

Abstract
Predicted highly expressed (PHX) genes in five currently available high G+C complete α-proteobacterial genomes are analyzed. These include: the nitrogen-fixing plant symbionts Sinorhizobium meliloti (SINME) and Mesorhizobium loti (MESLO), the nonpathogenic aquatic bacterium Caulobacter crescentus (CAUCR), the plant pathogen Agrobacterium tumefaciens (AGRTU), and the mammalian pathogen Brucella melitensis (BRUME). Three of these genomes, SINME, AGRTU, and BRUME, contain multiple chromosomes or megaplasmids (>1 Mb length). PHX genes in these genomes are concentrated mainly in the major (largest) chromosome with few PHX genes found in the secondary chromosomes and megaplasmids. Tricarboxylic acid cycle and aerobic respiration genes are strongly PHX in all five genomes, whereas anaerobic pathways of glycolysis and fermentation are mostly not PHX. Only in MESLO (but not SINME) and BRUME are most glycolysis genes PHX. Many flagellar genes are PHX in MESLO and CAUCR, but mostly are not PHX in SINME and AGRTU. The nonmotile BRUME also carries many flagellar genes but these are generally not PHX and all but one are located in the second chromosome. CAUCR stands out among available prokaryotic genomes with 25 PHX TonB-dependent receptors. These are putatively involved in uptake of iron ions and other nonsoluble compounds.
The complete genomes of five high G+C α-proteobacteria (α-genomes) have become available during the past 2 years. These are: Caulobacter crescentus (CAUCR, using the SWISSPROT nomenclature) (1), Sinorhizobium meliloti (SINME) (2), Mesorhizobium loti (MESLO) (3), Agrobacterium tumefaciens (AGRTU) (4, 5), and Brucella melitensis (BRUME) (6). SINME induces formation of root nodules on specific legumes. Bacterial nod genes are required for this developmental process. Bacterial endosymbionts (called bacteroids) fix nitrogen within the plant nodule. The bacteroids supply the plant with nitrogen, usually in the form of ammonia, in exchange for plant carbon. The SINME genome consists of three large circular replicons: the main replicon (chromosome) (7) is 3.65 Mb; a second genetic element is the replicon (megaplasmid) pSymB (8) ≈1.68 Mb; and a third replicon (megaplasmid) pSymA (9) is ≈1.35 Mb (see Table 1). pSymA carries the nod genes and most of the nitrogen-fixation genes. Although pSymA is required for nodulation, it can be cured without affecting viability, at least under laboratory conditions (10). pSymB carries genes required for successful infection and several essential genes (Arg tRNA and minCDE), making the classification of pSymB as a plasmid rather than a chromosome somewhat controversial (6). Both megaplasmids contain genes that may aid in soil survival (8, 9).
Table 1. General statistics of PHX and PA genes.
| SINME
|
MESLO
|
AGRTU
|
BRUME
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Characteristic | CAUCR | Chr | pSymA | pSymB | Chr | pMLa | pMLb | Circular | Linear | pAT | pTi | Chr1 | Chr2 |
| Genome size, kb | 4,017 | 3,654 | 1,354 | 1,683 | 7,036 | 352 | 208 | 2,842 | 2,075 | 543 | 214 | 2,117 | 1,178 |
| G+C content, % | 67.2 | 62.7 | 60.4 | 62.4 | 62.7 | 59.3 | 59.9 | 59.4 | 59.3 | 57.3 | 56.7 | 57.2 | 57.3 |
| No. of genes with ≥80 codons | 3,565 | 3,177 | 1,226 | 1,482 | 6,320 | 286 | 168 | 2,602 | 1,763 | 476 | 183 | 1,923 | 1,076 |
| PHX genes | |||||||||||||
| No. | 374 | 223 | 15 | 21 | 1039 | 6 | 3 | 183 | 35 | 8 | 3 | 188 | 47 |
| Percentage | 10.5 | 7.0 | 1.2 | 1.4 | 16.4 | 2.1 | 1.8 | 7.0 | 2.0 | 1.7 | 1.6 | 9.8 | 4.4 |
| Highest E(g) value | 2.39 | 1.93 | 1.67 | 1.36 | 2.05 | 2.03 | 1.21 | 2.26 | 2.27 | 1.31 | 1.15 | 2.39 | 1.81 |
| rpoC | fusA | groEL | chvE | rpoC | groEL | trbC | fus | rpsA = S1 | orf | orf | fus | groEL | |
| PA genes | |||||||||||||
| No. | 364 | 130 | 386 | 178 | 541 | 119 | 44 | 116 | 137 | 150 | 73 | 124 | 48 |
| Percentage | 10.2 | 4.1 | 31.5 | 12.0 | 8.6 | 41.6 | 26.2 | 4.5 | 7.8 | 31.5 | 39.9 | 6.4 | 4.5 |
Chr, chromosome
MESLO is a symbiotic nitrogen-fixing soil bacterium that infects several Lotus species. Like SINME, the MESLO genome contains nodulation and nitrogen-fixation genes, but instead of being megaplasmid borne, these are contained within a chromosomally located “symbiotic island.” The MESLO genome strain MAFF303099 consists of a 7-Mb chromosome and two plasmids, pMLa (352 kb) and pMLb (208 kb) (Table 1) (3).
The genome of AGRTU strain C58 (4, 5) consists of circular and linear chromosomes of lengths 2,842 and 2,075 kb, respectively, and two plasmids. The 214-kb Ti (tumor inducing) plasmid causes crown gall tumor in some plants, and the 543-kb AT plasmid may carry additional genes involved in pathogenesis.
BRUME (6), strain 16M, is a facultative intracellular pathogen that induces abortion in goats and sheep and Malta fever in humans. The bacteria enter the host through mucosal surfaces and can survive in macrophages. The 3,295-kb BRUME genome is distributed over two circular chromosomes of 2,117 and 1,178 kb. Potential virulence genes are distributed between both chromosomes. The recent sequencing of Brucella suis (11) and analysis reveals significant similarity to BRUME (not shown).
CAUCR is an innocuous aquatic bacterium with potential as a bioremediation agent (1). Unlike the previous four species, CAUCR is free-living at all stages of its life cycle. CAUCR divides asymmetrically: the stalked cell adheres to surfaces via a holdfast and gives rise to a new motile swarmer cell at each division. A swarmer cell does not initiate DNA replication until after its differentiation into a stalked cell. The 4-Mb CAUCR genome contains many genes essential for survival in dilute, and thus nutrient-poor, aquatic habitats; these include genes for chemotaxis, transport, and catabolic functions (1).
In this article, predicted highly expressed (PHX) genes are characterized for the above five α-genomes. There is a diversity of lifestyles among these species, yet the five α-genomes comprise a coherent group. Most, with the exception of CAUCR, are mammalian or plant pathogens or symbionts. The lifestyles are predominantly aerobic and the genomes are of high G+C content (Table 1). The bacteria SINME and MESLO are nitrogen-fixing symbionts of legume plants, whereas CAUCR, AGRTU, and BRUME do not fix nitrogen.
Materials and Methods
Our approach in estimating gene expression levels is related to codon usage differences between gene classes where codon usage reflects on the level of expression of a gene (12). Qualitatively, a gene is PHX if its codon frequencies are rather similar to those of the ribosomal protein (RP) genes, major transcription/translation factors (TF) genes, and the chaperone/ degradation (CH) genes, but deviate strongly from the average gene of the genome. Let G be a group of genes with average codon frequencies g(x, y, z) for the codon triplet (x, y, z) such that Σg(x, y, z) = 1 for each amino acid family. Similarly let {f(x, y, z)} indicate the average codon frequencies for the gene group F (F can be a single gene), normalized to 1 in each amino acid codon family. The codon usage difference of F with respect to G is calculated by the formula
 |
[1] |
where {pa(F)} are the average amino acid frequencies of the genes of F. Let B(g|G) denote the codon usage difference of the gene g relative to the gene class G. Formally, a gene g is PHX if the codon usage of g is very different from the codon usage of an average gene but quite similar to the codon usage of the gene classes RP, CH, and TF. Predicted expression levels with respect to individual standards can be based on the ratios ERP(g) = B(g|C)/B(g|RP), ECH(g) = B(g|C)/B(g|CH), and ETF(g) = B(g|C)/B(g|TF), where C is the totality of all genes of the genome. We introduce the overall expression measure
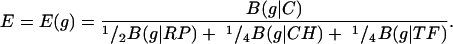 |
Other weights can also be used but the results do not qualitatively differ.
The gene classes (RP, CH, and TF) serve as representatives of highly expressed genes, and our method specifies genes with similar codon usages as PHX genes. These assignments are reasonable under fast growth conditions, where there is a need for many ribosomes, for proficient transcription and translation, and for many CH proteins to ensure properly folded, modified, and translocated protein products.
A gene is PHX if the following two conditions are satisfied: at least two among the three expression ratios, ERP(g), ECH(g), and ETF(g), exceed 1.05, and the overall expression level E(g) is >1.00. A gene g is putative alien (PA) if B(g|C), B(g|RP), B(g|TF), and B(g|CH) all exceed M(h) + 0.1, where M(h) is the median B(h|C) among all genes h of approximate similar length to g.
E(g) is an estimate of the expression level of the gene g. The criterion E(g) >1 and where at least two of the values ERP(g), ETF(g), or ECH(g) exceed 1.05 provides an excellent benchmark in reflecting high protein molar abundance in a rapid growth environment (see ref. 12).
It is instructive to plot B(g|C) versus B(g|RP), B(g|TF), or B(g|CH) for all genes g encoding proteins of ≥100-aa length (Fig. 1, which is published as supporting information on the PNAS web site, www.pnas.org). The distribution of points reveals two horns. The upper left horn corresponds to the PHX genes, and the upper right horn is designated PA genes. Examples of PHX protein classes in most prokaryotes include: (i) RPs; (ii) protein synthesis processing enzymes (RpoB, RpoC, RpoA, Tuf, Fus); (iii) major general chaperones and degradation proteins (GroEL, DnaK, Tig, FtsH, some peptidyl-prolyl cis-trans isomerases); (iv) polynucleotide phosphorylase (mRNA processing and degradation); (v) essential energy metabolism (glycolysis genes mainly in anaerobes, tricarboxylic acid (TCA) cycle genes mainly in aerobes, photosynthesis genes in cyanobacteria, methanogenesis genes in methanogens); and (vi) some enzymes of amino acid and nucleotide biosynthesis. Examples of protein classes PHX in some but not most genomes are: (i) fatty acid metabolism in Mycobacterium tuberculosis;(ii) urease only in Helicobacter pylori and Ureaplasma urealyticum; (iii) f lagellar proteins (α-proteobacteria (MESLO, CAUCR), spirochetes (Treponema pallidum, Borrelia burgdorferi); and (iv) many PHX detoxification genes in Deinococcus radiodurans. Our analysis of PHX genes is consistent with assessments of two-dimensional gel protein abundances in several prokaryotes (13); E(g) values empirically correlate with protein molar abundances. The data support the proposition that each genome has evolved a codon usage pattern reflecting “optimal” gene expression levels for most circumstances encountered in its lifestyle and habitat. Alien genes consist mostly of ORFs of unknown function but also include genes encoding transposases, cryptic prophage sequences, restriction or modification enzymes (which are often conjugatively transferred via plasmids), genes associated with lipopolysaccharide biosynthesis, and fimbrial-like genes (14, 15). PA genes have high codon bias relative to the gene classes C, RP, CH, and TF, and many have been acquired through lateral gene transfer (16).
A protein that belongs to the PHX class and performs several functions might be expected to show higher E(g) values than the average PHX gene. For example, polynucleotide phosphorylase is fundamental to RNA processing and mRNA degradation and attains the highest E(g) = 2.66 value among all Escherichia coli genes (12). Enolase, acting in energy metabolism (glycolysis) and RNA degradation, generally registers a significantly high E(g). Aconitase carries the highest E(g) value (2.56) in D. radiodurans and is PHX in many genomes (17). Aconitase not only interconverts citrate and isocitrate in the TCA cycle, but it also serves as a sensor detecting changes in the redox state and assaying iron content within the cell. Other multifunctional PHX proteins in many genomes include GAPDH, which acts primarily in the first step of the second phase of glycolysis. This gene product is multifunctional in that it possesses uracil DNA glycosylase activity, senses oxidative stress, and binds to RNA and DNA (18). Moreover, GAPDH serves as a source of reducing equivalents in mammalian cells. In contrast, proteins that are required in few molecules per cell cycle are not expected to be highly expressed. Thus, the following gene groups are seldom highly expressed: (i) specific regulatory proteins, (ii) specialized TFs, (iii) strict replication proteins, (iv) two component sensor proteins (e.g., histidine kinases, ref. 12), (v) most repair proteins, and (vi) vitamin biosynthesis enzymes.
Results and Discussion
The Statistics of PHX Genes in the High G+C α-Proteobacterial Genomes. Table 1 reports the numbers of PHX genes in the five α-proteobacterial genomes, indicating in the major chromosomes ≈7–10% PHX genes (exception, MESLO 16.2%), whereas in the secondary chromosomes (or megaplasmids) the count of PHX genes rarely exceeds 2.1% (BRUME 4.4%). By contrast, PA genes occupy a large fraction of secondary chromosomes and megaplasmids ranging from ≈8% to 40%. We speculate (cf. ref. 2) that the megaplasmids of SINME were originally acquired by the host bacterium via lateral gene transfer. Subsequently, many genes were lost from the megaplasmids and many utilitarian genes were transferred to the main chromosome. This was also speculated for the Ti and AT plasmids of AGRTU (4, 5). Also, in MESLO, the plasmids and symbiotic island have a lower G+C content than the rest of the chromosome. This seems to be a general phenomenon among all second or third chromosomes in bacterial genomes involving relatively few PHX genes [e.g., as occurs in the second chromosome of Vibrio cholerae and D. radiodurans (data not shown) and in bacterial plasmids (20)]. Another scenario to account for extra chromosomes/megaplasmids is that a DNA segment separated from the main chromosome and was converted into a megaplasmid or ancillary chromosome, which then lost and gained new genes. pSymA includes as PA genes most of the symbiotic genes that function in nodulation and nitrogen fixation. When pSymA is knocked out, SINME remains viable but no longer infects its plant host or engages in nitrogen fixation (10). The foregoing discussion may bear on the course of the evolution and development of multiple chromosomes.
Top PHX Genes of α-Proteobacterial Genomes. The giant RP gene S1 (generally >500 codons in length) has a PHX value among the top 10 E(g) values for all α-genomes except in MESLO in which S1 is still PHX but not among the top 10 (Table 5, which is published as supporting information on the PNAS web site). AGRTU S1 is encoded by the linear chromosome. The major translation/transcription processing factors EF-G and the β and β′ subunits of RNA polymerase (fus = EF-G, rpoC, rpoB, respectively) place among the top 10 PHX genes in all five α-genomes. EF-Tu invariably occurs in two PHX copies, and translation initiation factor infB ranks among the top 10 PHX genes of BRUME. All major TFs are encoded from the main chromosome of these genomes.
Both nitrogen-fixing plant symbionts SINME and MESLO have five copies of groEL, and MESLO features three groEL among its top 10 PHX genes. GroEL genes are often found in secondary chromosomes. Pnp is impressively PHX in all five α-genomes as it is in E. coli; pnp is a multifunctional protein fundamental to RNA processing and mRNA degradation. The glutamine synthetase I genes of CAUCR and MESLO are among the top 10 PHX genes in those genomes. Interestingly, MESLO also features glutamine synthatase II among the top 10.
In the five α-genomes there are many ABC transporter proteins that are distinctively PHX (Table 5). These include several transporters of small molecules, several ABC periplasmic binding proteins, peptidoglycan-associated lipoproteins, and glycerol-3-phosphate-binding proteins. An ammonium transporter (AmtB) protein is significantly PHX [CAUCR E(g) = 1.60, SINME 1.12, MESLO 1.69, AGRTU 1.72] except in BRUME (0.68). This finding is consistent with the fact that ammonium is the preferred nitrogen source when available. CAUCR is well adapted for survival in dilute nutrient environments. CAUCR has no OmpF-type porins that mediate passive diffusion of hydrophilic substrates across the outer membrane, but the CAUCR genome encodes 65 TonB-dependent receptors of which 25 are PHX and two show E(g) values among the top 10. The TonB protein in E. coli interacts with outer membrane receptor proteins and energizes uptake of specific substrates (e.g., iron and insoluble molecules). These substrates are either poorly permeable through the porin channels or are encountered at very low concentrations. TonB-dependent receptors are a diverse family with sequence similarity only for ≈100 aa near the carboxyl end of the protein. Studies show that CAUCR can convert mercury, copper, cadmium, cobalt, and other metals into chemical forms that are less soluble and less toxic to humans (1).
We observed that specialized regulatory proteins are seldom PHX (12, 13). However, among the α-genomes, the response regulator CtrA is significantly PHX [in CAUCR E(g) = 1.39, AGRTU 1.18, SINME 1.21, MESLO 1.24, and BRUME 1.05]. In CAUCR, CtrA is essential for cell cycle progression and directly regulates a diverse group of genes including those required for DNA methylation, cell division, flagella, and pili biogenesis (21).
Energy Metabolism, Detoxification, and Flagellar PHX Genes in α-Genomes. The α-genomes feature many PHX genes strongly dependent on aerobic conditions (Table 2). These include those encoding (i) the cytochrome c oxidase subunits I, II, and III; (ii) the ATP synthase (ATP synthase F1 subunits α, β, γ, and ε, and ATP synthase F0 subunits, A, B, and C); (iii) NADH dehydrogenase (Nuo) complex and associated enzymes of aerobic respiration; (iv) the pyruvate dehydrogenase subunits; and (v) the TCA cycle enzymes. The sucABCD of the TCA cycle are significantly PHX in all α-genomes. In contrast, most genes encoding glycolytic enzymes are not PHX in α-genomes. Exceptions are GAPDH and enolase, which are multifunctional (22); see also Materials and Methods.
Table 2. Predicted expression levels for glycolysis, TCA cycle, detoxification, and flagellar genes.
|
E(g)
|
|||||
|---|---|---|---|---|---|
| Gene | CAUCR | SINME | MESLO | AGRTU | BRUME |
| Energy metabolism: glycolysis | |||||
| Fructose-bisphosphate aldolase, class II (fba) | (0.99) | (0.97)*, (0.70)* | 1.06 | — | 1.48* |
| Fructose-bisphosphate aldolase, class I (fbaB) | — | (0.92) | 1.30 | 1.26* | — |
| Glyceraldehyde 3-phosphate dehydrogenase (gap), GAPDH | 1.54 | 1.66 | 1.75 | 1.83* | 1.90 |
| Phosphoglycerate kinase (pgk) | 1.46 | (0.82) | 1.29 | (0.85)* | 1.08 |
| Phosphoglycerate mutase (gpm) | (0.80) | (0.87) | 1.15 | — | 1.15* |
| Enolase (eno) | 1.28 | (0.89) | 1.42 | 1.53 | 1.22 |
| Pyruvate kinase (pyk) | (0.53) | (0.67) | 1.09 | (1.11)* | (0.81) |
| Energy metabolism: TCA cycle | |||||
| Citrate synthase (gltA) | 1.24 | (1.02) | 1.49 | 1.49 | 1.50 |
| Aconitate hydratase (acnA) | 1.55 | 1.14 | 1.61 | 1.32 | 1.50 |
| Isocitrate dehydrogenase (icd) | 1.52 | 1.31 | 1.46 | (0.76)† | 1.34 |
| 2-oxoglutarate dehydrogenase, E1 component (sucA) | 1.34 | 1.11 | 1.40 | 1.39 | 1.53 |
| Dihydrolipoamide succinyltransferase component (E2) of 2-oxoglutarate dehydrogenase complex (sucB) | 1.81 | 1.34 | 1.13 | 1.62 | 1.91 |
| Dihydrolipoamide dehydrogenase component (E3) of 2-oxoglutarate dehydrogenase complex (Ipd) | 1.24 | (0.96) | 1.45 | 1.12 | 1.22 |
| Succinyl-CoA synthetase, beta subunit (sucC) | 1.62 | (0.96) | 1.15 | 1.74 | 1.46 |
| Succinyl-CoA synthetase, alpha subunit (sucD) | 1.70 | 1.24 | 1.36, 1.18 | 1.79 | 1.70 |
| Succinate dehydrogenase, flavoprotein subunit (sdhA) | 1.31 | (0.94) | 1.38 | (0.90) | 1.45 |
| Succinate dehydrogenase, iron-sulfur protein (sdhB) | (1.00) | (0.77) | 1.12 | (0.92) | 1.08 |
| Succinate dehydrogenase, cytochrome b556 subunit (sdhC) | (0.75) | (1.01) | (0.99) | (0.79) | 1.08 |
| Succinate dehydrogenase, membrane anchor protein (sdhD) | (0.73) | (0.84) | (0.94) | 1.06 | (0.88) |
| Fumarate hydratase (fumC) | 1.22 | (0.71) | 1.13 | 1.08 | (0.55)* |
| Malate dehydrogenase (mdh) | 1.68 | 1.22 | 1.30 | 1.23 | 1.63 |
| Detoxification | |||||
| Superoxide dismutase (Mn, Fe) (sodB) | 1.31, (0.66) | 1.36 | 1.25 | 2.06, (0.75)*, (0.58)* | 1.11 |
| Alkyl hydroperoxide reductase, subunit c (ahpC) | 1.91 | (0.99)* | (0.88) | (0.71) | 1.45* |
| Alkyl hydroperoxide reductase, subunit f (ahpF) | 1.05 | — | — | — | — |
| Alkyl hydroperoxide reductase | 1.31 | 1.28, (0.79) | 1.13, (1.04) | (0.89), (0.81) | (0.91) |
| Catalase | 1.00 | (0.88)* | (0.93) | (0.78)* | — |
| Catalase C | — | 1.11, (0.57)* | (0.86) | (0.53) | (0.92) |
| Nonheme chloroperoxidase (cpo) | — | (0.94)*, (0.93)*, (0.81), (0.73)* | 1.45, (0.79), (0.68) | 1.09*, (1.08)*, (0.86)*, (0.83)*, (0.68)* | — |
| Organic hydroperoxide resistance protein (ohr) | 1.26 | (0.94), (0.82)*† | (1.95) | 1.29 | (0.72) |
| Glutathione S-transferase family protein | (0.74), (0.67)† | — | 1.11, (0.96), (0.86) | — | (0.68) |
| Glutathione S-transferase family protein | (0.67) | (0.72) | 1.10 | (0.65) | (0.82)* |
| Putative glutathione S-transferase | — | (0.64) | 1.03 | — | — |
| Glutathione transferase | — | — | 1.12 | — | (0.82)* |
| Flagellar proteins | |||||
| Flagellar motor protein (motA) | 1.35 | (0.82) | 1.10 | 1.02 | (0.82)* |
| Flagellar motor protein (motB) | — | (0.81) | 1.44 | (0.66)* | (0.79) |
| Flagellin (fla) | 1.62, 1.58, 1.20, 1.16, (0.74) | 1.41, 1.32, (0.95), (0.79) | 1.55, 1.26 | 1.85, 1.56, 1.08, (0.70) | 1.64* |
| Flagellin synthesis regulator (flaF) | (0.75) | (0.89) | 1.08 | (0.69) | (0.65)* |
| Flagellar hook-basal body protein (fliE) | 1.22 | 1.03 | 1.15 | (0.83) | 1.21* |
| Flagellar M-ring protein (fliF) | 1.01 | (0.63) | 1.29 | (0.61) | (0.93)* (0.76)* |
| Flagellar motor switch protein (fliN) | 1.07 | (0.86) | 1.13 | (0.75) | (0.91)* |
| Flagellar biosynthesis protein (fliP) | (0.83) | (0.91) | 1.33 | (0.74) | (0.83)* |
| Flagellar biosynthesis protein (fliQ) | 1.08 | (0.75) | 1.27 | (0.89) | (0.78)* |
| Flagellar biosynthesis protein (fliR) | (0.83) | (0.85) | 1.30 | (0.71) | (0.67)* |
| Flagellar biosynthesis protein (flhA) | (1.00) | (0.71) | 1.49 | (0.68) | (0.66)* |
| Flagellar protein (flgA) | — | (0.73) | 1.08 | (0.77) | (0.74)* |
| Flagellar basal-body rod protein (flgC) | (0.84) | (0.99) | 1.12 | (0.94) | (0.72)* |
| Flagellar hook assembly protein (flgD) | 1.08 | (0.91) | 1.09 | (0.76) | (0.77)* |
| Flagellar hook protein (flgE) | 1.52 | (0.92) | 1.05 | (0.78) | (0.72)* |
| Flagellar basal-body rod protein (flgF) | 1.10 | (0.95) | 1.23 | (0.84) | (0.65)* |
| Flagellar basal body distal rod protein (flgG) | 1.49 | (0.92) | 1.26 | 1.22 | (0.86)* |
| Flagellar L-ring protein (flgH) | 1.22 | (0.84) | (0.75) | 1.08 | (0.81)* |
| Flagellar P-ring protein (flgI) | 1.07 | (0.81) | 1.16 | (0.67) | (0.59)* |
| Flagellar hook-associated protein 3 (flgL) | — | (0.61) | 1.07 | (0.65) | (0.60)* |
| Flagellin synthesis repressor (flbT) | (0.80)† | (0.95) | 1.10 | (0.79) | (0.75)* |
Included are only genes that qualify as PHX in at least one of the genomes. Numbers in parentheses indicate the gene is not PHX; — indicates that the gene does not have a homolog in the genome
Gene located outside the largest chromosome
PA gene
Only MESLO and BRUME among the five α-genomes encode most glycolysis genes PHX. In MESLO, the proteins involved in the initial steps of glycolysis are near PHX levels [E(g) = 0.97 for glucose-6-phosphate isomerase and E(g) = 0.93 for 6-phosphofructokinase], whereas the other glycolysis enzymes are PHX at E(g) values in the range from 1.06 to 1.75. Strikingly, apart from the multifunctional GAPDH, the other glycolysis genes are not PHX in the symbiotic SINME. SINME and some other α-genomes lack an ATP-dependent phosphofructokinase although a complete Entner–Doudoroff pathway is present (but not PHX) that is the main route for glucose utilization in SINME (7). Presence of phosphofructokinase in MESLO coupled with PHX levels of most glycolytic enzymes suggests an enhanced role for the glycolysis pathway in MESLO compared with other α-genomes.
Genes encoding α and β subunits of ATP synthase are PHX in all five α-genomes. With respect to electron transport, flavoprotein α is a very high PHX gene in CAUCR and MESLO. Cytochrome c oxidase subunit I is among the top 30 PHX genes in all five α-genomes with E(g) values of 1.66, 1.43, 1.79. 1.37, and 1.55, respectively. Many of the NADH dehydrogenase I (nuoA–N) subunit genes are PHX and these are often encoded in a cluster. SINME has two copies of nuoA–N, one (PHX) encoded by the main chromosome and the other (not PHX) on megaplasmid pSymA.
BRUME stands out among the organisms we considered here by having the highest number of PHX genes involved in various energy metabolism pathways. Besides having the most TCA cycle and glycolysis genes PHX, BRUME contains 12 PHX subunits of NADH dehydrogenase (MESLO has eight, SINME six, AGRTU and CAUCR five each). Only BRUME among the five α-genomes has several PHX genes of anaerobic respiration, namely subunits of nitrate, nitrous oxide, and nitric oxide reductases. These genes in BRUME are located in the second chromosome, and their counterparts in AGRTU and SINME are found mostly in the linear chromosome and megaplasmids, respectively. It appears that BRUME has evolved to use efficiently alternative energy sources under both aerobic and anaerobic conditions. In these five α-genomes, the transaldolase and transketolase major genes of the pentose phosphate pathway are PHX. Other enticing PHX genes in the five α-genomes include ilvC, E(g) of 1.40–1.79; ndk (nucleotide diphosphate kinase, 1.40–1.91); and the gene encoding the cell division protein FtsZ, 1.33–2.07.
Table 3 describes the number of PHX glycolysis and TCA cycle genes in α-proteobacteria, γ-proteobacteria, and low G+C Gram-positive bacteria. Where the count of PHX TCA genes significantly exceeds the PHX glycolysis genes, the organism is predicted to prefer an aerobic lifestyle. For the opposite inequality, an anaerobic lifestyle is prevalent. Where the counts of PHX glycolysis are approximately the same, a facultative lifestyle is expected.
Table 3. Counts of PHX genes of glycolysis and of the TCA cycle pathways in different genomes.
| Genome | Glycolysis | TCA cycle | Predicted preferred lifestyle |
|---|---|---|---|
| Low G+C Gram+ | |||
| B. subtilis | 7 | 3 | Facultative/anaerobic |
| Bacillus halodurans | 4 | 8 | Facultative/aerobic |
| Listeria innocua | 9 | 1 | Anaerobic |
| Listeria monocytogenes | 9 | 1 | Anaerobic |
| Lactococcus lactis | 10 | 1 | Anaerobic |
| Streptococcus pyogenes | 9 | 0 | Anaerobic |
| Streptococcus pneumoniae | 11 | 0 | Anaerobic |
| Staphylococcus aureus | 8 | 1 | Anaerobic |
| Clostridium acetobutylicum | 8 | 0 | Anaerobic |
| Clostridium perfringens | 10 | 0 | Anaerobic |
| γ-proteobacteria | |||
| Shewanella oneidensis | 4 | 8 | Facultative/aerobic |
| V. cholerae | 8 | 3 | Facultative/anaerobic |
| E. coli | 10 | 10 | Facultative |
| Salmonella typhimurium | 9 | 6 | Facultative |
| Yersinia pestis | 8 | 3 | Facultative/anaerobic |
| Haemophilys influenzae | 8 | 1 | Anaerobic |
| Pasteurella multocida | 7 | 0 | Anaerobic |
| Pseudomonas aeruginosa | 1 | 9 | Aerobic |
| Buchnera sp. APS | 1 | 0 | Parasitic |
| α-proteobacteria | |||
| CAUCR | 3 | 11 | Aerobic |
| SINME | 1 | 6 | Aerobic |
| MESLO | 6 | 12 | Facultative/aerobic |
| AGRTU | 3 | 10 | Aerobic |
| BRUME | 5 | 12 | Facultative/aerobic |
Assembly of a flagellum, the motive organelle produced by many bacteria, requires export of protein subunits from the cytoplasm to the outer surface of the cell by a mechanism resembling type III secretion. Flagella generally consist of three main components: the basal body, hook, and filament. Flagellum biogenesis and chemotaxis occur in coordination with flagellum assembly and in response to environmental signals. Recent evidence shows that the flagellum regulon can influence bacterium–host interactions independent of motility (23). There is also an established selective connection of flagellar motion and chemotaxis responses. The flagellum secretion apparatus may be viewed as part of the chaperone family essential for bacterial viability. Flagella are generally absent in nonmotile prokaryotes. The five α-genomes contain many flagellar genes, many of which are encoded in clusters. Enigmatically, BRUME, although considered nonmotile (6), still contains many flagellar genes, but most are not PHX (Table 2). Has BRUME lost motility and converted the flagellar proteins to a secretion apparatus for delivery of toxins? Interestingly, all but one of the flagellar genes of BRUME are encoded on chromosome II, suggesting that these may be largely laterally transferred. MESLO features a plethora of flagellar and transport PHX genes. CAUCR is distinguished with 15 flagellar PHX genes, presumably because of the importance of motility in its life cycle.
Because these five α-proteobacteria function primarily in an aerobic environment, we might expect these genomes to express proteins that protect the cell from the toxic effects of oxygen radicals. We find that α-proteobacterial genomes generally contain several PHX detoxification genes. For example, GAPDH responds to oxidative stress and stimulates thioredoxin, glutathionine, and thioredoxin peroxidase (18). The alkyl hydroperoxide reductase genes ahpC and superoxide dismutase sodB are prominently PHX; consult Table 2 for other examples.
TCA Gene Clusters in α-Genomes. The TCA genes in the five α-genomes are organized into two four-gene clusters (possibly operons) encoded in the same orientation, namely, sucCDAB (succinyl-CoA synthetase and oxoglutarate dehydrogenase units) and sdhCDAB (succinate dehydrogenase units). The suc operon of the SINME, AGRTU, and BRUME genomes is augmented by the mdh (malate dehydrogenase) gene to a five-gene operon. The arrangement of the TCA genes in E. coli, Bacillus subtilis, and M. tuberculosis differs from the α-genomes. For example, the suc and sdh genes are combined into a single eight-gene cluster in E. coli, but are separated in B. subtilis and M. tuberculosis. The sucCD and sucAB operons in BACSU are separated by ≈400 kb. The three-gene cluster sdhCAB in BACSU is missing gene sdhD. The (gltA, icd, mdh) operon of BACSU is not observed among α-genomes.
Glycolysis genes are distributed widely on both strands of all five α-genomes, unlike in BACSU where all glycolysis genes are encoded from a single DNA strand. In contrast, glycolysis genes of E. coli switch strands six times.
Nodulation and Nitrogen Fixation Across α-Genomes. Of the five α-proteobacterial genomes, SINME and MESLO fix nitrogen symbiotically, whereas CAUCR, AGRTU, and BRUME do not, principally because they lack the nitrogenase complex essential for conversion of dinitrogen to ammonia. Nod genes are mainly of two kinds: those contributing to the formation of the signal molecule and those involved in communicating the signal to the plant. Nif genes encode nitrogenase cofactors, regulatory, or structural proteins. Fix genes appear to contribute accessory functions, mostly regulatory (some oxygen sensors), to the nitrogen-fixation process. All genes of nitrogen fixation and nodulation annotated fix, nif, or nod were examined. In this context, two major classes of genes stand out: (I) gene homologs exclusive to SINME and/or MESLO, and (II) genes present in all five α-genomes, which may contribute in an ancillary way to nitrogen fixation. In category I, nif, fix, or nod genes unique to SINME and/or MESLO are invariably encoded from the megaplasmid pSymA in SINME; these genes cluster in the order nodD*, fixK2*, noeA*, noeB*, fixU, nifB*, nifA*, fixX*, nodD3*, nodH*, fixC*, fixB*, fixA, nifH, nifD, nifK, nifE, nifX*, nodF, nodE, nodJ*, nodI*, nodC*, nodB*, nodA*, nodD1*, nifN*, nolF* (* signifies that the gene qualifies as PA; see Materials and Methods). Genes involved in nitrogen fixation or nodulation were largely verified by knockout experiments in SINME. Genes found in all five α-genomes that may function in nitrogen fixation or nodulation are about equally encoded from the main chromosome of SINME or from pSymA. Examples of category II proteins are described in Table 6, which is published as supporting information on the PNAS web site. The preponderance of PA genes related to nitrogen fixation is consistent with our previous speculations (21), that the megaplasmids were laterally transferred into SINME and MESLO, and touches on the larger question of whether nif genes in other species (even those with chromosomal locations) may have experienced lateral transfer.
Nitrogen-Fixation Proteins Unique to SINME or MESLO. SINME has nitrogen-fixation or nodulation-related genes that have no homologs in any of the other four α-genomes. All five are encoded on pSymA (fixQ3, noeA, noeB, nodH, nolF secretion protein). MESLO has 16 nitrogen-fixation or nodulation-related genes that have no homologs in any of the other four α-genomes: nifZ, nifW, nifS, nifQ, nifU, nitrogen assimilation control protein (mlr6097), noeI, nodZ, nodS, nolO, nolL, nolX, nolW, nolT, nolU, nolV. The differences between these two complements of genes are in large part caused by the two organisms making distinct signal molecules recognized by their separate plant hosts.
Genes of Unknown Function PHX in Several α-Genomes. ORFs of unknown function with high predicted expression levels may be attractive candidates for experimental characterizations because we assume that their PHX nature likely indicates that they have important functions in these organisms. Table 4 lists 11 families of homologous genes that are PHX in at least three of these genomes. Three families are of unknown function, three are putative ABC transporters of unknown specificity, and the remainder are annotated as putative transcription regulator, an exported protein, a GTP-binding protein, and two membrane proteins. The ABC transporter family, including genes SMc01605, mll3069, AGR_C_3890, and BMEII1120, stands out with rather high E(g) values in MESLO (1.73), AGRTU (1.57), and BRUME (1.47) and somewhat lower but still significantly PHX in SINME (1.18). There is no correspondingly strong homolog in CAUCR. Another intriguing family comprises genes CC3292, SMc04094, mll3765, AGR_C_4981, and BMEI0303. The AGRTU homolog is marginally PHX, whereas the representatives in the remaining four genomes are all PHX with E(g) of 1.10–1.49. These genes are unique to the α-genomes, having no homologs in other groups of prokaryotes (Clusters of Orthologous Groups database, ref. 19). This kind of analysis is valuable in helping to prioritize which of the many hundreds of genes of unknown function in these α-proteobacteria might be the more promising candidates on which to focus further experimental characterization.
Table 4. Hypothetical or poorly characterized proteins PHX in at least four genomes [three if E(g) > 1.40].
| CAUCR
|
SINME
|
MESLO
|
AGRTU
|
BRUME
|
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Gene | E(g) | Gene | E(g) | Gene | E(g) | Gene | E(g) | Gene | E(g) | Comments |
| CC0653 | 1.11 | SMc03874 | 1.21 | mlr3857 | 1.53 | AGR_C_5013 | (0.81) | BMEI0279 | 1.07 | Putative transcriptional regulator |
| SMb21642 | (0.91) | mlr3121 | 1.28 | AGR_L_1652 | 1.47 | BMEI1242 | 1.22 | Hypothetical exported protein | ||
| SMc03936 | (0.87) | mlr3122 | 1.07 | AGR_L_335 | 1.11 | |||||
| SMb21491 | (0.85) | |||||||||
| CC3663 | (0.91) | SMc03839 | 1.16 | mll4343 | 1.27 | AGR_C_4861 | 1.25 | BMEI1859 | 1.28 | Integral membrane protein |
| mlr0409 | 1.01 | |||||||||
| SMc02174 | 1.20 | mll8170 | 1.24 | AGR_C_1217 | 1.17 | BMEII1052 | 1.20 | Hypothetical membrane protein | ||
| SMb20597 | (0.86) | mll8168 | (0.96) | |||||||
| CC0479 | 1.01 | SMc02695 | 1.04 | mlr2695 | 1.47 | AGR_C_4063 | (1.02) | BMEI0479 | 1.23 | Probable GTP-binding protein |
| CC2148 | 1.30 | SMc04454 | (0.88) | mll1607 | 1.56 | AGR_C_3855 | 1.48 | BMEI0553 | (0.75) | ABC transporter |
| SMc01605 | 1.18 | mll3069 | 1.73 | AGR_C_3890 | 1.57 | BMEII1120 | 1.47 | ABC transporter binding protein | ||
| SMc03830 | 1.04 | AGR_C_4844 | 1.48 | BMEI0015 | 1.30 | Unknown; BMEI0015 annotated ABC transporter | ||||
| CC3292 | 1.10 | SMc04094 | 1.27 | mll3765 | 1.49 | AGR_C_4981 | (0.96) | BMEI0303 | 1.20 | Unknown |
| SMc03100 | 1.05 | mlr8117 | 1.17 | |||||||
| SMc00795 | (1.01) | mll9560 | 1.09 | AGR_C_4740 | 1.15 | BMEI0287 | 1.10 | Unknown | ||
| mlr3708 | 1.09 | AGR_pAT_235 | (0.86) | BMEI1214 | (0.96) | |||||
| SMc00950 | 1.18 | mll0513 | 1.26 | AGR_C_3259 | 1.25 | BMEII0692 | 1.44 | Unknown | ||
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health Grants 5ROIGM10452-38 and 5ROIHG00335.14.
Abbreviations: CAUCR, Caulobacter crescentus; SINME, Sinorhizobium meliloti; MESLO, Mesorhizobium loti; AGRTU, Agrobacterium tumefaciens; BRUME, Brucella melitensis; PHX, predicted highly expressed; RP, ribosomal protein; TF, transcription/translation factor; CH, chaperone/degradation; PA, putative alien; TCA, tricarboxylic acid.
References
- 1.Nierman, W. C., Feldblyum, T. V., Laub, M. T., Paulsen, I. T., Nelson, K. E., Eisen, J. A., Heidelberg, J. F., Alley, M. R., Ohta, N., Maddock, J. R., et al. (2001) Proc. Natl. Acad. Sci. USA 98, 4136-4141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Galibert, F., Finan, T. M., Long, S. R., Puhler, A., Abola, P., Ampe, F., Barloy-Hubler, F., Barnett, M. J., Becker, A., Boistard, P., et al. (2001) Science 293, 668-672. [DOI] [PubMed] [Google Scholar]
- 3.Kaneko, T., Nakamura, Y., Sato, S., Asamizu, E., Kato, T., Sasamoto, S., Watanabe, A., Idesawa, K., Ishikawa, A., Kawashima, K., et al. (2000) DNA Res. 7, 331-338. [DOI] [PubMed] [Google Scholar]
- 4.Goodner, B., Hinkle, G., Gattung, S., Miller, N., Blanchard, M., Qurollo, B., Goldman, B. S., Cao, Y., Askenazi, M., Halling, C., et al. (2001) Science 294, 2323-2328. [DOI] [PubMed] [Google Scholar]
- 5.Wood, D. W., Setubal, J. C., Kaul, R., Monks, D. E., Kitajima, J. P., Okura, V. K., Zhou, Y., Chen, L., Wood, G. E., Almeida, N. F., Jr., et al. (2001) Science 294, 2317-2323. [DOI] [PubMed] [Google Scholar]
- 6.DelVecchio, V. G., Kapatral, V., Redkar, R. J., Patra, G., Mujer, C., Los, T., Ivanova, N., Anderson, I., Bhattacharyya, A., Lykidis, A., et al. (2002) Proc. Natl. Acad. Sci. USA 99, 443-448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Capela, D., Barloy-Hubler, F., Gouzy, J., Bothe, G., Ampe, F., Batut, J., Boistard, P., Becker, A., Boutry, M., Cadieu, E., et al. (2001) Proc. Natl. Acad. Sci. USA 98, 9877-9882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Finan, T. M., Weidner, S., Wong, K., Buhrmester, J., Chain, P., Vorholter, F. J., Hernandez-Lucas, I., Becker, A., Cowie, A., Gouzy, J., et al. (2001) Proc. Natl. Acad. Sci. USA 98, 9889-9894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barnett, M. J., Fisher, R. F., Jones, T., Komp, C., Abola, A. P., Barloy-Hubler, F., Bowser, L., Capela, D., Galibert, F., Gouzy, J., et al. (2001) Proc. Natl. Acad. Sci. USA 98, 9883-9888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Oresnik, I. J., Liu, S. L., Yost, C. K. & Hynes, M. F. (2000) J. Bacteriol. 182, 3582-3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Paulsen, I. T., Seshadri, R., Nelson, K. E., Eisen, J. A., Heidelberg, J. F., Read, T. D., Dodson, R. J., Umayam, L., Brinkac, L. M., Beanan, M. J., et al. (2002) Proc. Natl. Acad. Sci. USA 99, 13148-13153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Karlin, S. & Mrázek, J. (2000) J. Bacteriol. 182, 5238-5250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Karlin, S., Mrázek, J., Campbell, A. & Kaiser, D. (2001) J. Bacteriol. 183, 5025-5040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mrázek, J., Bhaya, D., Grossman, A. R. & Karlin, S. (2001) Nucleic Acids Res. 29, 1590-1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mrázek, J. & Karlin, S. (1999) Ann. N.Y. Acad. Sci. 870, 314-329. [DOI] [PubMed] [Google Scholar]
- 16.Karlin, S. (2001) Trends Microbiol. 9, 335-343. [DOI] [PubMed] [Google Scholar]
- 17.Karlin, S. & Mrázek, J. (2001) Proc. Natl. Acad. Sci. USA 98, 5240-5245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sirover, M. A. (1999) Biochim. Biophys. Acta 1432, 159-184. [DOI] [PubMed] [Google Scholar]
- 19.Tatusov, R. L., Natale, D. A., Garkavtsev, I. V., Tatusova, T. A., Shankavaram, U. T., Rao, B. S., Kiryutin, B., Galperin, M. Y., Fedorova, N. D. & Koonin, E. V. (2001) Nucleic Acids Res. 29, 22-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Campbell, A., Mrázek, J. & Karlin, S. (1999) Proc. Natl. Acad. Sci. USA 96, 9184-9189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Laub, M. T., Chen, S. L., Shapiro, L. & McAdams, H. H. (2002) Proc. Natl. Acad. Sci. USA 99, 4632-4637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carpousis, A. J. (2002) Biochem. Soc. Trans. 30, 150-155. [PubMed] [Google Scholar]
- 23.Young, G. M., Schmiel, D. H. & Miller, V. L. (1999) Proc. Natl. Acad. Sci. USA 96, 6456-6461. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}