

Abstract
Microtubule (MT) ensemble polarity is a diagnostic determinant of the structure and function of neuronal processes. Here, polarized MT structures are selectively imaged with second-harmonic generation (SHG) microscopy in native brain tissue. This SHG is found to colocalize with axons in both brain slices and cultured neurons. Because SHG arises only from noninversion symmetric structures, the uniform polarity of axonal MTs leads to the observed signal, whereas the mixed polarity in dendrites leads to destructive interference. SHG imaging provides a tool to investigate the kinetics and function of MT ensemble polarity in dynamic native brain tissue structures and other subcellular motility structures based on polarized MTs.
Microtubules (MTs) are a major cytoskeletal element of neuronal cell processes and are responsible for structural support and active intracellular transport. MTs exhibit an intrinsic axial polarity, defined by different + and - ends, giving an overall noninversion symmetric structure. This polarity determines the self-assembly characteristics of the MT polymer from its tubulin subunits (1, 2) and the directionality of vesicle and organelle movements via the unidirectional molecular motors (3). The MT ensemble polarity has been implicated in determining the unique morphological and compositional features of axons and dendrites in culture (4).
Research on the role of MT ensemble polarity in the dynamical development of neuronal processes, growth cones, and injury response has been hindered by the lack of suitable techniques. The only previous technique capable of determining MT polarity is the elegant electron microscopy “hook method” (5, 6) in thin fixed sections. However, now a noninvasive imaging modality for thick living tissue (≈300- to 400-μm imaging depth) that is capable of recording MT ensemble polarity information is described here. This technique is provided by second-harmonic generation (SHG) microscopy.
SHG microscopy has emerged as an effective tool in biology (7–10) 40 years after its discovery in the early days of the laser (11) and 30 years after the discovery of SHG from collagen (12). Like two-photon fluorescence (TPF) microscopy (13), the SH signal is restricted to the focal volume (≈1 μm3) and is easily separated from the near-IR excitation. In the SH case, however, the signal is generated at exactly half the wavelength of the incident light and relies on noninversion symmetric structures for its production. Both TPF and SHG are generally carried out by using trains of ≈100-fs laser pulses with high peak intensities at the focus (≈1012 W/cm2). Addition of styryl dyes has generated the asymmetry and optical nonlinearity needed for SHG in membranes (14, 15), and recently it was found that the biologically intrinsic asymmetric structures of mitotic spindles and contractile filaments in muscle generate SH signals (16).
Here we describe intrinsic sources of SHG from cultured neurons and acute slices from the hippocampus and other brain regions of rat. SHG was observed from axons, but not from dendrites and somata. In acute hippocampal slices, we found the SHG prominently localized in the dense unmyelinated axon bundle known as the mossy fibers, with smaller signals from individual axons originating from CA3 pyramidal neurons. Additionally, we describe the SHG imaging of native, functioning cilia in the aqueductus cerebri, a brainstem ventricle, and provide SHG time series movies of mitotic spindle development in RBL cells. We establish that these signals arise only from uniform polarity MT ensembles.
Materials and Methods
Imaging. SHG and TPF microscopy were simultaneously performed on either a Bio-Rad MRC1024 or Radiance scan head on a modified inverted Olympus IX-70 microscope (Fig. 1). The excitation source was a mode-locked Ti:Sapphire laser (≈100-fs pulses at 80 MHz) (Spectra-Physics) pumped by a 5-W solid state Millennia laser (Spectra-Physics). The laser polarization was controlled via a Berek polarization compensator (New Focus, San Jose, CA) and the beam focused into the sample with one of the following (overfilled back aperture) objectives: Zeiss C-Apochromat ×10/0.45 numerical aperture (NA), Olympus UApo ×20/0.7 NA, Zeiss Fluar ×20/0.75 NA, Olympus UApo ×40/1.15 NA, Zeiss Fluar ×40/1.3 NA. The resultant SHG was collected in the transmitted (forward) direction with an Olympus XLUMPlanFl ×20/0.95 NA objective, whereas the TPF was epi-collected through the excitation objective. A combination of dichroic mirrors, band-pass and blue-glass filters (Chroma Technology, Brattleboro, VT), and polarization analyzers (Newport, Fountain Valley, CA) separated the signals for detection with Bialkali photo-multiplier tubes (PMTs) (Hammamatsu, Ichinocho, Japan). The excitation wavelength, average excitation intensity (<I>) at the sample, and two-photon 1/e radial Gaussian beam waist approximation (w) (17) are given with the figures. The average power at the sample is (<I>·π·w2). The images are temporally summed over several scans at dwell times ≈10–100 μs/μm2. Confocal imaging was performed on the same microscope, but using an argon-krypton laser for excitation.
Fig. 1.

TPF and SHG microscope. SHG and TPF images are simultaneously recorded. The key elements of the excitation are a pulsed Ti:Sapphire laser (TS) operated between 760 and 880 nm, a Berek polarization compensator (BC), a scan box (XYS), and a focusing objective (FO) (0.45–1.3 NA). For collection, TPF is epi-collected from the sample (S) and split into appropriate wavelength channels with dichroic mirrors (DM) and emission filters (EF). The predominately forward propagating SHG is collected in the transmitted light direction with a dipping objective (DO) (0.95 NA) and a plano-convex lens (L) and separated from the excitation and fluorescence light through the use of band pass and blue glass filters (F). Bialkali PMT (PMT) detectors are connected to a computer for acquisition (the SHG PMT signal is amplified before acquisition).
SHG spectra were recorded by stage scanning the sample and coupling the transmitted light into a liquid N2-cooled, fiber-coupled spectrometer (Jobin-Yvon, Edison, NJ). Relative effective SHG cross sections were corrected for power at the sample, transmission of optics, pulse width, etc. Propagation direction distributions were deduced from images of SHG from mossy fibers in both forward and epi propagation directions with fluorescence calibration for absorption and scattering in the tissue and collection efficiency differences of the optics and detectors in each direction.
Acute Hippocampal Slices. Transverse hippocampal slices 250–400 μm thick were prepared from 14- to 20-day-old Sprague–Dawley rat pups by using a vibratome and were incubated at 34°C in artificial cerebrospinal fluid (ACSF) containing 118 mM NaCl, 3 mM KCl, 1 mM KH2PO4, 1 mM MgSO4, 20 mM glucose, 1.5 mM CaCl2, and 25 mM NaHCO3. The ACSF was oxygenated with 95% O2 and 5% CO2. Imaging was performed in ACSF-filled glass-bottom culture dishes (World Precision Instruments, Sarasota, FL) at room temperature or 34°C.
Cell Cultures. Embryonic day 18 rat hippocampi obtained from BrainBits (South Illinois University School of Medicine, Springfield) were dissociated and plated in gridded glass coverslip bottom culture dishes (MatTek, Ashland, MA) at ≈5,000–50,000 cells per cm2 in phenol red free Neurobasal medium supplemented with B27, 0.5 mM GlutaMAX (GIBCO), and 25 μM glutamate (GIBCO). Neurons were incubated at 37°C in 5% CO2. Half of the medium was replaced every 3–4 days with Neurobasal medium supplemented with B27 and 0.5 mM GlutaMAX. Neurons were imaged in buffer solution containing 135 mM NaCl, 5 mM KCl, 1 mM MgCl2, 1.8 mM CaCl2, and 20 mM Hepes. RBL-2H3 cells were grown in MEM (GIBCO) supplemented with 10% heat-inactivated FBS and GlutaMAX. Stocks were kept in antibiotic free media.
Immunochemistry. Brains from Sprague–Dawley 20-day-old rat pups were perfusion fixed with 4% paraformaldehyde in PBS, postfixed overnight in the same buffer, and embedded in paraffin. Slices 10 μm thick were cut and processed for labeling with one of the following primary antibodies: β-tubulin (1:100; Sigma), MAP2 (1:200; Chemicon), Tau1 (1:200; Chemicon), SMI 312 (1:200; Sternberger–Meyer, Jarrettsville, MD), or the dynein intermediate chain (1:200; Abcam, Cambridge, U.K.). The sections were incubated with secondary antibodies conjugated to the fluorophore cy3 (1:200; Jackson ImmunoResearch).
Cultured neurons were fixed in 3.7% paraformaldehyde in PBS containing 4% sucrose at 37°C. The neurons were processed for labeling with the primary antibodies MAP2 (1:100; rabbit, Chemicon) and Tau1 (1:200; mouse, Chemicon) and incubated with rhodamine-conjugated anti-rabbit (50 μg/ml; Jackson ImmunoResearch) and coumarin-conjugated anti-mouse (6.25 μg/ml; Jackson ImmunoResearch) secondary antibodies.
Pharmacology. The slices were transferred from the static incubation bath to a static ACSF bath on the microscope stage. The bath was constantly equilibrated with 95% O2 and 5% CO2 and warmed by using an objective heater set to 34°C. Baseline images (six optical z-sections, 20 μm apart) of SHG from the mossy fibers in all slices were taken every 5 min for 20 min. Subsequently, four slices were continuously treated with 25 μM nocodazole (Sigma), four slices were continuously treated with 1 μM cytochalasin D (Sigma), and four slices (control) received continuous treatment only with solvents (DMSO + 0.05% pluronic). Imaging of the SHG from the same region continued every 5 min for 70 min. The average intensities from the three groups were calculated and compared for statistical significance with a t test.
Physics of SHG
Assuming a uniaxial molecule (as needed here) whose first hyperpolarizability tensor, 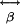 , is dominated by a single component, β0, the radiated SHG intensity (I2ω) at twice the illumination frequency is proportional to the square of the illumination intensity (Iω) (14):
, is dominated by a single component, β0, the radiated SHG intensity (I2ω) at twice the illumination frequency is proportional to the square of the illumination intensity (Iω) (14):
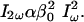 |
[1] |
This SHG stems from the power series expansion of the nonlinear induced electric dipole moment (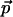 ) of a molecule driven by an excitation optical electric field (
) of a molecule driven by an excitation optical electric field (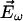 ):
):
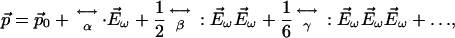 |
[2] |
where 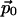 is the static electric dipole moment,
is the static electric dipole moment, 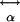 is the linear polarizability responsible for linear optics, and
is the linear polarizability responsible for linear optics, and 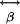 and
and 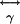 are the nonlinear first and second hyperpolarizability tensors (18, 19), respectively. SHG arises from generation arise from
are the nonlinear first and second hyperpolarizability tensors (18, 19), respectively. SHG arises from generation arise from 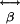 , and TPF and third-harmonic
, and TPF and third-harmonic 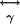 (18, 19). Molecules lacking inversion symmetry with highly inducible dipole moment changes along the asymmetric axes lead to large
(18, 19). Molecules lacking inversion symmetry with highly inducible dipole moment changes along the asymmetric axes lead to large 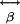 element values. Excitation electric fields interacting with such molecules induce strong SH dipoles that generate SH electromagnetic radiation. A detectable coherent SH signal is generated when scattering molecules of noninversion symmetry are spatially ordered, giving an overall asymmetry (lack of an inversion center) to the collective structure, allowing phase matching to take place between the individual scatterers. Because SHG requires coherent summation of the local SH radiation fields, I2ω depends quadratically on the number of SH scattering molecules in the focal volume (14).
element values. Excitation electric fields interacting with such molecules induce strong SH dipoles that generate SH electromagnetic radiation. A detectable coherent SH signal is generated when scattering molecules of noninversion symmetry are spatially ordered, giving an overall asymmetry (lack of an inversion center) to the collective structure, allowing phase matching to take place between the individual scatterers. Because SHG requires coherent summation of the local SH radiation fields, I2ω depends quadratically on the number of SH scattering molecules in the focal volume (14).
Results
SHG in the Hippocampus. Fig. 2A shows the observed SH signal from a hippocampal slice. The large curved white bundle corresponds to the mossy fiber axons from the dentate gyrus (DG) granular neurons that innervate the CA3 pyramidal neurons (20). Fig. 2B shows a magnified image of processes emanating from the CA3 pyramidal neurons. These processes appear to correspond to the axons leaving the hippocampus to the fimbria and may also give rise to the Schaffer collaterals, which innervate CA1 (21). We find a similar morphological correspondence between SHG and axons in other brain regions, i.e., the superior colliculus, cortex, and spinal cord (images not shown).
Fig. 2.

SHG and immunohistochemistry in hippocampal brain slices. (A) SHG from an acute slice. This mosaic is formed by 10 images, each a projection of three optical z-sections taken ≈30 μm apart. The dense mossy fiber axon bundle between the DG and CA3 is clearly seen. (B) Zoom of CA3 region of A shows individual axons emanating from the pyramidal neurons (arrowheads). Circularly polarized excitation at 880 nm with average laser intensity (<I>) = 16.3 MW/cm2 (w = 0.44 μm. (C–E) Confocal images of fixed sections immunostained for tau, MAP2, and β-tubulin, respectively (no SHG was seen with confocal imaging). The tau and SHG morphology is similar (arrows), whereas the MAP2 and SHG are not. This finding suggests an axonal origin of the SHG. Similarly, magnified regions of CA3 stained for axonal neurofilaments (F) and MAP2 (G) reveal that the individual processes seen in B are axons (arrowheads in F). A and C–E are different slices. (Scale bars = 200 μm, A; 100 μm, B, F, and G; 400 μm, C–E.)
Immunological stainings further prove the colocalization of SHG with axons. Tau immunostaining (Fig. 2C) marks axons (22) whereas MAP2 immunostaining (Fig. 2D) predominantly marks dendrites in slice preparations (23). The tau and SHG signals have almost identical morphological patterns whereas the MAP2 and SHG do not, confirming the axonal and not dendritic origin of the SHG. Note that the tau and SHG signals reveal the mossy fibers, but not other axon bundles whose trajectories are out of the plane. β-Tubulin immunostaining (Fig. 2E) reveals both axons and dendrites, but as expected, only the axonal portion colocalizes with the SHG. The SHG processes emanating from CA3 pyramidal neurons (Fig. 2B) are similar in morphology to the axonal neurofilament immunostaining of this region (Fig. 2F) (24), which reveals these axons better than the tau immunostaining. In contrast, the magnified MAP2 dendritic stain of this region (Fig. 2G) does not reveal these processes. We note that fixation leads to disappearance of the SHG signal.
Neuron Culture SHG and Immunohistochemistry. SHG originates from certain processes of hippocampal neurons in cell culture (Fig. 3A). MAP2 immunostaining [marking dendrites and somata in culture (25)] and tau immunostaining [marking axons (25)] (Fig. 3B) performed after SHG and TPF imaging reveal that the SHG and tau signals colocalize, further confirming the axonal origin, whereas dendrites and somata do not lead to SHG.
Fig. 3.

Primary hippocampal neuron cultures at 2 weeks. (A) Green pseudocolor is SHG, and red pseudocolor is intrinsic fluorescence. (B) Same location as A, but cultures now fixed and doubly immunostained for MAP2 (red) and tau (green); yellow is a colocalization of MAP2 and tau (no SHG or intrinsic fluorescence signal is present). Because tau marks axons, it is clear that the SHG emanates from axons (white arrows). Because MAP2 marks dendrites and somata, it is also clear that the SHG does not stem from either of these structures (blue arrows). (A) Elliptically polarized excitation (white elliptical arrow) at 880 nm with <I> = 30.0 MW/cm2 (w = 0.26 μm). (B) TPF images. Excitation = 780 nm. Both A and B are projections of 16 optical z-sections taken 0.5 μm apart. (Scale bars = 50 μm.)
Pharmacology: MTs Are the SHG Source. Because SHG requires spatially ordered noninversion symmetric structures, we hypothesized that the signal arises from one or more of the polymerized filaments in axons. MTs and actin filaments satisfy this requirement, but inversion symmetric neurofilaments do not (26). To test the dependence of the SHG on MTs and actin filaments, we used pharmacological agents known to depolymerize them selectively.
Nocodazole depolymerizes MTs in neuronal cell types (27). Fig. 4 shows the effect of nocodazole on the SHG from the mossy fibers. After the addition of the drug, the SHG decreased to ≈39% of its original value, as expected for this short duration treatment (28), and the control remained at ≈87% of its original value. The slight SHG drop during the control experiment can be explained by the spontaneous depolymerization of MTs in the ex vivo environment. The control and nocodazole experiments are significantly different (t test: P = 0.006). Given the specificity of nocodazole to MTs, the decrease in signal shows a direct correlation between MTs and the SHG.
Fig. 4.

Effects of cytochalasin D and nocodazole on SHG from mossy fibers. Mean SHG from mossy fibers versus time. At 20 min, control, cytochalasin D, or nocodazole solutions were added. Cytochalasin D did not affect the SHG; however, the effect of nocodazole is drastic, showing the dependence of SHG on MTs and not on actin. Error bars represent standard deviation of the four trials for each drug; 880-nm excitation with <I> = 11.4 MW/cm2 (w = 0.44 μm).
Cytochalasin D depolymerizes the actin cytoskeleton in neuronal cell types (29). Fig. 4 shows that this drug had no significant effect on the SHG compared with the control. The two data sets are not significantly different (t test: P = 0.49). Given the specificity of cytochalasin D to actin filaments, this finding shows that there is no correlation between actin and the SHG.
SHG from MTs in Non-Neuronal Structures. Non-neuronal cell structures with well-organized MT ensembles provide further evidence that SHG arises from MTs. It has previously been shown that mitotic spindles from developing Caenorhabditis elegans embryos generate SH signals under similar experimental conditions to ours (16). We find that distinct SH signals are also generated from the mitotic spindles of M-phase RBL cells and MTs nucleated from their centrosomes during interphase (Fig. 5A). Additionally, in brainstem slices we find that cilia lining the inner walls of the ventricles lead to SHG (Fig. 5B) (see Movie 1, which is published as supporting information on the PNAS web site, www.pnas.org, for time series movie of cilia motion imaged via SHG).
Fig. 5.

Non-neuronal SHG structures. (A) SHG (green pseudocolor) is seen from mitotic spindles (orange arrows) and from interphase MT ensembles (blue arrow). Red pseudocolor is intrinsic fluorescence. Projection of nine optical z-sections 0.5 μm apart. Horizontally polarized excitation at 880 nm with <I> = 81.6 MW/cm2 (w = 0.16 μm). (B) SHG (green pseudocolor) is seen from the cilia lining the walls of the aquaductus cerebri in brainstem slices. Red pseudocolor is intrinsic fluorescence. Horizontally polarized excitation at 780 nm with <I> = 31.0 MW/cm2 (w = 0.39 μm). (Scale bars: 10 μm, A; 100 μm, B.)
Signal Characterization as SHG. To establish the SH nature of the brain signal, we first noted the quadratic (2.01 ± 0.05) dependence of the signal on Iω. This finding is consistent with two-photon processes including SHG and TPF. Next, spectra were taken from a 100-μm-by-100-μm square region of the mossy fibers with varying excitation wavelengths (Fig. 6A). The emission peaks occur at exactly half of the excitation wavelength. This finding is consistent with SHG, and it excludes TPF emission, which is typically Stokes shifted by many tens of nm from the SH wavelength and remains constant regardless of substantial variation in excitation wavelength. Additionally, with an excitation bandwidth of ≈10 nm, the SHG bandwidth should be 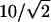 nm, typically 5 to 10 times less broad than fluorescence emission. The relative effective SHG cross section increases as the excitation is tuned toward shorter wavelengths (Fig. 6A).
nm, typically 5 to 10 times less broad than fluorescence emission. The relative effective SHG cross section increases as the excitation is tuned toward shorter wavelengths (Fig. 6A).
Fig. 6.

Spectral and polarization characterization of signal. (A) Normalized spectra from the mossy fibers of an acute hippocampal slice show peaks always at exactly half the excitation wavelength and of similar bandwidth as the excitation, proving the SHG nature of the emission. The relative effective SHG cross section increases as the excitation is moved to shorter wavelengths (error bars represent standard deviation of three trials at each wavelength). (B and C) DG axons from the same location, but different excitation polarizations (gray arrows). No emission analyzer was used. (D and E) Mossy fibers from the same location with the same excitation polarization (gray arrows), but different orientations of an emission analyzer (white arrows). (B–E) 880 nm excitation with <I> = 17.9 MW/cm2 (w = 0.44 μm). (Scale bars = 50 μm.)
We find that the SHG propagates mostly in the forward direction. Taking collection efficiencies of objectives (×10/0.45 NA focusing and ×20/0.95 NA condensing objectives), filters, and detectors into account, the ratio of forward to epi propagating SHG is 9.8 ± 2.9 and 9.6 ± 1.8 with the focusing objective changed to a ×40/1.15 NA. Such an obvious anisotropic emission is characteristic of SHG, but in stark contrast to the isotropic emission of TPF.
Finally, we find that the SHG from axons is strongly polarization dependent, maximum with the excitation light polarized parallel to the axons (Fig. 6 B and C), and essentially no SHG with the excitation polarized perpendicular to the axons (Fig. 6 B and C). We find a similar effect in the SHG emission polarization. SHG is observed with an analyzer in front of the detector oriented parallel to the excitation polarization and excited axons (Fig. 6D). As the analyzer is rotated toward a perpendicular configuration, the detected signal disappears (Fig. 6E).
Discussion
Having established the MT origin of the SHG, we can understand the observed results. Specifically, why SHG is seen only from axons, although dendrites and, to a lesser extent, somata (30) also contain MTs. The differences lie in the polarity of the MTs. Electron microscopy hook method studies of the orientation of the MT ensembles in differentiated cultured hippocampal neurons have shown that the MTs in the axons have uniform polarity. They are 95% aligned with their plus ends distal to the soma, giving an overall noninversion symmetric ensemble structure (31). In contrast, MTs in the dendrites have mixed polarity with ≈50% with their + ends distal to the soma and ≈50% oppositely oriented. No concentrations of uniform polarity MTs are found in the dendrites, giving an overall inversion symmetric ensemble structure (31). In fact, the SH-generating regions of interphase RBL cells, cilia, mitotic spindles, and axons have only one thing in common: uniform polarity MTs (see below). Therefore, we see that uniform polarity MT ensembles support SHG and are responsible for the SHG seen here in axons.
MT-Associated Proteins (MAPs) and Molecular Motors Are Not the SHG Source. The nocodazole depolymerization, immunostaining, and morphology experiments that indicate the MT origin of the SH signal do not rigorously exclude MAPs or molecular motors as possible SH generators. These molecules could, in principle, gain the orientation necessary to support SHG through their binding to the polar MTs, leading to SHG in regions where MTs are aligned with uniform polarity. However, the (tubulin)/(MAP or molecular motor) number ratio is ≈10–100 (32, 33). Therefore, the quadratic dependence of the SHG on the number of scatterers makes MTs the effective signal generators. Although the MT structures generating a SH signal contain various different MAPs and molecular motors, the only elements common to all of these structures are uniform polarity MTs and dynein (3, 31, 33–38). In mitotic spindles the dynein is quite sparse and is known to be localized in specific regions; therefore it cannot account for the observed pattern of SHG (34). The immunological stains of hippocampal brain slices for dynein (not shown) reveal a distribution having no resemblance to the SHG morphology. These facts show that MAPs and molecular motors do not significantly participate in the SHG.
Axons Versus Dendrites in SHG. Individual MTs contain an overall axial asymmetry; however, the elements of the hyperpolarizability tensor, 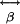 , are not known. Because MTs are aligned with axons and dendrites (31), the observed excitation polarization effect (Fig. 6 B and C) shows that SH dipole radiation is produced when the MTs and the excitation polarization vector are parallel. Excitation polarization perpendicular to MTs yields no SHG. The emission polarization experiments (Fig. 6 D and E) show that SH dipoles are induced along the asymmetric axial direction of the MT polymer, proving the existence of a dominant diagonal element of
, are not known. Because MTs are aligned with axons and dendrites (31), the observed excitation polarization effect (Fig. 6 B and C) shows that SH dipole radiation is produced when the MTs and the excitation polarization vector are parallel. Excitation polarization perpendicular to MTs yields no SHG. The emission polarization experiments (Fig. 6 D and E) show that SH dipoles are induced along the asymmetric axial direction of the MT polymer, proving the existence of a dominant diagonal element of 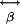 . We presume that the loss of SHG after fixation is caused by protein cross-linking disrupting the electronic configuration and increasing the inversion symmetry of the MTs.
. We presume that the loss of SHG after fixation is caused by protein cross-linking disrupting the electronic configuration and increasing the inversion symmetry of the MTs.
To understand physically the difference in SHG between axons and dendrites, the phase of the induced SH electric dipole in each MT must be considered. The relative phase between neighboring MT SH dipoles is determined by the relative polarity of the MTs. If MTs are aligned with the same axial polarity (axons), the induced SH dipoles will be in phase, and if they are aligned with opposite axial polarity (dendrites) the SH dipoles will be 180° out of phase. The MTs in axons and dendrites are typically separated by a subresolution distance of ≈90 nm, corresponding to one-fifth of a wavelength for 440-nm SH light (31, 39–41). Because of this extreme proximity, the SH dipole radiation produced by neighboring MTs in axons will constructively combine to produce coherent propagating SH radiation whereas the radiation from MTs in dendrites will destructively interfere producing no SH radiation. Although this explanation is qualitatively sufficient, a detailed calculation of the radiated SH signal generated by a focused laser beam is complicated (42), requiring coherent summation of the SH radiation from each MT driven at the local phase of the focused excitation illumination.
Photodamage Minimization. Because the SHG is relatively weak, it is necessary to use relatively high <I> at the sample and temporal summing for an acceptable signal-to-background ratio (≈2) in imaging. The high <I> used here (≈10–100 MW/cm2) may cause photodamage through absorption, but not through SHG because of its nonabsorptive scattering photophysics. Fortunately, the long wavelength excitation (≈880 nm) predominantly used here is relatively benign because its absorption in tissue is negligible. We find that with 880-nm excitation with <I> = 50.2 MW/cm2 (w = 0.16 μm), and 118 μs/μm2 dwell time, it is possible to watch RBL cells dividing while imaging their mitotic spindles with SHG (see Movie 2, which is published as supporting information on the PNAS web site). These cells divide and fully “pinch off,” showing their tolerance of the incident laser intensity and full functioning of the mitotic machinery. This two-photon excitation dose (<I>2 times dwell time) is ≈10 times the dose used during our deep brain tissue imaging experiments (i.e., Fig. 2 A). High <I> at 880 nm appears to be acceptable for repeated imaging of MT polarity development in living neuronal samples. The same cell division experiments repeated with 780 ± 20-nm excitation showed no signs of mitotic activity. Continuous SHG imaging at ≈780 nm (i.e., Fig. 7) allows ≈10 scans in tissue and neuron cultures before photoinduced morphological changes are seen and SHG begins to disappear. Considering these problems, it may be desirable to take “snapshots” of the MT polarity at shorter wavelengths to increase signal-to-background ratio (Fig. 6A) or simultaneously image fluorophores with TP cross sections or fluorescent emissions incompatible with SHG from ≈880-nm excitation. For repeated imaging of morphological development of MTs in living samples by SHG, the longer wavelengths are clearly preferred.
Fig. 7.

SHG should allow for following neuronal polarity development in living brain tissues. Experiments on hippocampal neurons in culture suggest it is possible to follow ensemble MT polarity development in living neurons over time. (A) Neuron after 5 days in culture; at this development stage, MT polarity is uniform not only in the nascent axon (blue arrow), but also the proto-processes (40) (orange arrow). Green pseudocolor is SHG, and red pseudocolor is intrinsic fluorescence. Approximately circularly polarized excitation at 760 nm with <I> = 109.3 MW/cm2 (w = 0.14 μm). (B) Neuron after 7 days in culture (neuron different from A); at this stage in development, the MTs in dendrites (not seen in this image, but present in wide field illumination) have attained a mixed polarity but the axon (blue arrow) remains with uniform polarity MTs (40). Green pseudocolor is SHG, and red pseudocolor is intrinsic fluorescence. Circularly polarized excitation at 800 nm with <I> = 48.6 MW/cm2 (w = 0.16 μm). (C) Image of axons funneling into the mossy fibers in the DG in an acute hippocampal slice. Axonal morphology seen is similar to that of the neuron in B, indicating the possibility of investigating the development of ensemble MT and neuronal polarity with SHG in vivo or ex vivo. Green pseudocolor is SHG, red pseudocolor is intrinsic fluorescence, and the dark band is the somatic layer of the DG granular neurons. Horizontally polarized excitation at 780 nm with <I>= 24.8 MW/cm2 (w = 0.39 μm). (Scale bars = 20 μm.)
Applications of SHG Imaging of MTs. Our studies show that SHG is well suited for imaging uniform polarity MT ensembles deep in living brain tissue and will find many applications. For example, MTs have been at the heart of many neuronal polarity development studies, involving transport, ensemble polarity, stabilization and density (3, 43–45). Nearly all such studies have been performed in vitro or in fixed samples, but many questions remain about the bearing their results have on neuronal development in vivo (46). Fig. 7 demonstrates the possibility of following such development in unfixed in vitro and ex vivo samples with SHG. Similarly, little is known about MT ensemble polarity in active growth cones, neuronal repair, the dynamics of migrating cells, and neurodegenerative diseases. SHG should prove valuable for studies on these and other dynamic MT structures in living tissue, offering insights into MT ensemble polarity in systems currently beyond previous techniques.
Supplementary Material
Acknowledgments
We thank M. Levene and R. Molloy for helpful discussions, R. Williams and W. Zipfel for advice and instrumentation help, K. Fitch for technical support, and K. Hodgson for figure design. This work used Science Technology Center shared experimental facilities supported by the National Science Foundation under Agreement ESC-9876771. The work at Cornell University was supported by National Institutes of Health–National Center for Research Resources Biomedical Resource Grant P412RR04224 and National Institutes of Health Grant GM08267. The work at Harvard Medical School was supported by National Institutes of Health Grant AG08487, the Hellmuth Hertz Foundation, the Wenner-Gren Foundations, and the Alzheimer's Association (Pioneer Award).
Abbreviations: MT, microtubule; SH, second harmonic; SHG, SH generation; TPF, two-photon fluorescence; NA, numerical aperture; PMT, photo-multiplier tube; DG, dentate gyrus; <I>, average excitation intensity; w, two-photon radial beam waist; MAP, MT-associated protein.
References
- 1.Bergen, L. G. & Borisy, G. G. (1980) J. Cell Biol. 84, 141-150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Binder, L. I., Dentler, W. L. & Rosenbaum, J. L. (1975) Proc. Natl. Acad. Sci. USA 72, 1122-1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baas, P. W. (1999) Neuron 22, 23-31. [DOI] [PubMed] [Google Scholar]
- 4.Baas, P. W. (2000) Microsc. Res. Tech. 48, 75-84. [DOI] [PubMed] [Google Scholar]
- 5.Heidemann, S. R. & Euteneuer, U. (1982) Methods Cell Biol. 24, 207-216. [DOI] [PubMed] [Google Scholar]
- 6.Heidemann, S. R. & Mcintosh, J. R. (1980) Nature 286, 517-519. [DOI] [PubMed] [Google Scholar]
- 7.Moreaux, L., Sandre, O., Charpak, S., Blanchard-Desce, M. & Mertz, J. (2001) Biophys. J. 80, 1568-1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roth, S. & Freund, I. (1982) J. Appl. Crystallogr. 15, 72-78. [Google Scholar]
- 9.Bouevitch, O., Lewis, A., Pinevsky, I., Wuskell, J. P. & Loew, L. M. (1993) Biophys. J. 65, 672-679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wilson, T. & Sheppard, C. (1984) Theory and Practice of Scanning Optical Microscopy (Academic, London).
- 11.Franken, P. A., Weinreich, G., Peters, C. W. & Hill, A. E. (1961) Phys. Rev. Lett. 7, 118-119. [Google Scholar]
- 12.Fine, S. & Hansen, W. P. (1971) Appl. Opt. 10, 2350-2353. [DOI] [PubMed] [Google Scholar]
- 13.Denk, W., Strickler, J. H. & Webb, W. W. (1990) Science 248, 73-76. [DOI] [PubMed] [Google Scholar]
- 14.Moreaux, L., Sandre, O. & Mertz, J. (2000) J. Opt. Soc. Am. B 17, 1685-1694. [Google Scholar]
- 15.Campagnola, P. J., Wei, M. D., Lewis, A. & Loew, L. M. (1999) Biophys. J. 77, 3341-3349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Campagnola, P. J., Millard, A. C., Terasaki, M., Hoppe, P. E., Malone, C. J. & Mohler, W. A. (2002) Biophys. J. 82, 493-508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Richards, B. & Wolf, E. (1959) Proc. R. Soc. London A 253, 358-379. [Google Scholar]
- 18.Chemla, D. S. & Zyss, J. (1987) Nonlinear Optical Properties of Organic Molecules and Crystals (Academic, Orlando, FL).
- 19.Bloembergen, N. (1996) Nonlinear Optics (World Scientific, Singapore).
- 20.Claiborne, B. J., Amaral, D. G. & Cowan, W. M. (1986) J. Comp. Neurol. 246, 435-458. [DOI] [PubMed] [Google Scholar]
- 21.Finch, D. M., Nowlin, N. L. & Babb, T. L. (1983) Brain Res. 271, 201-216. [DOI] [PubMed] [Google Scholar]
- 22.Binder, L. I., Frankfurter, A. & Rebhun, L. I. (1985) J. Cell Biol. 101, 1371-1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Di Stefano, G., Casoli, T., Fattoretti, P., Gracciotti, N., Solazzi, M. & Bertoni-Freddari, C. (2001) J. Histochem. Cytochem. 49, 1065-1066. [DOI] [PubMed] [Google Scholar]
- 24.Ulfig, N., Nickel, J. & Bohl, J. (1998) Cell Tissue Res. 291, 433-443. [DOI] [PubMed] [Google Scholar]
- 25.Mandell, J. W. & Banker, G. A. (1995) Neurobiol. Aging 16, 229-237. [DOI] [PubMed] [Google Scholar]
- 26.Lee, M. K. & Cleveland, D. W. (1996) Annu. Rev. Neurosci. 19, 187-217. [DOI] [PubMed] [Google Scholar]
- 27.Baas, P. W. & Ahmad, F. J. (1992) J. Cell Biol. 116, 1231-1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baas, P. W. & Black, M. M. (1990) J. Cell Biol. 111, 495-509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bradke, F. & Dotti, C. G. (1999) Science 283, 1931-1934. [DOI] [PubMed] [Google Scholar]
- 30.Yu, W. Q., Centonze, V. E., Ahmad, F. J. & Baas, P. W. (1993) J. Cell Biol. 122, 349-359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Baas, P. W., Deitch, J. S., Black, M. M. & Banker, G. A. (1988) Proc. Natl. Acad. Sci. USA 85, 8335-8339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hirokawa, N., Bloom, G. S. & Vallee, R. B. (1985) J. Cell Biol. 101, 227-239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hyams, J. S. & Lloyd, C. W. (1994) Microtubules (Wiley–Liss, New York).
- 34.Sharp, D. J., Rogers, G. C. & Scholey, J. M. (2000) Nature 407, 41-47. [DOI] [PubMed] [Google Scholar]
- 35.Euteneuer, U. & Mcintosh, J. R. (1981) Proc. Natl. Acad. Sci. USA 78, 372-376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bloom, G. S., Luca, F. C. & Vallee, R. B. (1984) J. Cell Biol. 98, 331-340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Avila, J. (1990) Microtubule Proteins (CRC, Boca Raton, FL).
- 38.Huber, G., Alaimobeuret, D. & Matus, A. (1985) J. Cell Biol. 100, 496-507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Peters, A., Palay, S. L. & Webster, H. D. (1991) The Fine Structure of the Nervous System: Neurons and Their Supporting Cells (Oxford Univ. Press, New York).
- 40.Baas, P. W., Black, M. M. & Banker, G. A. (1989) J. Cell Biol. 109, 3085-3094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yu, W. Q. & Baas, P. W. (1994) J. Neurosci. 14, 2818-2829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mertz, J. & Moreaux, L. (2001) Opt. Commun. 196, 325-330. [Google Scholar]
- 43.Baas, P. W. (2002) Int. Rev. Cytol. 212, 41-62. [DOI] [PubMed] [Google Scholar]
- 44.Rakic, P., Knyihar-Csillik, E. & Csillik, B. (1996) Proc. Natl. Acad. Sci. USA 93, 9218-9222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Teng, J. L., Takei, Y., Harada, A., Nakata, T., Chen, J. G. & Hirokawa, N. (2001) J. Cell Biol. 155, 65-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Craig, A. M. & Banker, G. (1994) Annu. Rev. Neurosci. 17, 267-310. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.