

Abstract
Although the surface proteins of human influenza A virus evolve rapidly and continually produce antigenic variants, the internal viral genes acquire mutations very gradually. In this paper, we analyze the sequence evolution of three influenza A genes over the past two decades. We study codon usage as a discriminating signature of gene- and even residue-specific diversifying and purifying selection. Nonrandom codon choice can increase or decrease the effective local substitution rate. We demonstrate that the codons of hemagglutinin, particularly those in the antibody-combining regions, are significantly biased toward substitutional point mutations relative to the codons of other influenza virus genes. We discuss the evolutionary interpretation and implications of these biases for hemagglutinin's antigenic evolution. We also introduce information-theoretic methods that use sequence data to detect regions of recent positive selection and potential protein conformational changes.
Influenza A virus is a negative-stranded RNA virus that infects roughly one-fifth of the human population each year, causing significant mortality and morbidity worldwide (1). The surface glycoproteins hemagglutinin (HA) and neuraminidase (NA) are the most important targets for the human immune system. Gradual mutations to HA continually produce immunologically distinct strains of the virus that cause annual outbreaks. An influenza infection brings lasting immunity to the infecting strain, but most people are susceptible to reinfection by a new strain within a few years.
The HA protein consists of two chains, HA1 and HA2, respectively 329 and 175 residues long. Phylogenetic reconstructions (2, 3) reveal that modifications to HA1, the immunogenic part of HA, accrue at a dramatic rate. Those sites of HA1 involved in antigen determination exhibit significantly more nonsynonymous than synonymous nucleotide substitutions (4, 5), whereas the remaining sites show the more common pattern of primarily synonymous variation. These observations suggest that HA1 is undergoing diversifying, or “positive,” Darwinian selection (6). Because immunity to an infecting strain is longlasting, and because influenza infects a large proportion of its host population each year, the antigenic regions of HA1 experience strong frequency-dependent selection for novel functional variants.
Although the influenza A NA gene also acquires substitutions rapidly, NA is not considered as important an antigenic determinant as HA (7) and is less prevalent than HA on the surface of the viral particle (8). Moreover, antibodies to NA do not neutralize the virus as do HA antibodies (9–11).
The mechanism of influenza A's antigenic plasticity, that is, how the virus continually evades immunity by producing variant strains, remains an outstanding evolutionary problem with obvious practical implications. The structure of the HA heterotrimer solved for a 1968 virus strain (12, 13), along with matrices of immunological crossreaction assays (14), has led to the identification of five antibody-combining regions, or epitopes, of the HA protein. Epitopic residues exhibit greater variability, higher ratios of replacement to silent mutations, and greater correlation with future phylogenetic trajectory (15).
Nonepitopic sites of HA do not evolve as rapidly as epitopic sites. Similarly, internal viral proteins such as matrix (M1, M2), polymerase (PB1, PB2), nucleoprotein (NP), and nonstructural protein accrue mutations very gradually, presumably because, compared to epitopic residues of HA, (i) they are hidden from antibodies and thus under less selective pressure to change, and (ii) they are structurally and functionally more fragile and cannot sustain significant mutation. As a result, influenza faces an intragenomic conflict over the mutation rate: certain genes, and specific residues within those genes, experience frequency-dependent selection to change, whereas other genes experience purifying selection to remain fixed.
In this paper, we address influenza's gene- and site-specific requirements for antigenic plasticity. We discuss the notion of codon usage biased toward substitutional or stereochemical diversification. We report that codons of HA, and particularly epitopic regions of HA, are significantly biased toward diversification relative to other influenza virus genes. We discuss the importance of these biases for HA evolution. We also introduce information-theoretic methods to detect regions of recent positive selection and potential protein conformational changes, on the basis of sequence data alone.
Codon Bias Across Taxa
Synonymous mutation was long considered neutral with respect to selection (16, 17). After all, synonymous mutations have no effect on translated gene products, so it is difficult to imagine how selection could discriminate among synonymous codons. Yet large-scale DNA sequencing has revealed a surprising amount of statistically significant codon bias, that is, the unequal usage of synonymous codons, in genomes across a wide range of taxa, such as Escherichia coli, Saccharomyces cerevisiae, and Drosophila (17–19).
The explanation of genomewide codon biases requires either that some nucleotide mutations are more frequent than others, or that selection can discriminate among synonymous codons, or both. It has been reported, for example, that mutational bias toward G/C determines codon usage patterns in Drosophila (20). Other scientists reject this explanation (17, 21, 22) because codon biases are preferentially seen at exonic regions, or because there is little correlation between intronic and exonic base usage. The most common selective explanation of bias posits that codon usage is optimized to match the relative abundances of isoaccepting tRNAs, thereby increasing translational efficiency (23, 24). Others discuss codon bias as the result of selection for regulatory function mediated by ribosome pausing (25) or selection against pretermination codons (26, 27). In RNA viruses, codon bias may also result from selection for RNA secondary structure (26, 28) or, in the case of HIV envelope, from selection to mutate nonsynonymously in hypervariable regions (29).
Measures of Codon Bias and Diversification
The most common measure of codon bias, called the effective number of codons (ENC), is analogous to the effective number of alleles in population genetics. ENC ranges from 20, when only one codon is used for each amino acid, to 61, if all synonymous codons are used in equal frequency. ENC does not describe whether codons are biased in a certain direction but rather measures the (inverse of the) probability that two randomly chosen synonymous codons are identical.
In the context of influenza virus evolution, we are interested in codon usage biased toward increasing or decreasing the effective amino acid substitution rate. Each codon has nine single-nucleotide mutational neighbors, some proportion of which correspond to silent mutations, and the remainder to substitutional mutations (or to a stop codon). We define the volatility of a codon c,
 |
[1] |
as the sum over all one-point neighboring codons ci of the distances between corresponding amino acids. The volatility of a codon measures the degree to which a random nucleotide mutation will change the corresponding amino acid. The definition of volatility requires us to choose a metric, d, that quantifies the distance between amino acids (see below).
Several amino acid metrics, d, may be used in Eq. 1. The simplest choice, called the Hamming metric, equals zero or one, depending on whether two amino acids are identical. When using the Hamming metric, the volatility of a codon is denoted by νH(c) and quantifies the degree to which a random point mutation will cause an amino acid substitution. As an important alternative, the Miyata metric weighs differences between amino acids according to their hydrophobicity and volume (30). Under the Miyata metric, the volatility of a codon is denoted by νM(c) and quantifies the degree to which a random point mutation will change the stereochemical properties of the corresponding acid.
We will use the measures νH and νM to query whether HA codons are biased toward amino acid changes, presumably as a mechanism for (or effect of) escaping frequency-dependent selection mediated by antibodies. In this context, a nonsense mutation will certainly not be selectively advantageous for the virus. Hence we define the distance between any amino acid and a stop codon as zero in both the Hamming and Miyata metrics. As a result, νH and νM also detect codon biases that result from pretermination avoidance (26, 27).
Several amino acids, especially those encoded by six codons, exhibit variation in the volatility of the codons by which they are encoded. For example, both AGG and CGG encode arginine, but νH(AGG) = 7, whereas νH(CGG) = 5. Hence, with a constant per-base mutation rate, AGG is 1.4 times more likely to undergo a substitution than is CGG. This property is not unique to arginine; four amino acids, comprising 22 codons, exhibit variation in the Hamming volatility of synonymous codons. Under the Miyata metric, 12 amino acids exhibit variable volatility among synonymous codons. Thus, even for a fixed amino acid sequence, codon usage may bias a sequence toward, or away from, future substitutional or stereochemical changes.
Detecting Codon Bias
Even for homologous genes, an interspecies comparison of codon usage is difficult to interpret, due to a variety of confounding species-specific factors (e.g., mutational biases, tRNA abundances, etc.). Few authors have attempted to disentangle the relative contributions of species-specific factors affecting codon usage (but see refs. 31 and 32). By contrast, in this study we compare codon usage between and within genes of the same viral species circulating in the same host species. As a result, there is little chance that mutational biases or differences in tRNA pools drive differential codon usage. All influenza A viral genes are replicated by the same polymerase (PB1 and PB2) and translated by the same human tRNAs.
We will compare codon usage in the HA, NA, and NP genes of human influenza A virus, subtype H3N2. We have compiled 525 HA sequences, each 987 nt long; 46 NA sequences, each 1,407 nt long; and 216 NP sequences, each 456 nt long. Each HA sequence consists of the HA1 chain alone. The strains were isolated from patients infected from 1968 through 2000, with the majority of isolates occurring after 1985. All sequences were obtained from a public database (ref. 33; Los Alamos National Laboratory database, www.flu.lanl.gov) and were easily aligned without gaps.
We desire a method to compare codon usage between two genes, controlling for their amino acid sequences. For example, we will not report that gene X is biased toward diversification relative to gene Y if gene X simply contains more amino acids, such as methionine, that can be encoded only by highly volatile codons. In addition, we must control for the fact that the multiple aligned sequences of each gene are highly related to each other by descent. For example, a biased codon found at a particular residue in every sequence of an alignment for gene X should not, in itself, be tallied as compelling evidence that gene X preferentially uses biased codons, because that codon may be common to all sequences by descent. Therefore, we develop below a bootstrap methodology to compare codon usage between gene alignments, controlling for their amino acid sequences.
Consider a multiple sequence alignment of a gene X that contains m residues. We start by producing a list of the unique codons used at each residue. We call this the “codon list” of the alignment, denoted LX. The list LX contains every codon with multiplicity equal to the number of different alignment offsets at which that codon appears. The first several codons in the list LX correspond to the set of (unique) codons found at the first offset in the alignment of gene X. The next several codons in the list correspond to the set of codons found at the second offset, and so on. The length of the list LX may vary from m, if all sequences are identical, to 61 × m, if every codon occurs at least once at each offset. We define the volatility of gene X as the sum of the volatilities of each codon LX(i) in its codon list: ν(LX) = Σ ν(LX(i)). Because we have disregarded redundant codons at each offset, ν(LX) does not overrepresent codons that are identical by descent.
To compare codon usage in gene X to usage in gene Y, we will compare the volatility of gene X to the volatility of bootstrapped alternative versions of gene X that share the same amino acid sequence but that follow Y's codon usage patterns. More specifically, we compare the observed volatility of X, ν(LX) to the volatility of a null-distribution of codons 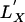 , which (i) translate into the same amino acids as LX, and (ii) are drawn according to the codon usage in the list LY. For each of the 20 amino acids, we measure the observed relative frequencies of synonymous codons used in the codon list LY. The relative frequencies provide a (discrete) distribution of Y's codon usage for each amino acid. In each bootstrap trial, we produce a random codon remapping,
, which (i) translate into the same amino acids as LX, and (ii) are drawn according to the codon usage in the list LY. For each of the 20 amino acids, we measure the observed relative frequencies of synonymous codons used in the codon list LY. The relative frequencies provide a (discrete) distribution of Y's codon usage for each amino acid. In each bootstrap trial, we produce a random codon remapping,  , of the true codons by replacing each codon in LX with a synonymous codon drawn from the distribution measured in gene Y. We will say that the codons of gene X are biased toward diversification compared to the codons of gene Y if the observed volatility ν(LX) is greater than some two-sided P value proportion of the bootstrap trials (
, of the true codons by replacing each codon in LX with a synonymous codon drawn from the distribution measured in gene Y. We will say that the codons of gene X are biased toward diversification compared to the codons of gene Y if the observed volatility ν(LX) is greater than some two-sided P value proportion of the bootstrap trials ( ). This method controls for both the amino acid composition of the two genes and identity by descent within the alignment of each gene.
). This method controls for both the amino acid composition of the two genes and identity by descent within the alignment of each gene.
Results on Codon Bias
Table 1 summarizes a comparison of codon usage among sequences of influenza A HA, NA, and NP genes. In each of the seven cases reported, we compare the observed codon volatility of one gene to a null distribution generated by 10,000 Monte Carlo trials based on another gene's codon usage, as described above. All sequences have the potential for synonymous changes that greatly alter their overall volatility. For example, each HA1 sequence has ≈100 amino acids with variable νH and ≈230 acids with variable νM.
Table 1. A comparison of codon usage among sequences of influenza A HA, NA, and NP genes.
| Miyata | Hamming | |
|---|---|---|
| HA vs. NP | 0.998** | 1.000** |
| HA vs. NA | 0.997** | 0.987* |
| HA+ vs. NP | 0.999** | 1.000** |
| HA+ vs. NA | 0.999** | 1.000** |
| HA- vs. NP | 0.706 | 0.211 |
| HA- vs. NA | 0.692 | 0.053 |
| NA vs. NP | 0.390 | 0.841 |
We write X vs. Y to denote that the codon usage in gene X was compared to a null distribution of 10,000 Monte Carlo trials generated from usage in gene Y. (We do not count a tie as one of the 10,000 trials.) We report the proportion of these Monte Carlo trials for which the volatility was exceeded by the observed volatility of X in the Miyata or Hamming metric. HA+ denotes 130 epitopic residues of HA1; HA- denotes the remaining 199 nonepitopic residues. A single asterisk indicates statistical significance for a two-tailed test at the 5% confidence level; a double asterisk indicates significance at the 1% confidence level. Note that the codon usage of HA, especially in the epitopic residues, is significantly biased toward diversification relative to the usage of NA and NP, but the codon usage of the nonepitopic residues of HA is not significantly biased relative to NA or NP, nor is NA biased relative to NP
The observed volatility of HA1 codons exceeds 99.8% of the Monte Carlo trials based on NP codon usage in both the Hamming and Miyata metrics. In other words, HA codons are significantly biased toward substitutional and stereochemical-altering point mutations relative to NP codons. This bias toward diversification reflects the fact that the surface protein HA is under much stronger frequency-dependent selection to change than the internal NP.
Similarly, we find that HA codons are also biased toward diversification relative to NA codons in both Miyata and Hamming metrics. This bias indicates that HA is under stronger frequency-dependent selection than is NA. Although both HA and NA are antibody targets, diversifying selection is stronger on HA, likely because (i) antibodies to HA, but not to NA, neutralize the virus (9–11), and (ii) HA is 5-fold more prevalent and uniform than NA on the viral particle (8, 34). These results support the idea that HA is the primary protein responsible for antigenic variation (7).
We do not find a statistically significant difference in volatility between NA and NP codons under either the Hamming or Miyata metrics (Table 1). Even though NA is less immunogenic than HA, it is nevertheless a surface protein. Hence, it is somewhat surprising that we do not find a difference in codon usage between NA and NP.
Of the 329 residues in HA1, 130 lie in the five main epitopes, labeled A–E (15). Table 1 also compares codon usage at HA epitopic residues to codon usage of other genes. We denote the 130 epitopic residues of HA (which all fall within HA1) by HA+ and the remaining 199 HA1 residues by HA-. We find that HA+ codons are extremely biased toward diversification under both metrics relative to NA or NP. On the other hand, HA- codons are not significantly biased in either metric relative to NA or NP. In other words, virtually all codon bias seen in HA1 is due to biases at the immunogenic residues. This observation strongly supports the interpretation that HA1 codon bias results from frequency-dependent selection to escape antibody pressure.
Table 2 summarizes codon usage comparisons between regions within HA1. We separate HA1 residues into epitopes A–E and the remaining 199 sites, HA- (15). In the Hamming metric, the individual epitopes A, C, D, and E are each significantly biased toward diversification relative to the nonepitopic sites, HA-. Similarly, all epitopic sites taken together, HA+, are biased toward diversification relative to HA-. In other words, even within HA1, the immunogenic residues use codons with a greater proportion of substitutional neighbors than the nonepitopic residues. Codon usage in HA is so precise that those particular residues involved in antibody combination are biased relative to the other residues of the same gene.
Table 2. A comparison of codon usage between regions of HA1: epitopes A-E and the remaining residues, HA-.
| Miyata | Hamming | |
|---|---|---|
| Ep A vs. HA- | 0.927 | 1.000** |
| Ep B vs. HA- | 0.852 | 0.962 |
| Ep C vs. HA- | 0.917 | 0.998** |
| Ep D vs. HA- | 0.974 | 1.000** |
| Ep E vs. HA- | 0.816 | 0.982* |
| HA+ vs. HA- | 0.999** | 1.000** |
| P vs. HA- | 0.993* | 1.000** |
| HA- vs. HA- | 0.506 | 0.506 |
| HA+ vs. HA+ | 0.498 | 0.496 |
We also compare codon usage in the 18 residues of positive selection, denoted by P, identified by Bush et al. (15). Asterisks indicate statistical significance, as in Table 1. Note that, in general, epitopes are significantly biased relative to nonepitopes in the Hamming but not the Miyata metric. As a consistency check for our bootstrap methodology, we also compare HA+ and HA- to themselves and find they lie within two standard deviations of the 50th percentile, as expected.
However, no individual epitope is significantly biased relative to HA- under the Miyata metric. This result likely indicates that epitopic residues of HA1 are under strong pressure to accrue substitutions (hence the common bias under the Hamming metric) but not to accrue stereochemically dramatic substitutions (hence the lack of a strong bias under the Miyata metric). The epitopic residues presumably must walk a fine line between generating enough diversity to evade existing antibodies without dramatically changing their stereochemical properties or functionality. Codon biases under the Hamming and Miyata metrics reflect the opposing forces of antibody-mediated diversifying selection and structurally mediated purifying selection.
Interpretation of Codon Bias
We have seen that codons of HA are significantly biased toward substitutional or stereochemical change, compared to codons of NA and NP. Moreover, the HA residues involved in antibody combination are significantly biased relative to the nonimmunogenic HA residues.
A naive interpretation of our results posits that HA codon biases are selectively advantageous at immunogenic residues because they allow the virus to respond efficiently, through substitution, to escape antibody pressure. But this explanation violates causality: a viral gene cannot know in advance that a volatile codon will be beneficial for producing future antigenic variants. The selective advantage of a volatile codon is realized only on mutation, whereupon the codon changes. Thus selection cannot favor the genotype with volatile codons in ipso.
There is, however, a simple retrospective interpretation of codon bias that does not violate causality: codon biases result from prior frequency-dependent selection. Because the mutational process is symmetric in time, if a codon has a large proportion of substitutional one-point neighbors, we may conclude that the previous mutation to that codon was likely a substitution (just as we can also conclude that the next mutation to that codon will likely cause a substitution). Hence, if a residue has experienced frequency-dependent selection to alter its amino acid, we would expect to see the footprint of this diversifying selection in the form of biased codons at that residue. In this interpretation, observed codon biases are the remnants of strong prior frequency-dependent selection.
The retrospective view provides the most parsimonious explanation of observed codon biases in influenza A. Nevertheless, the prospective interpretation, namely that codon biases are of direct adaptive utility, cannot be entirely dismissed. Indeed, individual genotypes cannot themselves be selected for biased codons, because the adaptive utility of the bias is realized only when the genotype mutates. Nevertheless, if codon bias toward substitution is itself heritable and preserved on mutation, then it may provide a selective advantage to a lineage of viral sequences. Lineage-level selection for codon bias requires that if a codon c is highly volatile (in either the Hamming or Miyata metric), the one-point neighbors of codon c are also, on average, highly volatile, so that the bias is heritable along a mutating lineage. In Fig. 1 we demonstrate, based on the properties of the standard genetic code, that volatility is indeed a heritable trait of codons.
Fig. 1.

The relationship between the volatility of a codon and the average volatility of its one-point neighbors. Stop codons are not counted as neighbors. Codon bias is preserved on mutation under both the Miyata (Upper; r2 = 0.73, P < 10-5) and the Hamming (Lower; r2 = 0.38, P < 10-7) metrics. As a result, codon bias is heritable along a mutating viral lineage.
The prospective and retrospective interpretations both provide evolutionary frameworks for understanding observed codon composition of influenza A viral genes. The retrospective interpretation is certainly part of the story, but the prospective interpretation may also be important. Further analysis, including mathematical modeling, is required to determine the strength of lineage-level selection for codon bias in viral lineages.
Evolution of HA Epitopes
Whether interpreted prospectively or retrospectively, codon biases in the epitopic residues of HA indicate that these residues are under strong selection for substitutional or stereochemical change. The commonly used (refs. 7, 13, and 15; www.flu.lanl.gov) definitions of the five antibody-combining regions of HA are based on the solved structure of a 1968 influenza A strain. But HA has evolved considerably since 1968. In fact, analysis of HA1 sequences shows that in the past several years, increasingly more substitutions have occurred outside of the 130 sites classically considered as epitopes (35). These results suggest that the locations of epitopic sites on the HA trimer may have changed since their original characterization.
Given the extent of HA sequence and potential structural evolution, we naturally desire a method, based on sequence data alone, to detect (new) residues that are involved in antibody combination or, more generally, that are under diversifying selection. With a large enough database, consistent codon bias toward volatility at a residue may by itself indicate that the residue is under selection to change. But the current publicly available database of HA sequences is not sufficient to detect diversifying selection at an individual residue on the basis of codon bias alone.
Nevertheless, as Fig. 2 demonstrates, an information-theoretic analysis of HA sequence variation can aid in the identification of residues under diversifying selection. For each of the 329 residues in HA1, we plot the diversity of codons found at that residue against the diversity of amino acids found at that residue. To quantify the “diversity” of codons at a particular residue of HA1, we compute a variant of Simpson's index:  , where pi denotes the relative frequency of the ith codon at the residue in the multiple sequence alignment. We use the same formula to compute the diversity of amino acids at each residue. The measure D yields results that are extremely similar to the classical Shannon–Weaver measure of entropy.
, where pi denotes the relative frequency of the ith codon at the residue in the multiple sequence alignment. We use the same formula to compute the diversity of amino acids at each residue. The measure D yields results that are extremely similar to the classical Shannon–Weaver measure of entropy.
Fig. 2.

The relationship between codon and amino acid diversity at each of the 329 residues in a 525-sequence alignment of the HA1 gene. The lower scatterplot shows an enlarged portion of the upper scatterplot. Diversity is quantified by using the modified Simpson index. Points are colored according to the epitope in which each residue lies: red (A), orange (B), green (C), light blue (D), and dark blue (E). The nonepitopic residues are shown in black. Epitopic residues generally lie along the diagonal x = y, whereas nonepitopic residues generally lie along the x axis. Exceptions to this pattern are discussed in the text. The top 25 diagonal residues are identified in Table 3.
The residues of HA1 shown in Fig. 2 fall into essentially two categories: residues with little amino acid diversity but a large diversity of codons, which lie along the x axis; and residues with as much codon diversity as amino acid diversity, which lie along the diagonal x = y. In other words, most residues are either functionally constrained and vary only synonymously, or they are functionally plastic and exhibit amino acid variation.
The residues along the diagonal of Fig. 2, particularly those far from the origin, exhibit the trademarks of diversifying selection: a large number of mutations, almost all of which are substitutions. As we might expect, the top 25 residues along the diagonal all belong to antibody-combining regions of the HA protein (Table 3). Conversely, the residues of neutral variation lying along the x axis in Fig. 2 are predominantly drawn from sites outside of the classical epitopes. All 11 residues primarily responsible for the overall stability of the HA trimer (36) also fall along the x axis. Hence the essential pattern shown in Fig. 2 coincides with the our intuition: diversifying selection occurs at antigen-combining regions, whereas neutral variation predominates elsewhere (including sites of structural importance).
Table 3. The top 25 residues on the diagonal in Fig. 2 listed by residue number, corresponding epitope, diversity of codons, and diversity of amino acids.
| Residue | Epitope | D(codons) | D(acids) |
|---|---|---|---|
| 135 | A | 0.713 | 0.711 |
| 226 | D | 0.713 | 0.705 |
| 124 | A | 0.699 | 0.685 |
| 262 | E | 0.672 | 0.669 |
| 133 | A | 0.640 | 0.639 |
| 121 | D | 0.634 | 0.630 |
| 276 | C | 0.616 | 0.615 |
| 172 | D | 0.552 | 0.551 |
| 156 | B | 0.544 | 0.536 |
| 278 | C | 0.537 | 0.528 |
| 145 | A | 0.529 | 0.524 |
| 197 | B | 0.514 | 0.505 |
| 189 | B | 0.467 | 0.466 |
| 190 | B | 0.459 | 0.449 |
| 157 | B | 0.453 | 0.451 |
| 196 | B | 0.418 | 0.345 |
| 193 | B | 0.384 | 0.375 |
| 158 | B | 0.365 | 0.359 |
| 144 | A | 0.343 | 0.343 |
| 62 | E | 0.373 | 0.308 |
| 142 | A | 0.341 | 0.332 |
| 131 | A | 0.307 | 0.294 |
| 275 | C | 0.291 | 0.284 |
| 83 | E | 0.288 | 0.276 |
| 299 | C | 0.278 | 0.275 |
We consider a residue to be on the diagonal of Fig. 2 if the difference between its codon and amino acid diversity is <10% of their sum. In total, there are 78 diagonal residues in Fig. 2. Residues in the table are sorted by the sum of their codon and amino acid diversities. Fourteen of these 25 sites were previously identified as being under positive selection (15)
But there are several striking counterexamples to the general pattern seen in Fig. 2. Several epitopic residues (see Table 4, which is published as supporting information on the PNAS web site, www.pnas.org) exhibit mainly synonymous variation, indicative of functional or structural constraints at those sites (36). Such residues, which are involved in antibody binding and yet conserved, should be considered in the design of potential broadly effective influenza vaccines. Conversely, several residues that have not been characterized as antigenic, shown in black in Fig. 2, nevertheless exhibit the footprint of diversifying selection: the equality of codon and amino acid diversity. In Fig. 3 we show seven examples of these sites, residues 271-Asp, 220-Arg, 112-Val, 31-Asp, 5-Gly, 3-Leu, and 2-Asp, on the crystal structure of a 1968 HA. [Amino acids indicated for each residue correspond to the crystalized strain (12).]
Fig. 3.

The solved structure (12) of a 1968 strain of HA, consisting of two chains 329 and 175 residues long, shown here in its monomer form. The commonly used (15) definitions of the five antibody-combining regions are shown in red (A), orange (B), green (C), light blue (D), and dark blue (E). Residues 271-Asp, 220-Arg, 112-Val, 31-Asp, 5-Gly, 3-Leu, and 2-Asp are shown in yellow. These seven residues have not previously been characterized as positively selected but nevertheless show the same genomic pattern of codon variation as many epitopic residues (Fig. 2). The residues in yellow may represent sites that, in the current HA, are directly involved in antibody combination, comutate with epitopic residues, or determine the conformation of epitopes.
The seven residues identified above may be new sites that, through conformational changes in HA since 1968, are now involved in antibody combination. Indeed, six of these sites lie on the exposed surface of the protein (Fig. 3). The α carbon of residue 220-Arg lies within 4 Å of epitope D (the α carbon of 219-Ser) and is likely now involved in antibody combination with the current shape of epitope D. Similarly, residue 271 is extremely close to the classical resides of epitope C, and residue 112 is close to epitope E.
Although residues 31, 5, 3, and 2 are on the surface of the HA trimer, they lie far away from regions of antibody combination. These four residues may represent sites of obligate comutation with epitopic residues. Alternatively, they may be sites not directly involved in antibody combination, but that determine the conformation of the epitopes. Extensive structural study, through simulation or crystallization of a modern HA, will be required to determine the function of these sites. In the meantime, we may conclude only that, despite their position on the protein, these four residues are under strong diversifying selection.
The methods developed in this section provide a powerful alternative to a replacement-to-silent ratio for detecting residues undergoing diversifying selection. Because such selection is mediated by neutralizing antibodies binding to the protein surface, one may inquire whether there is a correlation between evidence of diversifying selection and the solvent accessibility of a residue. We therefore calculated the solvent-accessible surface area of each HA1 residue in the solved structure of the 1968 HA trimer (12) by using the computer program SURFV¶ at a probe radius of 1.4 Å. If surface residues are more likely to experience diversifying selection, we would expect a correlation between a residue's accessibility and the ratio of the diversity of amino acids to the diversity of codons found at that residue, denoted by τ = D(acids)/D(codons). Among the 304 sites with at least two codons, there is a moderate (r2 = 0.175) but statistically significant (P < 10-13) correlation between τ and solvent-accessible surface area of a residue. Among the same residues, there is a similar correlation (r2 = 0.175, P < 10-14) between solvent accessibility and the ratio of the number of amino acids to number of codons at a residue.
We may further inquire about solvent accessibility for the two basic categories of residues seen in Fig. 2: those residues that are closer to the diagonal, D(acids) ≥ D(codons)/2, and those that are closer to the x axis, D(acids) < D(codons)/2. The mean ± standard error solvent accessibility in the first group is 62.6 ± 4.5 Å, and in the second group is 16.6 ± 3.3 Å. In other words, the residues falling closer to the diagonal of Fig. 2 are, on average, much more accessible to solvents in the folded trimer than residues near the x axis. Thus, surface residues show much stronger signs of diversifying selection than residues buried inside the folded trimer.
Discussion
The tremendous amount of sequence data that have become available in the last few years opens up new research possibilities and calls for new analytical techniques. In this paper, we have developed and applied two techniques to influenza sequences from a public database to gain perspective on both interactions between codon usage and Darwinian selection and the evolution of HA in particular.
We have explored codon usage biased toward diversification by comparing different regions of the same genome, thus controlling for a variety of confounding factors. We expected to find a bias in HA1 codons caused by frequency-dependent selection. We not only find such a bias, but we also find that within HA1, codons involved in antibody combination are biased with respect to nonantigenic HA1 codons. To our surprise, we detect no evidence that NA codons are biased relative to the internal protein NP.
We have argued that the observed biases toward substitutional or stereochemical change result from previous selection to evade HA antibodies. Nevertheless, we have also shown that such biases can be passed on to offspring even after a mutation. This observation opens the possibility that codon bias may itself be selected in genes under strong diversifying selection, a possibility that requires further research.
Additionally, we have found surprising differences between codon bias measured by the Hamming metric, which quantifies the number of substitutions, and the Miyata metric, which accounts for stereochemical differences. The biases in epitopic regions of HA are systematically weaker in the Miyata metric, which is likely the result of important stereochemical constraints on amino acid variation near the receptor-binding pocket.
Whereas our bootstrap methodology detects codon bias in epitopic regions of HA as a whole, we have also developed techniques for analysis of codon variation at individual residues, where a more complex picture emerges. We find that not all of the residues in the classically identified epitopic regions show signatures of diversifying selection. Conversely, we find some residues outside the defined epitopes that do show diversifying selection, including some far from the known antibody-combining regions. It is possible that such residues have immunologically important effects on the overall conformation of HA.
Supplementary Material
Acknowledgments
We are grateful for valuable input from S. Levin, D. Krakauer, M. Weigert, D. Hartl, A. Murray, and two anonymous referees. We also thank the Los Alamos National Laboratory and those who have contributed to the Influenza Sequence Database, including contributors of unpublished sequences: N. Komadina, A. Hapson, N. Cox, C. Bender, O. Hungnes, and M. Brytting. This work was supported by National Institutes of Health Grant 1-R01-GM60729-01 (to Simon A. Levin). J.B.P. acknowledges support from the National Science Foundation, the Burroughs Wellcome Fund, and the Porter Ogden Jacobus Fellowship.
Abbreviations: HA, hemagglutinin; NA, neuraminidase; NP, nucleoprotein.
Footnotes
Sridharan, S., Nicholls, A. & Honig, B. (1992) Biophys. J. 6, A174.
References
- 1.Hayden, F. G. & Palese, P. (1997) in Clinical Virology, eds. Richman, D. D., Whitley, R. J. & Hayden, F. G. (Churchill Livingstone, New York), pp. 911-942.
- 2.Fitch, W. M., Bush, R. M., Bender, C. A. & Cox, N. J. (1997) Proc. Natl. Acad. Sci. USA 94, 7712-7718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fitch, W. M., Bush, R. M., Bender, C. A., Subbarao, K. & Cox, N. J. (2000) J. Hered. 91, 183-185. [DOI] [PubMed] [Google Scholar]
- 4.Ina, Y. & Gojobori, T. (1994) Proc. Natl. Acad. Sci. USA 91, 8388-8392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bush, R. M., Fitch, W. M., Bender, C. A. & Cox, N. J. (1999) Mol. Biol. Evol. 16, 1457-1465. [DOI] [PubMed] [Google Scholar]
- 6.Fitch, W. M., Leiter, J. M. E., Li, X. Q. & Palese, P. (1991) Proc. Natl. Acad. Sci. USA 88, 4270-4274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wilson, I. A. & Cox, N. J. (1990) Annu. Rev. Immunol. 8, 737-771. [DOI] [PubMed] [Google Scholar]
- 8.Wrigley, N. G. (1979) Br. Med. Bull. 35, 35-38. [DOI] [PubMed] [Google Scholar]
- 9.Webster, R. G. & Laver, W. G. (1967) J. Immunol. 99, 49-55. [PubMed] [Google Scholar]
- 10.Kilbourne, E. D., Laver, W. G., Schulman, J. L. & Webster, R. G. (1968) J. Virol. 2, 281-288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zebedee, S. L. & Lamb, R. A. (1988) J. Virol. 62, 2762-2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wilson, I. A., Skehel, J. J. & Wiley, D. C. (1981) Nature 289, 366-373. [DOI] [PubMed] [Google Scholar]
- 13.Wiley, D. C., Wilson, I. A. & Skehel, J. J. (1981) Nature 289, 373-378. [DOI] [PubMed] [Google Scholar]
- 14.Chakraverty, P. (1971) Bull. W. H. O. 45, 755-766. [PMC free article] [PubMed] [Google Scholar]
- 15.Bush, R. M., Bender, C. A., Subbarao, K., Cox, N. J. & Fitch, W. M. (1999) Science 286, 1921-1925. [DOI] [PubMed] [Google Scholar]
- 16.King, J. & Jukes, T. (1969) Science 164, 788-798. [DOI] [PubMed] [Google Scholar]
- 17.Powell, J. R. & Moriyama, E. N. (1997) Proc. Natl. Acad. Sci. USA 94, 7784-7790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sharp, P. M. & Li, W. H. (1987) Nucleic Acids Res. 15, 1281-1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ikemura, T. (1985) Mol. Biol. Evol. 2, 13-34. [DOI] [PubMed] [Google Scholar]
- 20.Marais, G., Mouchiroud, D. & Duret, L. (2001) Proc. Natl. Acad. Sci. USA 98, 5688-5692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sharp, P. M. & Li, W. H. (1986) J. Mol. Evol. 24, 28-38. [DOI] [PubMed] [Google Scholar]
- 22.Bulmer, M. (1991) Genetics 129, 897-907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zuckerkandl, E. & Pauling, L. (1965) J. Theor. Biol. 8, 357-366. [DOI] [PubMed] [Google Scholar]
- 24.Ikemura, T. (1981) J. Mol. Biol. 146, 1-21. [DOI] [PubMed] [Google Scholar]
- 25.Lawrence, J. G. & Hartl, D. L. (1991) Genetica 84, 23-29. [DOI] [PubMed] [Google Scholar]
- 26.Fitch, W. M. (1980) J. Mol. Evol. 16, 153-209. [DOI] [PubMed] [Google Scholar]
- 27.Modiano, G., Battistuzzi, G. & Motulsky, A. G. (1981) Proc. Natl. Acad. Sci. USA 78, 1110-1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miyata, T., Yasunaga, T. & Nishida, T. (1980) Proc. Natl. Acad. Sci. USA 77, 7328-7332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stephens, C. R. & Waelbroeck, H. (1999) J. Mol. Evol. 48, 390-397. [DOI] [PubMed] [Google Scholar]
- 30.Miyata, T., Miyazawa, S. & Yashunaga, T. (1979) J. Mol. Evol. 12, 219-236. [DOI] [PubMed] [Google Scholar]
- 31.Hartl, D. L., Moriyama, E. N. & Sawyer, S. A. (1994) Genetics 138, 227-234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Akashi, H. (1997) Gene 205, 269-278. [DOI] [PubMed] [Google Scholar]
- 33.Macken, C., Lu, H., Goodman, J. & Boykin, L. (2001) in Options for the Control of Influenza IV, eds. Osterhaus, A. D. M. E., Cox, N. & Hampson, A. W. (Elsevier, Amsterdam), pp. 103-106.
- 34.Murti, K. G. & Webster, R. G. (1986) Virology 149, 36-43. [DOI] [PubMed] [Google Scholar]
- 35.Plotkin, J. B., Dushoff, J. & Levin, S. A. (2002) Proc. Natl. Acad. Sci. USA 99, 6263-6268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Isin, B., Doruker, P. & Bahar, I. (2002) Biophys. J. 82, 569-581. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.