parasites, an alternate strategy to that described (13) was required to obtain culture supernatant containing EBA-175. The supernatants from synchronized parasites at 8–10% parasitaemia were collected after schizont rupture, were centrifuged at 1,500 rpm (Beckman GS-6KR), and were stored at −70°C. Before use, supernatants were centrifuged at 13,000 rpm (Eppendorf 5415C), to remove cellular debris. Metabolic labeling of parasites was carried out in a similar manner. Synchronized parasites were grown to late trophozoite stage, at which time the medium was replaced with methionine-deficient medium (GIBCO/BRL) supplemented with [3H]isoleucine (50 μCi/ml; Amersham Pharmacia) and Expre35S35S-protein labeling mix (100 μCi/ml; NEN). Culture supernatants were collected 18 h after addition of the radiolabel. Transfection of W2-mef parasites with 100 μg of CsCl purified pHH1-Δeba was carried out as described (19, 21).
Antibody Production.
To obtain polyclonal antibodies directed toward regions III–V of the EBA-175 protein sequence, oligonucleotide primers 5′-caagaagcagttcctgaggaaaac-3′ and 5′-cccagaatttcccccccgatcttgtc-3′ were used to amplify the DNA sequence encoding amino acids 761–1271 and were cloned into pGEX-4T-2 (Amersham Pharmacia) in-frame with the glutathione S-transferase sequence. Fusion protein was affinity-purified according to standard procedures. Polyclonal antibodies were purified from rabbit sera from which glutathione S-transferase specificities had been removed by affinity chromatography using purified EBA-175/glutathione S-transferase fusion protein coupled to CNBr-activated Sepharose (Amersham Pharmacia). Polyclonal antibodies were also generated to region VI of the EBA-175 protein sequence. This region was amplified by using oligonucleotide primers 5′-aataatattccaagtagatataatttatatg-3′ and 5′-atgaaaaagcctcctttctgaaacatg-3′ and was cloned into the pProEX-HTb vector (GIBCO/BRL) with a histidine affinity tag. Washed inclusion bodies containing the recombinant fusion protein were solubilized in 8 M urea, 20 mM Tris (pH 8), 1.4 mM cystamine, and 2.4 mM 2-mercaptoethylamine and were refolded by dialysis into 20 mM Tris (pH 8.25), 150 mM NaCl, 1.4 mM cystamine, and 2.4 mM 2-mercaptoethylamine. The refolded protein was further purified through Ni-NTA chromatography (Qiagen, Chatsworth, CA).
Immunofluorescence.
Smears were fixed in methanol (−20°C) and were processed for indirect immunofluorescence (22). Affinity-purified αEBA-175 (middle) antibodies were used at 1:500 followed by FITC-conjugated anti-rabbit IgG (Silenus, Paris).
Erythrocyte Binding Assays.
Binding of metabolically labeled supernatant material to uninfected erythrocytes was according to established protocols (23) except that, before salt elution, centrifugation of erythrocytes through silicon oil was performed twice to minimize carryover of unbound material. Trypsin and Nm treatment of washed erythrocytes was performed as described (4) except that 1 mg/ml trypsin (Sigma) and 1 unit of Vibrio cholerae Nm (Calbiochem) were used to treat 4 × 109 erythrocytes. Soybean trypsin inhibitor (Sigma) was added to the same number of washed, trypsin-treated erythrocytes at 0.5 mg/ml.
Immunoprecipitation.
Erythrocyte binding assays were carried out with metabolically labeled culture supernatants. After pelleting and washing in serum free medium, erythrocytes were lysed with 0.5 ml of PBS containing 1% TX-100 and were centrifuged briefly at 13,000 rpm (Eppendorf 5415C), and the supernatant was incubated with affinity-purified αEBA-175 (middle) antibodies (1:100) for 2 h at 4°C. Immune complexes were precipitated with protein G-Sepharose (Amersham Pharmacia) for 2 h at 4°C. After washing with 1% TX-100/PBS, the beads were resuspended in reducing sample buffer. After SDS/PAGE, precipitated antigens were detected by fluorography.
Invasion Assays.
The inability to purify W2-mef schizont-infected erythrocytes by Percoll-gradient centrifugation necessitated a novel method to measure erythrocyte invasion. This method relies on the inability of merozoites to invade RBCs treated with both Nm and trypsin. Forty-eight hours before inclusion in invasion assays, parasites were synchronized by sorbitol lysis. On the day of the assay, ring stage parasites were adjusted to 2% parasitaemia (4% hematocrit) and were synchronized for a second time before being placed back into culture for 3 h. After this time, approximately 109 erythrocytes containing 2 × 107 parasites were washed twice with serum-free RPMI-1640/Hepes medium (0.2% NaHCO3), resuspended in the same buffer and mixed with 0.33 units of Nm for 1 h at 37°C. After washing twice, the parasites were resuspended and treated with 1 mg/ml trypsin for 1 h at 37°C. The cells were washed, resuspended, and treated for 10 min at room temperature with 0.5 mg/ml soybean trypsin inhibitor. Finally, the parasites were washed and resuspended in complete medium and were incubated overnight under standard conditions. To set up the invasion assays, 2 × 107 uninfected “target” erythrocytes (normal or enzyme treated) were added to 2 × 107 double enzyme-treated erythrocytes (4 × 105 schizont-infected) contained within 96-well microtiter trays in a final volume of 200 μl of culture medium. After 24 h of incubation, the percentage of ring-stage parasites present within 1,000 erythrocytes was determined from Giemsa-stained thin blood smears. Two independent assays were performed, each in triplicate. Control wells in which only the double enzyme-treated parasitized erythrocytes were present were included in each assay to enable background correction.
Results
Targeting and Truncation of the eba-175 Gene.
To generate a parasite line expressing a truncated form of EBA-175, a vector (pHH1-Δeba) for transformation of P. falciparum was constructed that incorporated most of regions III–V (exon 1; nucleotides 2,345–3,789) of the eba-175 gene within the pHH1 transfection vector (18). W2-mef parasites (24) were chosen in this study because they depend on sialic acid residues present on glycophorin A for normal rates of invasion. Hence, it was thought that this parasite background relied on EBA-175 for normal invasion of erythrocytes. After transformation, the 3′cys-rich domain (region VI) as well as the transmembrane and cytoplasmic domains (region VII) would be predicted to be deleted after a single crossover event between the eba-175 gene and the partial eba-175 insert in pHH1-Δeba. Conversely, regions I–V (exon 1), including the signal peptide and 5′cys-rich domain (region II), would be expected to be expressed within these parasites (Fig. 1A).
Figure 1.

Truncation of the eba-175 locus. (A) Schematic representations of the pHH1-Δeba transfection plasmid (7.1 kb) and eba-175 genetic locus (wild-type and transgenic) are shown. The four exons of eba-175 are indicated. The features represented are human dihydrofolate reductase selectable marker (hDHFR); P. berghei dhfr-ts 3′ untranslated region (Pb-DT 3′); duplicate 5′ cysteine rich domains (F1, F2); 3′ cysteine rich domain (3′cys); transmembrane domain (TM). The bracket indicates the approximate boundaries between which two copies of pHH1-Δeba have integrated. SacI (S) and MfeI (M) restriction sites used in mapping the plasmid integration events are shown. Regions III–V against which antisera was raised are indicated. (B) Southern blot analysis of MfeI- and SacI-restricted genomic DNA isolated from W2-mef and the two truncation mutants W2-m/Δc.1 and W2-m/Δc.2 indicating complete disruption of the endogenous eba-175 locus. The Southern blot was probed with a fragment of eba-175 internal to the pHH1-Δeba insert. The presence of the approximately 7.1-kb fragment indicates that two copies of pHH1-Δeba have co-integrated. (C) Southern blot analysis of HindIII-restricted genomic DNA probed with the repetitive probe, rep20, demonstrates that each of the truncation clones is derived from W2-mef.
The structure of the eba-175 locus after integration of pHH1-Δeba was confirmed for two independent clones by PCR (data not shown) and Southern blot analysis of MfeI- and SacI-restricted genomic DNA probed with a fragment of eba-175 (Fig. 1B). The loss of the hybridization signal corresponding to the endogenous eba-175 locus, concurrent with the appearance of additional fragments of expected number and size, confirmed the nature of the integration events for the two clones, W2-m/Δc.1 and W2-m/Δc.2. In addition, the presence of the 7.1-kb hybridization signal in both the MfeI and SacI digests indicates that two copies of pHH1-Δeba have co-integrated into the same site (Fig. 1B).
To confirm that the W2-m/Δc.1 and W2-m/Δc.2 clones were derived from the W2-mef parent as expected, a highly repetitive genetic element (rep20) that is contained within the telomeric regions of most P. falciparum chromosomes (25) was used to probe HindIII-restricted genomic DNA. Previous studies have used rep20 to successfully type P. falciparum strains (26). In this manner, we were able to confirm that W2-m/Δc.1 and W2-m/Δc.2 are both subclones of W2-mef (Fig. 1C).
Expression of the Truncated EBA-175 Protein.
For analysis of EBA-175 protein expression within W2-mef and each of the transfectant lines, polyclonal antisera was raised in rabbits against amino acids 761-1271 (regions III–V) of the EBA-175 sequence. This region includes the entire eba-175 insert sequence present in pHH1-Δeba. Hence, given the structure of the integration events in W2-m/Δc.1 and W2-m/Δc.2 (Fig. 1A), the antibodies would be expected to react with each of the three parasite lines examined in this study. Affinity purified αEBA-175 (middle) antibodies were incubated with Western blots of parasite-associated proteins isolated after saponin lysis of infected RBCs as well as parasite proteins released into the culture supernatant. As expected, the αEBA-175 (middle) antibodies react with an approximately 190-kDa protein within the W2-mef parasite pellet and an approximately 175-kDa protein within the W2-mef culture supernatant (Fig. 2A) (13). A smaller, less abundant protein was also detected in both samples and presumably represents either a specific cleavage event or a degradation product. For the protein samples derived from W2-m/Δc.1 and W2-m/Δc.2, a smaller protein of approximately 160 kDa was detected in both the parasite pellet and culture supernatant. Given that EBA-175 expressed by W2-m/Δc.1 and W2-m/Δc.2 is predicted to be missing approximately 25 kDa from the C terminus, the 160-kDa molecule corresponds with the size of the protein expected to result from the truncation event (Fig. 2A). Unlike EBA-175 of the W2-mef parent, there is no obvious size difference between the pellet and supernatant forms of truncated EBA-175 as it is released into the culture supernatant. There is, however, a minor cleavage or degradation product present in the supernatant reactive with the EBA-175 antibodies.
Figure 2.

Western blot analysis of EBA-175 protein expression. (A) Western blot analysis (5% SDS/PAGE) of parasite pellet and culture supernatant proteins isolated from W2-mef and the clones W2-m/Δc.1 and W2-m/Δc.2. The membrane was probed with antibodies directed toward regions III–V of EBA-175 and indicates that the approximately 160-kDa truncated form of EBA-175 is expressed and released into the culture supernatant at similar levels to the wild-type protein. Before its release into the culture supernatant, wild-type EBA-175 has an approximate molecular weight of 190 kDa. The soluble form of EBA-175 present within culture supernatant has an apparent molecular weight of 175 kDa. (B) Western blot analysis of parasite pellet proteins probed with antibodies raised against the 3′cys-rich domain confirms the absence of this region within the truncation mutants. The arrow indicates the position of wild-type EBA-175 detected with these antibodies.
Within both the cell pellet and supernatant samples, the level of expression of EBA-175 by the W2-mef parent and clones W2-m/Δc.1 and W2-m/Δc.2 appears to be approximately equal. This is not surprising, given that the truncated EBA-175 is still expressed from the endogenous eba-175 promoter. To confirm that equal amounts of protein were present in each of the cell pellet samples, Western blots were also probed with antibodies against heat shock protein 70 (data not shown).
It is expected that the 3′cys-rich region and everything downstream would not be expressed in W2-m/Δc.1 or W2-m/Δc.2. To confirm this, rabbit polyclonal antibodies directed against amino acids 1,284–1,394 (including the 3′cys-rich domain; region VI) were used to probe Western blots of parasite pellet proteins obtained after saponin lysis (Fig. 2B). It is clear from this experiment that W2-m/Δc.1 and W2-m/Δc.2 do not express the 3′cys-rich region of EBA-175.
Immunolocalization of the Truncated EBA-175 Protein.
In each of the three lines, the punctate fluorescence pattern observed through the use of the αEBA-175 (middle) antibodies in indirect immunofluorescence was characteristic of antigens localized within the apical complex of merozoites (27) (Fig. 3). This is consistent with the reported micronemal localization of EBA-175. No significant difference in fluorescence intensity was noted between W2-mef and either W2-m/Δc.1 or W2-m/Δc.2 parasites. In each case, staining was most intense in the apical region of developing merozoites within segmenting schizonts and also free merozoites released after schizont rupture. In some instances, the outline of free merozoites appears to fluoresce, suggestive of a surface localization. This latter finding remains to be confirmed. After careful examination of a large number of fields within several immunofluorescence assays, it was concluded that deleting the 3′cys-rich, transmembrane, and cytoplasmic domains has no obvious effect on EBA-175 localization or the degree of protein expression.
Figure 3.

Immunolocalization analysis of wild-type and mutant EBA-175. Representative images of indirect immunofluorescence labeling using antibodies directed toward regions III–V of EBA-175 are presented for W2-mef and W2-m/Δc.1 parasites. In each case, staining was most intense in the apical region of developing merozoites within segmenting schizonts and also free merozoites released after schizont rupture. This punctate fluorescence pattern is characteristic of antigens localized within the apical complex of merozoites.
Erythrocyte Binding Activity of Truncated EBA-175.
Metabolically labeled parasite proteins released into the culture supernatant after schizont rupture and merozoite release were used in binding assays to define the erythrocyte binding specificity of truncated EBA-175 expressed by W2-m/Δc.1 and W2-m/Δc.2. To confirm that each of the parasite lines was radiolabeled to an equivalent degree, equal volumes of culture supernatant derived from metabolically labeled parasites were analyzed by SDS/PAGE and fluorography (Fig. 4A). This result confirmed that wild-type EBA-175 is not expressed in W2-m/Δc.1 or W2-m/Δc.2. Instead, the expected 160-kDa truncated form of EBA-175 is expressed and radiolabeled in these two clones. Aside from a slightly increased level of protein within the W2-mef supernatant (which was adjusted before the assays), no other differences were readily detectable on inspection of the profiles of the labeled parasite proteins (Fig. 4A).
Figure 4.
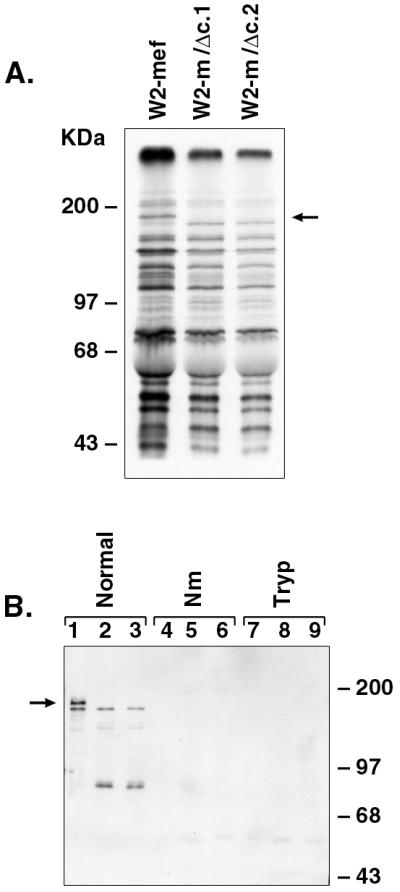
Erythrocyte binding assays of metabolically labeled parasite proteins. (A) Metabolically labeled culture supernatant proteins derived from merozoites released after schizont rupture were analyzed by SDS/PAGE (7%) and fluorography. The absence of wild-type EBA-175 within the culture supernatants obtained from W2-m/Δc.1 and W2-m/Δc.2 is indicated by an arrow. (B) Erythrocyte binding assays were carried out by incubating metabolically labeled parasite proteins present in culture supernatant with either normal, neuraminidase-treated (Nm), or trypsin-treated (Tryp) erythrocytes. After salt elution, labeled proteins were analyzed by Western blotting using αEBA-175 antibodies. Lanes 1–9 are as follows: W2-mef (lanes 1, 4, and 7), W2-m/Δc.1 (lanes 2, 5, and 8), W2-m/Δc.2 (lanes 3, 6, and 9). This result confirms that wild-type and truncated EBA-175 binds normal erythrocytes, although binding of truncated EBA-175 appears to be consistently less than that of the wild-type EBA-175. Neither wild-type nor truncated EBA-175 binds Nm- or trypsin-treated erythrocytes. Soybean trypsin inhibitor alone has no effect on protein binding (not shown). The arrow indicates the position of wild-type EBA-175.
As reported (13), relatively few of the parasite proteins released into the culture supernatant bind RBCs efficiently, and by far the major erythrocyte binding protein able to be identified in this manner is EBA-175. The Western blot analysis result clearly demonstrates that both full-length EBA-175 and the 160-kDa truncated form expressed by W2-m/Δc.1 and W2-m/Δc.2 bind normal human erythrocytes (Fig. 4B). An approximately 90-kDa cleavage product is also detected within the W2-m/Δc.1 and W2-m/Δc.2 samples. As predicted, neither full-length or truncated EBA-175 binds RBCs that have been treated with Nm or trypsin. When the parental line, W2-mef, was compared with the W2-m/Δc.1 and W2-m/Δc.2 clones, no consistent differences were observed in the parasite proteins eluted from either Nm or trypsin treated erythrocytes, as determined by SDS/PAGE and fluorography (data not shown). Similarly, when EBA-175-derived proteins are discounted, there were no novel proteins able to be detected within the W2-m/Δc.1 or W2-m/Δc.2 samples bound to normal RBCs (data not shown).
It is apparent from the Western blot that wild-type EBA-175 may be binding normal erythrocytes to a greater degree than the truncation mutant (Fig. 4B). To explore this possibility further, immunoprecipitation was carried out on parasite proteins bound to normal erythrocytes by using the affinity-purified αEBA-175 (middle) antibodies. The extent to which erythrocyte binding was diminished through truncation of EBA-175 was even more pronounced in these experiments (Fig. 5) and is consistent with the hypothesis that removal of the 3′ cys-rich domain has an adverse effect on erythrocyte binding in vitro. Importantly, both wild-type and mutant EBA-175 are able to be immunoprecipitated directly from supernatant to an equivalent extent (data not shown). The 90-kDa EBA-175 cleavage product was again detected within the W2-m/Δc.1 and W2-m/Δc.2 samples. It is not yet clear whether the approximately 140-kDa molecule co-precipitated from each of the samples represents either a degradation product or a protein that specifically interacts with EBA-175 during erythrocyte binding.
Figure 5.
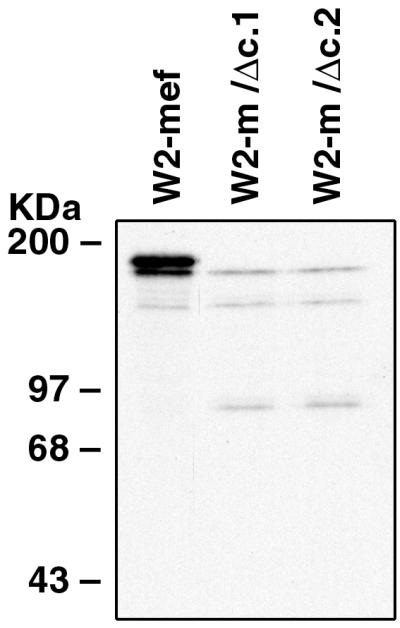
Immunoprecipitation analysis of EBA-175 bound to erythrocytes. Erythrocytes to which labeled parasite proteins had been bound were lysed with TX-100 and were incubated with αEBA-175 antibodies. Immune complexes were precipitated with Protein G-Sepharose and were analyzed by SDS/PAGE (7%) followed by fluorography. Erythrocyte binding of truncated EBA-175 to RBCs is significantly less than that of wild-type EBA-175. It is unclear whether the approximately 140-kDa protein co-precipitated in each sample represents a specific EBA-175 binding protein.
Invasion of Erythrocytes by W2-m/Δc.1 and W2-m/Δc.2.
The invasion phenotype of the Dd2 clone derived from W2-mef is well described (4) and was confirmed for the W2-mef line used in the present study (Table 1). These parasites are unable to invade Nm-treated erythrocytes to any significant extent and, hence, completely rely on sialic acid for invasion. On the other hand, W2-mef are able to invade trypsin treated cells, albeit at reduced efficiency (approximately 60% of invasion into normal cells). From this information, we are able to deduce that W2-mef parasites are capable of invading erythrocytes via sialic acid residues present on both glycophorins A and B and at least partially rely on EBA-175 binding to glycophorin A for normal rates of invasion. Hence, W2-mef provided a good model with which to test the ability of EBA-175 truncation mutants to invade RBCs via glycophorin A. Unlike the W2-mef parent, W2-m/Δc.1 and W2-m/Δc.2 are able to invade erythrocytes in a sialic acid-independent manner, as demonstrated by their ability to invade Nm-treated RBCs (Table 1). In these experiments, the efficiency of invasion into Nm treated cells was 85% of invasion into normal erythrocytes. The fact that the sialic acid-independent pathway that operates for W2-m/Δc.1 and W2-m/Δc.2 is inactive after trypsin treatment is consistent with these parasites invading via the so-called trypsin sensitive “X-receptor” (4). Invasion into RBCs treated with trypsin alone does not appear to have been significantly affected through truncation of EBA-175.
Table 1.
Effect of EBA-175 truncation on invasion phenotypes
| Strain | Relative invasion (percent of control)
|
||
|---|---|---|---|
| Neuraminidase | Trypsin | Neuraminidase + trypsin | |
| W2-mef | 0.0 | 62.0 ± 14.4 | 0.0 |
| W2-m/Δc.1 | 83.2 ± 11.1 | 59.9 ± 7.0 | 0.0 |
| W2-m/Δc.2 | 85.7 ± 2.0 | 73.7 ± 9.1 | 0.0 |
Values are the percent invasion rates relative to invasion in normal, untreated cells and represent the average of two independent assays, each performed in triplicate. Values for re-invasion into normal, untreated cells ranged from 3.8–5.5% parasitaemia as compared to 0–0.3% for background controls.
Discussion
In the present study, the ability to truncate the eba-175 gene clearly demonstrates that full length EBA-175 is not required for merozoite invasion or parasite growth. Truncating the eba-175 gene also has no effect on either the level of EBA-175 protein expression or its subcellular localization. Similarly, there appears to be no impairment in the ability of soluble EBA-175 to be released into the culture supernatant after schizont rupture. Each of these results would seem to argue against the idea that the transmembrane and cytoplasmic domains participate in intracellular trafficking and sequestration of EBA-175, as has been suggested (28). Moreover, this disruption is associated with a switch in invasion pathway from a sialic acid-dependent pathway to one that is independent of sialic acid.
The results described herein are particularly interesting in light of recent findings that demonstrate that deletion of the cytoplasmic tail of the Plasmodium berghei thrombospondin-related adhesive protein (TRAP) does not alter either its micronemal or surface localization within sporozoites. These TRAP mutants, however, are incapable of invading mammalian cells or mosquito salivary glands and exhibit an atypical pattern of motility (29). Hence, given that RBC invasion via the EBA-175/glycophorin A route appears to have been affected within the W2-m/Δc.1 and W2-m/Δc.2 mutants, it is possible that the EBA-175 and TRAP cytoplasmic domains play analogous roles in invasion that are not directly related to adhesion. Consistent with this hypothesis, the cytoplasmic tail of EBA-175, like TRAP and TRAP-related proteins from other Apicomplexan parasites, is found to be rich in acidic amino acid residues (22%) (29). These cytoplasmic domains, for example, may transmit and/or receive external signals obtained on interaction of the extracellular binding regions with receptors on target cells. Alternatively, both the EBA-175 and TRAP cytoplasmic tails may interact with elements of the parasite cytoskeleton.
The phenotypic switch of the transfectants away from the sialic acid-dependent invasion pathway of the W2-mef parental line to sialic acid independence supports the contention that truncation of EBA-175 has functionally inactivated the protein. The parental line W2-mef is able to invade via sialic acid residues located on the RBC receptors glycophorin A and B, and removal of the ability to utilize one of these receptors may have compromised the ability of these parasites to invade efficiently. Therefore, we suggest that functional inactivation of EBA-175 has forced switching or up-regulation of a sialic acid-independent pathway to allow survival of these parasites.
The observation that truncated EBA-175 expressed by clones W2-m/Δc.1 and W2-m/Δc.2 appears to bind erythrocytes less efficiently than wild-type EBA-175 is surprising because the region II binding domain is present in each case. It is possible that significant conformational differences exist between wild-type and mutant EBA-175 resulting in inefficient RBC binding. Support for this idea comes from the apparent increase in protease sensitivity of truncated EBA-175 after binding to erythrocytes (Fig. 4B).
The appearance of a sialic acid-independent pathway of invasion for W2-m/Δc.1 and W2-m/Δc.2 provides support for the associations between EBA-175 and merozoite invasion via glycophorin A (13, 14). The ability of this pathway to account for approximately 85% of invasion into normal cells mirrors the phenotypic switch that occurs in Dd2 parasites that have been up-selected for invasion into Nm-treated RBCs (11). Unlike the up-selected Dd2 parasites, however, the switch in invasion phenotype observed in the present study occurred within parasites grown continuously in normal, human O+ erythrocytes. Similar to the Dd2/NM line, the W2-m/Δc.1 and W2-m/Δc.2 phenotype has thus far remained stable over a period of 12 weeks in the absence of any selection pressure. As with the W2-mef parent, the W2-m/Δc.1 and W2-m/Δc.2 parasites are also able to invade RBCs via trypsin-insensitive glycophorin B. The ability of P. falciparum to utilize alternate pathways for invasion is likely to provide a huge survival advantage in terms of combating immune pressure or host receptor heterogeneity. For instance, an age-dependent increase in the frequency of glycophorin A mutations has been described for humans (30). The potential for enormous diversity in invasion specificity was recently described for Plasmodium yoelii yoelii in which merozoites originating from a single schizont were each found to express a distinct member of the py235 protein family (31). This family of rhoptry proteins comprises at least 11 novel members and is thought to determine the subset of erythrocytes that the parasites invade (32). Evidence in support of the idea that P. falciparum has the capacity to generate considerable invasion diversity comes from a recent Indian field study indicating that 12 of 15 isolates were able to invade via diverse pathways independent of sialic acid residues present on glycophorin A (10).
Acknowledgments
We thank Dr. Robin Anders for assistance with antisera preparation and Deborah Baldi for providing pHC1-Δeba. This work was supported by the National Health and Medical Research Council of Australia. M.B.R. is the recipient of a National Health and Medical Research Council of Australia “Peter Doherty” PostDoctoral Fellowship.
Abbreviations
- EBA-175
erythrocyte binding antigen 175
- RBC
red blood cell
- Nm
neuraminidase
- TRAP
thrombospondin-related adhesive protein
References
- 1.Wu Y, Kirkman L A, Wellems T E. Proc Natl Acad Sci USA. 1996;93:1130–1134. doi: 10.1073/pnas.93.3.1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Crabb B S, Cowman A F. Proc Natl Acad Sci USA. 1996;93:7289–7294. doi: 10.1073/pnas.93.14.7289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Crabb B S, Cooke B M, Reeder J C, Waller R F, Caruana S R, Davern K M, Wickham M E, Brown G V, Coppel R L, Cowman A F. Cell. 1997;89:287–296. doi: 10.1016/s0092-8674(00)80207-x. [DOI] [PubMed] [Google Scholar]
- 4.Dolan S A, Proctor J L, Alling D W, Okubo Y, Wellems T E, Miller L H. Mol Biochem Parasitol. 1994;64:55–63. doi: 10.1016/0166-6851(94)90134-1. [DOI] [PubMed] [Google Scholar]
- 5.Cartron J P, Prou O, Luilier M, Soulier J P. Br J Haematol. 1983;55:639–647. doi: 10.1111/j.1365-2141.1983.tb02846.x. [DOI] [PubMed] [Google Scholar]
- 6.Hadley T J, Klotz F W, Pasvol G, Haynes J D, McGinniss M H. J Clin Invest. 1987;80:1190–1193. doi: 10.1172/JCI113178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Breuer W V, Ginsburg H, Cabantchik Z I. Biochim Biophys Acta. 1983;755:263–271. doi: 10.1016/0304-4165(83)90213-1. [DOI] [PubMed] [Google Scholar]
- 8.Hadley T J, Miller L H. Prog Allergy. 1988;41:49–71. [PubMed] [Google Scholar]
- 9.Mitchell G H, Hadley T J, McGinniss M H, Klotz F W, Miller L H. Blood. 1986;67:1519–1521. [PubMed] [Google Scholar]
- 10.Nnaemeka Okoyeh J, Pillai C R, Chitnis C E. Infect Immun. 1999;67:5784–5791. doi: 10.1128/iai.67.11.5784-5791.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dolan S A, Miller L H, Wellems T E. J Clin Invest. 1990;86:618–624. doi: 10.1172/JCI114753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Adams J H, Sim K L, Dolan S A, Fang X, Kaslow D C, Miller L H. Proc Natl Acad Sci USA. 1992;89:7085–7089. doi: 10.1073/pnas.89.15.7085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Camus D, Hadley T J. Science. 1985;230:553–556. doi: 10.1126/science.3901257. [DOI] [PubMed] [Google Scholar]
- 14.Sim B K L, Chitnis C E, Wasniowska K, Hadley T J, Miller L H. Science. 1994;264:1941–1944. doi: 10.1126/science.8009226. [DOI] [PubMed] [Google Scholar]
- 15.Chitnis C E, Miller L H. J Exp Med. 1994;180:497–506. doi: 10.1084/jem.180.2.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ranjan A, Chitnis C E. Proc Natl Acad Sci USA. 1999;96:14067–14072. doi: 10.1073/pnas.96.24.14067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Crabb B S, Triglia T, Waterkeyn J G, Cowman A F. Mol Biochem Parasitol. 1997;90:131–144. doi: 10.1016/s0166-6851(97)00143-6. [DOI] [PubMed] [Google Scholar]
- 18.Reed M B, Saliba K J, Caruana S R, Kirk K, Cowman A F. Nature (London) 2000;403:906–909. doi: 10.1038/35002615. [DOI] [PubMed] [Google Scholar]
- 19.Fidock D A, Wellems T E. Proc Natl Acad Sci USA. 1997;94:10931–10936. doi: 10.1073/pnas.94.20.10931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Trager W, Jensen J B. Nature (London) 1978;273:621–622. doi: 10.1038/273621a0. [DOI] [PubMed] [Google Scholar]
- 21.Triglia T, Wang P, Sims P F G, Hyde J E, Cowman A F. EMBO J. 1998;17:3807–3815. doi: 10.1093/emboj/17.14.3807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bianco A E, Crewther P E, Coppel R L, Stahl H D, Kemp D J, Anders R F, Brown G V. Am J Trop Med Hyg. 1988;38:258–267. doi: 10.4269/ajtmh.1988.38.258. [DOI] [PubMed] [Google Scholar]
- 23.Haynes J D, Dalton J P, Klotz F W, McGinniss M H, Hadley T J, Hudson D E, Miller L H. J Exp Med. 1988;167:1873–1881. doi: 10.1084/jem.167.6.1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oduola A M, Milhous W K, Weatherly N F, Bowdre J H, Desjardins R E. Exp Parasitol. 1988;67:354–360. doi: 10.1016/0014-4894(88)90082-3. [DOI] [PubMed] [Google Scholar]
- 25.Corcoran L M, Forsyth K P, Bianco A E, Brown G V, Kemp D J. Cell. 1986;44:87–95. doi: 10.1016/0092-8674(86)90487-3. [DOI] [PubMed] [Google Scholar]
- 26.Triglia T, Foote S J, Kemp D J, Cowman A F. Mol Cell Biol. 1991;11:5244–5250. doi: 10.1128/mcb.11.10.5244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sim B, Orlandi P A, Haynes J D, Klotz F W, Carter J M, Camus D, Zegans M E, Chulay J D. J Cell Biol. 1990;111:1877–1884. doi: 10.1083/jcb.111.5.1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sim B K L. Parasitol Today. 1995;11:213–217. doi: 10.1016/0169-4758(95)80080-8. [DOI] [PubMed] [Google Scholar]
- 29.Kappe S, Bruderer T, Gantt S, Fujioka H, Nussenzweig V, Menard R. J Cell Biol. 1999;147:937–943. doi: 10.1083/jcb.147.5.937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Akiyama M, S, K, Hirai Y, Kusunoki Y, Iwamoto K S, Nakamura N. Mutat Res. 1995;338:141–149. doi: 10.1016/0921-8734(95)00019-3. [DOI] [PubMed] [Google Scholar]
- 31.Preiser P R, Jarra W, Capiod T, Snounou G. Nature (London) 1999;398:618–622. doi: 10.1038/19309. [DOI] [PubMed] [Google Scholar]
- 32.Holder A A, Freeman R R. Nature (London) 1981;294:361–364. doi: 10.1038/294361a0. [DOI] [PubMed] [Google Scholar]