


Abstract
The coexistence of multiple codes in the genome of human immunodeficiency virus type 1 (HIV-1) was analyzed. We explored factors constraining the variability of the virus genome primarily in relation to conserved RNA secondary structures overlapping coding sequences, and used a simple combination of algorithms for RNA secondary structure prediction based on the nearest-neighbor thermodynamic rules and a statistical approach. In our previous study, we applied this combination to a non- redundant data set of env nucleotide sequences, confirmed the conservative secondary structure of the rev-responsive element (RRE) and found a new RNA structure in the first conserved (C1) region of the env gene. In this study, we analyzed the variability of putative RNA secondary structures inside the nef gene of HIV-1 by applying these algorithms to a non-redundant data set of 104 nef sequences retrieved from the Los Alamos HIV database, and predicted the existence of a novel functional RNA secondary structure in the β3/β4 regions of nef. The predicted RNA fold in the β3/β4 region of nef appears in two forms with different loop sizes. The loop of the first fold consists of seven nucleotides (positions 494–500), with consensus UCAAGCU appearing in 79% of sequences. The other has a five-base loop (positions 495–499) with consensus CAAGC. The difference in size between these two loops may reflect the difference between respective counterparts in the hairpin recognition. This may also have an adaptive biological significance.
INTRODUCTION
It is well known that genomic sequences frequently contain overlapping biological messages (1–3). Viral genomes, in general, and retroviral genomes, in particular, present the most striking examples of overlapping codes. A few examples are the overlapping genes env, tat and rev occupying all three reading frames, the overlapping coding region of the pol gene and the sophisticated mechanism of ribosomal frameshifting in this region. Viruses regularly use RNA secondary structures that are located and conserved within protein-coding regions of genomic sequences. The famous case is the rev-responsive element (RRE) (4,5), located in the transmembrane part of the coding region for the Env protein, directly downstream from the cleavage site in the protein. Konings analyzed the multiple-coding constraints on the RRE (6), and mentioned that the overlap may be viewed as a factor of the high conservation both in the RRE and in the transmembrane part of the Env protein. While Konings analyzed a known case of the overlapping codes, we tried to reveal regions in human immunodeficiency virus type 1 (HIV-1) with a high potential for coexistence of multiple codes. In this study, we concentrated our efforts on the coding sequence for the Nef protein.
The negative factor of lentiviruses is a 27–35 kDa myristoylated accessory protein (7). The Nef protein plays a key role in the pathogenesis of lentiviruses. Although the Nef protein structure and functions have been investigated, they are still poorly understood. It is involved in CD4 endocytosis, decreases the expression of CD4 and major histocompatibility complex (MHC) I antigens, alters the cellular signaling pathways by interacting with tyrosine and serine/threonine kinases, and enhances HIV-1 replication in primary T cells (7–9). The structure–function relationships in HIV-1 Nef have been reviewed by Geyer et al. (10). In addition to the features of signaling and trafficking, it appeared that nef also has an RNA-binding capacity (11).
Specific amino acid motifs along the Nef protein sequence together with structural motifs of α-helices and β-sheets dictate a protein conservation pattern that inevitably influences the DNA conservation (12). However, the conservation of a protein sequence is not the only reflection of evolutionary pressure. Additional factors such as protein–DNA and protein–RNA binding sites, RNA motifs, etc. can influence the conservation of DNA sequences. Tremendous efforts revealed signals, function and secondary structure of the Nef protein (10,13–15). However, little is known about the secondary structure of nef RNA. One of the few investigations carried out was a computer prediction of an RNA motif of a selenocysteine insertion sequence element located at the end of env and the start of nef in HXB2 (16). Although HXB2 is known as the representative subtype for HIV-1, predictions based on one subtype alone are far from satisfactory. Several well-known RNA secondary structures along the HIV-1 genome play functional roles during the virus life cycle. The best-known structures are the transactivation responsive elements (17) and the RRE (4,5). Interestingly, in elucidation of these structures, both the statistical treatment of variability and RNA secondary structure predictions played a crucial role (18,19). Lately, we have demonstrated how high conservation of the third codon position presumes highly conserved RRE RNA secondary structure, and a stem–loop structure in the C1 region of the env gene (20). This third position conservation also implies the existence of a biological function for these RNA secondary structures. Predictions of both RRE and C1 secondary structure of RNA were indicated by very strong signals. In contrast, here we are demonstrating how rather weak but clear signals of secondary structure in the nef gene of HIV-1 can also be predicted on the basis of conservation in the third positions.
RNA secondary structures overlapping the nef gene of HIV-1
In this study, we did not try to predict the minimum free energy RNA secondary structure for the entire HIV-1 genome (21). Instead, our efforts focused on the prediction of conserved functional RNA secondary structures located in the nef gene. These structures may exist either at the stage of vRNA, or at the stage of mRNA, or both. In order to elucidate features and positions of such structures, we analyzed all available variants of the sequences coding for the complete Nef protein and applied various computational methods to predict common RNA folds within the nef gene. One approach for predicting common RNA folds involves the evolution of RNA molecules (22–24). Schuster and co-workers introduced the notion of a structure density surface and computed it as the conditional probability of two structures having distance t given that their sequences have distance h. Using this approach, one can compare RNA secondary structures by counting the minimal number of point mutations required to convert the sequences into each other. They found that the vast majority of possible minimum free energy secondary structures occur within a fairly small neighborhood of any typical (random) sequence. Another approach based on probability models is the ‘covariance models’ method (25). This method is based on an algorithm for learning the consensus of sequences and assessing the covariation of point mutations. It may be applied to any family of small RNA sequences. However, the alleles of the nef gene are too long to utilize this method. RRE is the best example of how the selection for the RNA secondary structure can be as important as the amino acid sequence in the overall env genomic conservation. The region containing RRE is conserved for at least two reasons: to preserve the gp120–gp41 consensus cleavage motif and to preserve the functional RRE RNA secondary structure. The presence of this conserved RNA secondary structure is a major contributor to the conservation of this genomic sequence (20).
Thermodynamic RNA structure predictions
In general, the use of thermodynamic calculations for searching a common RNA fold in evolutionarily related sequences is, to say the least, controversial. In one of the early studies, Trifonov and Bolshoi (26) used multiple alignments for predicting base matching in 5S rRNA by superimposing triangular base pairing diagrams on each other. Later, the RRE secondary structure was revealed by calculation of the difference between the lowest free energy of real HIV-1 sequences and randomly shuffled sequences, using Monte Carlo simulation (18,21,27–32). A remarkable innovation is the Alifold program, a method for computing the consensus structure of a set of aligned RNA sequences taking into account both thermodynamic stability and sequence covariation (33). However, sequence covariation analysis is possible only if the aligned sequences are variable enough. Our goal was to use the advantages of a combination of the energy minimization methods together with the multiple alignment approach. Lück et al. (34) used different but similar methodology, based on thermodynamic structure predictions combined with the information obtained from the phylogenetic alignment of sequences. However, Lück’s Construct software demands large computation resources and is suitable for short fragments or a limited number of sequences. The software is still unable to give a good indication of the biological significance of the structure. In our approach, the presence of the biological function of the RNA secondary structure regions can be indicated when the following criteria are met simultaneously: signals of the nucleotide conservation overlap with signals of RNA fold conservation and also overlap with conservation of the third position along the translatable open reading frame.
MATERIALS AND METHODS
The nef data set
We used the data set of 140 complete HIV-1 genome sequences retrieved from the Los Alamos HIV sequence database. A pool of 140 nef sequences was obtained by extraction of nef genes encoding the Nef protein. This pool was subjected to cleaning and redundancy control utilizing the following steps. (i) Cleaning by size and valid features of coding sequences: only sequences larger than 600 bases, starting with ‘ATG’ and ending with a stop codon, possessing coding region length modulo 3, were retained. (ii) Myristoyl ation signal and SH3 domain: only sequences starting with Met–Gly and containing the PxxPxR signal for the SH3 domain of src family kinases were kept. (iii) Redundancy control: in order to obtain a data set in which the similarity between any two sequences is below a certain threshold, we used CLEANUP, a fast computer program for removing redundancies from nucleotide sequence databases (35). A subset of 106 sequences with pairwise similarity of <95% nucleotide identity was selected. (iv) Alignment consistency: two nef sequences, having a few non-mutational insertions, probably resulting from recombination, caused large gaps in the alignment file and were discarded. This resulted in an improved alignment, leaving a final pool of 104 sequences.
Information content of multiple alignments
To find the relevant signal in the multiple alignments, we used the Kullback–Leibler measure of information content (36,37). This measure quantifies the contrast between an actual and an expected distribution of amino acids and nucleotides, respectively. This is used to calculate the total amount of information per position in the alignment. In general, the information content for position i in the alignment may be written as:

where index k sums either over all possible amino acids or over all possible nucleotides, when in both cases k may also mean a gap. Thus, index k varies from 1 to 5 for DNA (A, C, G, T, –) and from 1 to 21 for proteins. The quantity qik is the observed fraction of amino acid/base/gap k at position i, and pk (pA, pC, pG, pT, p_) is the expected value. Frequently, in information content calculations, the columns with gaps are not taken into account, nor are uniform expected values used. These assumptions reduce the Kullback–Leibler entropy measure to the Shannon information measure [see discussion in Schneider et al. (38), and Schneider and Stephens (39)]. Note that for gaps, we used the background probability p_ = 1 as discussed in the references above [see also Gorodkin et al. (40)]. In the case of the Shannon information, the maximum information in bits per position is log220 ≈ 4.3 for amino acids and log24 = 2 for nucleotides. The quantifier pk used here for nucleotides is pN = 0.25.
Analysis of multiple alignments of RNA fold predictions
The multiple alignments were in all cases made by CLUSTALW (41,42) for the Nef protein sequences. After that, every protein sequence was back-translated, and further analysis was performed by searching for RNA secondary structures of corresponding back-translated candidates. RNA folds were provided by the computer programs Mfold [the software is described in Zuker (43,44–47)] and RNAfold (Vienna RNA Package version 1.4; 28,30,31). The output of RNAfold is a string of dots ‘·’ for bases not involved in complementary contacts, and brackets ‘(’ and ‘)’ for 5′- and 3′-complementary bases, respectively. Gaps were inserted according to their positions in the aligned amino acid sequences. Thus, the alignment of the RNA structures was made in order to reveal structural RNA motifs common for many of the nef sequences. Conservation of an RNA secondary structure element in position i is calculated again using a relative information measure:

where the index k runs over all RNA secondary structure elements and gaps. The quantities qi,‘(’, qi, ‘)’, qi,‘.’ and qi, ‘_’ are the observed fraction of 5′ds, 3′ds, ss and gaps, respectively at position i. The expected probability that the base belongs to a single-stranded section at every position i p‘·’ has been found empirically to equal 0.5, consequently ds probabilities p‘(’, p‘)’ are equal to 0.25, and the gap background pi‘–’ is equal to 1. The incorporation of gaps in the alignments was performed as proposed by Hertz et al. (48).
Back-translation
In order to demonstrate the significance of the analysis of the nucleotide sequence and the analysis of RNAfold multiple alignments, a two-step back-translation procedure was performed. The first step was replacement of each amino acid by one of the triplets encoding this amino acid. The amino acid was replaced either by a correct genomic codon or by a suitable randomly selected codon. The selection procedure was either uniformly random or took into account the codon usage of HIV-1. We used the EMBOSS program ‘Backtranseq’ and the HIV-1 codon-usage file. This EHuman_immunodeficiency_virus_type_1.cut file was kindly provided by Dr Alan Bleasby and can be retrieved from the database ftp://ftp.ebi.ac.uk/pub/databases/cutg/. The second step was carried out by insertion of gaps in the nucleotide sequence according to the gaps appearing in the corresponding amino acid sequence. At each position in which a gap appeared in the amino acid sequence, three gaps were added to the generated randomized DNA sequence.
Visualization of RNA folds
To illustrate the most conserved features of the putative RNA secondary structures in the β3/β4 (469–534) fragment, we presented the mRNA fold predictions. To illustrate the common features of the folds, we randomly chose 10 sequences from our cleaned database and folded them by the Mfold program. The accession numbers of the sequences are: AF067159, AF075702, AF75702, AF289548, U71182, AF286226, AY008718, M62320, AF286223 and AF179368.
Visualization by logo
To display the information content of nucleotide and amino acid sequences of the restricted β3/β4 region, we have used an extension of the sequence logo by Schneider and Stephens (39) in the form presented by Gorodkin et al. (40).
RESULTS
Conservation of the nef DNA
The multiple alignment was made at the amino acid level following back-translation to the nucleotide level as described above. For the analysis of variability, we used the information content measure. Figure 1 presents superposition of the information content curves related to the three different types of back-translated nucleotide sequences. This figure serves as an essential sequence conservation background to compare it with the following RNA secondary structure conservation. Although the randomized back-translated DNA sequences are more variable than the real genomic DNA, the conservation pattern of the back-translated sequences still follows the pattern of the real genomic DNA. This is a result of the conservation of the first and the second positions in the codon. Surprisingly, the information content of the set of codon usage-related back-translated sequences is far higher than the two others, implying higher conservation. We can explain this in the following way. The program Backtranseq was made in order to predict the most probable DNA sequence back-translated from a single amino acid string. This feature inevitably leads to a reduction of sequence variability when it comes to comparison of multiple back-translated sequences. Although this value of information content of the codon usage back-translation is higher than expected, we decided to use this value as a ‘high hurdle’ threshold.
Figure 1.

DNA conservation of nef DNA. To compare the information content of nef DNA sequences with encoded Nef proteins, we used a back-translation technique. The correct back-translation was made by replacing every amino acid by the corresponding codon from the real, known gene sequence; the randomized back-translation was made by replacing every amino acid by one of the corresponding randomly selected codons of its own repertoire (equiprobable within the group). The codon usage-related back-translation was made by using EMBOSS Backtranseq according to the HIV-1 codon usage file: EHuman_immunodeficiency_virus_type_1.cut. The curves showing the information content were smoothed by a running average with a window size equal to six. The green line represents the DNA conservation of the nef sequences retrieved from the original database. The blue line describes the conservation of the codon usage-related back-translated nef sequences, and the orange line describes the conservation of the random back-translation. Conservation of DNA at every position was computed according to Equation 1.
RNA secondary structure conservation of the nef gene
Comparative methods are very reliable in determining the RNA secondary structures common to a set of related RNA sequences (27,29,49–51). We took the three above-mentioned sets of back-translated sequences, calculated optimal RNA secondary structures for each sequence, and replaced each nucleotide by one of the symbols ‘(’, ‘)’ or ‘·’ using outputs of RNAfold software. RNA secondary structure conservation was computed according to Equation 2 as described above. The distribution of RNA secondary structure conservation along the nef gene is demonstrated in Figure 2. There is only one region consisting of a highly conserved putative RNA secondary structure, which is more conserved in the set of genomic sequences than in the codon usage back-translated set (the introduced threshold), located between positions 480 and 510 and covering the β3/β4 region of the protein. This region is also one of the regions characterized by the highest conservation of the third positions in the respective codon (data not shown). Moreover, the bases involved in base pairing are much more conserved than the bases that are not involved in such interactions. This lends additional support to the presumed biological significance of this structure.
Figure 2.

Conservation of predicted RNA folds. RNA secondary structures of the three different data sets of nucleotide sequences mentioned in Figure 1 were predicted by the Vienna package. The outputs of these predictions were aligned and the gaps were inserted according to the alignments of Nef amino acid sequences. Conservation of RNA secondary structure at every position was computed as the information contribution of the stem or loop relative to their expected distribution according to the structure information equation (Equation 2). The colors of the lines refer to the same databases as in Figure 1.
Prediction of two forms of RNA secondary structure within the β3/β4 region
The visualization of the RNA secondary structure alignments reveals an apparently common RNA secondary structure in the β3/β4 region of the nef gene. In Figure 3, we demonstrate 10 different randomly chosen Mfold predictions of the β3/β4 region. Superimposing these structures reveals a common core structure in the region around positions 480–520. The predicted structures are located from position 469 to 534. It seems that they consist of one defined bulged stem–loop structure with a free energy of –10.6 kcal/mol. There are only two hairpin-loop sizes: five and seven. The five-base loop has a consensus sequence YAARY appearing in 21 sequences (21%). This loop is associated with the consensus 5′-UA–loop–UR-3′. The larger loop has a consensus UYVAVYU. This loop appeared in 83 out of 104 sequences (79%) and associated with the consensus 5′-NU–loop–YR-3′.
Figure 3.
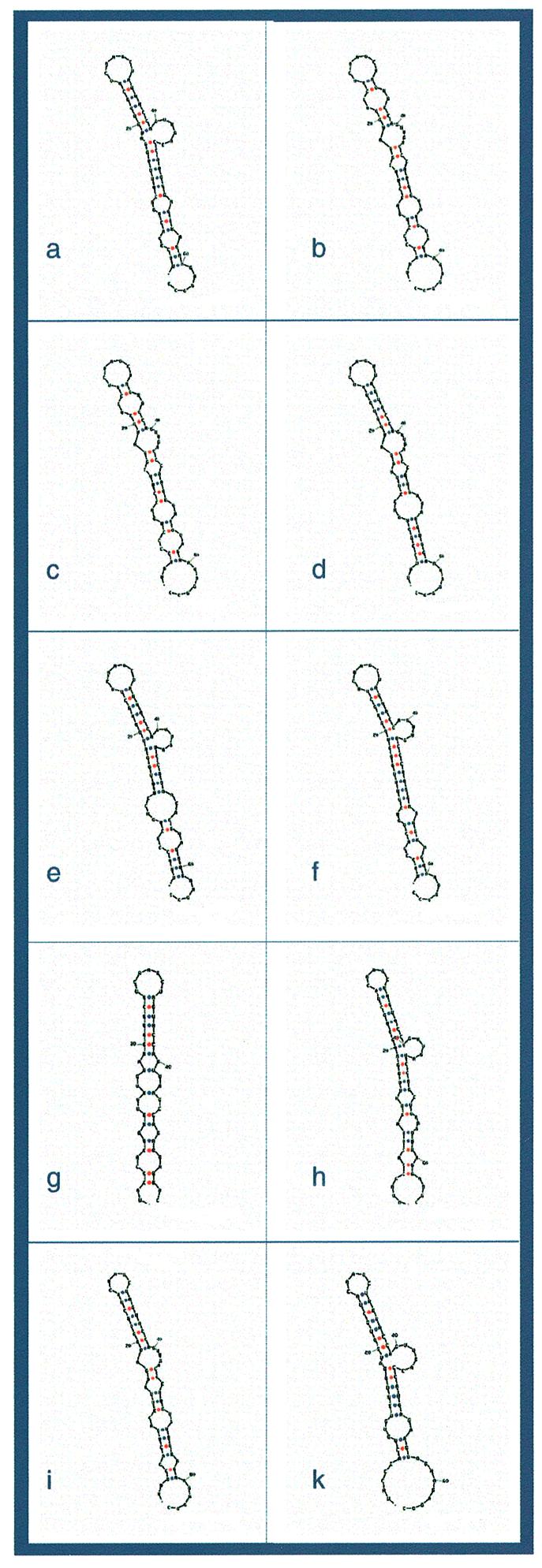
Examples of predicted RNA secondary structures in the β3/β4 region from a few randomly selected nef sequences. The β3/β4 RNA region of nef (positions 469–534 of the multiple alignment notation) was retrieved from 10 different randomly chosen sequences. The retrieved fragments were folded using the Mfold program. An approximate 60 bp bulged hairpin with interior loops appears in all chosen sequences. It is the main feature of the putative common RNA secondary structure. The seven-base hairpin loop appears in (a–g) and the five-base hairpin loop appears in (h–k).
In Figure 4, we present a conservation analysis of these two folds in a logo form. A comparison between Figure 4A and B reveals that the difference between a seven-base loop size and a five-base loop size lies in the substitution of uracil for adenine at position 494 (U494→A494). U494 is the second nucleotide in the triplet coding for the amino acid Phe165. The substitution (U494→A494) changes triplets UUc/u to UAc/u and causes the replacement of amino acid Phe165 by Tyr165 (see Fig. 4C and B). On the opposite 3′ side of the loop, Leu167 is encoded by c/uUA codons. A501 is very conserved in spite of being the third position of the Leu167 codon. Any substitution of A501 is a silent mutation, i.e. without changing the amino acid. An obvious explanation of the high conservation of A501 is its pairing with U493. Val168 is fully conserved. Although its potential third position repertoire is four bases, position 504 is over-conserved, with a domination of adenine but also with a minor guanine presence. The reasonable explanation for this conservation pattern is the pairing of A504 or G504 with U490. On the complementary strand, Pro169 is fully conserved. Again, although its potential third position repertoire is four bases, position 507, the third position of Pro169, is over-conserved and dominated by adenine. The only explanation for this high conservation is the pairing with U487 in the opposite strand. The codon for Phe161 in the 5′ side of the stem is highly conserved. If amino acid conservation were the only factor constraining the variability of nucleotides, we would expect a mixture of uracil and cytosine in position 483. However, in position 483, uracil is dominant and in the case of the five-base loop, the uracil is fully conserved. A good explanation for this conservation pattern is, again, the pairing of U483 with G511.
Figure 4.

(A and B) Structural RNA logo analysis of the hairpin loop. Two different common loops were detected in position 493–501 in the multiple alignment file: seven bases long (((…….))) and five bases long (((…..))). By composing a correct regular expression pattern for each loop, removing the gaps and including 16–17 bases expanding from each side of the loop, we were able to distinguish between 83 large loop RNA structures and 21 small loop structures. All alignments were analyzed by structural RNA logo analysis. The results are presented in bits, reflecting the information content of each base. The base pair fraction of each position has been calculated and appears below the logo. (A) refers to the large stem–loop structure and (B) refers to the small one. (C and D) Protein logo analysis of the amino acid sequence encoded by the β3/β4 hairpin loop region. The two nucleotide data sets (large and small loop), described in (A) and (B), were translated to amino acid sequences and analyzed by protein logo analysis. The results are presented in bits, reflecting the information content of each amino acid. (C) Refers to the large loop hairpin sequence and (D) refers to the small loop hairpin sequence.
DISCUSSION
The aim of our study was to check whether the conservation of regions in the nef gene of HIV-1 could be due to RNA secondary structures located in it. We predicted conserved RNA secondary structures in the nef region using a simple combination of the thermodynamic and the genomic approach. We believe that the structures are functional and, as a result, conserved. A related approach for detection of biologically significant RNA secondary structures was recently applied to positive-stranded RNA viruses (52,53). The authors paid special attention to the conservation of the third codon position, and, as a result, reduction of the variability in synonymous sites. Unlike the covariance analysis for revealing RNA secondary structures as in the above works, we applied the Kullback–Leibler surprise measure of information content of the dot and bracket presentation of predicted RNA folds. If fact, this method, fortified by observation of the conservation of the third codon position, gave us the precise location of the nef β3/β4 RNA secondary structure. Biologically relevant RNA secondary structures encoded as a hidden message within protein-coding sequence increase the base conservation so that covariance analysis becomes inapplicable (see Table 1) even if the parsimony-based algorithm is used. Table 1 presents the combined results of the nef data set multiple alignment (CLUSTALW software), RNA secondary structure predictions (RNAfold software) and base pair content examination using the Alidot software. The conservation of base pairing in the RNA secondary structure in the β3/β4 region is shown as was detected by Alidot.
Table 1. Variants of base pairing in the common nef β3/β4 RNA fold.
| Base position | Base-pair content | Occurrence | % base pairing | Comments |
|---|---|---|---|---|
| 494–500 | AU | 21 | 20.2 | Only in the 5-base loop |
| 493–501 | UA | 101 | ||
| UG | 3 | 100 | ||
| 492–502 | CG | 93 | ||
| UG | 5 | |||
| UA | 1 | 95.2 | ||
| 491–503 | GU | 97 | 93.3 | |
| 490–504 | UA | 90 | ||
| UG | 11 | |||
| GC | 1 | 98.1 | ||
| 489–505 | GC | 104 | 100 | |
| 488–506 | GC | 104 | 100 | |
| 487–507 | UA | 102 | ||
| UG | 1 | 99 |
RNA secondary structure within the β3/β4 region of nef
Several pieces of evidence support the assumption of the existence of RNA secondary structure in the β3/β4 region of HIV-1 nef. Not only the amino acids, but also the codons are highly conserved there. As a result of the conservation of the third position in the codons, precisely in this region, the predicted maximum of the RNAfold plot of the natural sequences in this region (Fig. 2) is higher than the predicted plot of the RNA fold of the codon usage-related back-translation sequences. The RNAfold secondary structure predictions reinforce the assumption of the biological significance of this secondary structure due to the low free energy of the structure and the repetition of conserved RNA secondary structure motifs. The high conservation of the third position in this region apparently indicates that this structure has a biological function, although what exactly is the functional role of the conserved RNA secondary structure remains unclear. Unlike the case of the β3/β4 RNA structure, for both RBE and TAR, specific protein-binding features are detected and well studied. Recently, a non-specific feature of binding of these secondary structures to the chromosomal protein HMG-D has been discovered (54). HMG-D is known to bind preferentially to DNA of irregular structure with little or no sequence specificity. The HMD-D can also bind to double-stranded RNA. It appears that this feature of non-specific binding to HMG-D plays a role in the development of HIV-1 in the host cell. It may be that the stem–loop RNA structures in the β3/β4 region are an evolutionary design for such non-specific binding.
Back-translation
The difference between the peaks of the RNA structure conservation derived from the real back-translation and the random back-translation is very dramatic. In fact, in the case of the random back-translation, the peak is not actually seen. This indicates a high likelihood of the presence of the RNA secondary structure in the β3/β4 region. Moreover, the peak of the RNA secondary structure derived from the real back-translated DNA is even higher than that obtained from the codon usage-related back-translated sequences. Since we suspect that the codon usage back-translation is biased and does not reflect the real variability of the nef gene, this outcome is more meaningful. On the other hand, the pattern of RNA secondary structure conservation of the codon usage back-translated RNA fragments is quite similar to the pattern of the natural database. This implies that both the amino acid sequence and the codon usage have an important influence on the conservation pattern of RNA secondary structures that share the same genomic sequence in HIV-1.
Third codon position conservation in the nef gene
It is well known that the third position in a codon is often free for synonymous substitution. Therefore, over-conservation in this position along any sequence can indicate the existence of a message other than an amino acid code, at this particular location. The high conservation of the third positions in β3/β4 regions indicates such hidden messages in this region. Higher normalized information content in the third position revealed that these third positions are over-conserved. A strikingly high conservation was indicated in Phe161, Leu167, Val168 and Pro169 (the numbering corresponds to gapped multiple alignment of nef). The third codon positions of these amino acids are remarkably conserved even though the protein conservation leaves the third position of leucine, valine and proline completely variable. Moreover, the plot of normalized conservation of the third codon position indicates a maximum exactly in the region between 480 and 520 (data not shown).
Putative RNA secondary structures within the nef gene
Straightforward alignment of thermodynamically optimal RNA folds of the entire sequence of nef indicates the existence of RNA structures. A highly conserved RNA secondary structure is located between positions 480 and 520 in exactly the same region, which was indicated by the conservation of the third position. The structure has stable features although the optimal RNA secondary structures for individual sequences possess potentially significant differences. RNA secondary structures in other regions of nef are less conserved than β3/β4.
Two forms of the end loop
The central hairpin loop of the β3/β4 region appears in two sizes: a majority of seven bases and a minority of five bases. The reason for this dual appearance is the change of U494 to adenine. This conversion results in the change of F165 to Y. Apparently, there is a pressure to retain only aliphatic amino acids in this particular position, since only adenine and thymine participate in this conversion. On the other hand, the importance of the loop at this site is demonstrated by the hyper-conservation of A501 of the third position in the codon for Leu167 that base-pairs with U493. The difference between the sizes of the two hairpin loops probably reflects a difference between respective counterparts (probably proteins) in the hairpin recognition.
Y to F conversion
Tyrosine to phenylalanine substitutions are well known in retroviruses. Some of them have a known phenotypic effect. Such substitution in reverse transcriptase (RT) is more AZT resistant (55,56). Y115F mutation of RT is critical for enzyme activity (57). Y183→F183 mutation in the YXDD motif of RT results in loss of polymerase activity (58). The Y712→F712 mutation, located in the dominant endocytosis motif of HIV-1 gp41, increased infectivity of the HIV-1 MA30LE virions 11.1-fold (59). The Phe165 to Tyr165 mutation in this region of the nef gene is the only observed mutation that probably is of adaptive evolutionary importance to the Nef protein. The biological significance of the site is also supported by hyper-conservation of Leu167. The adenine in the third position in the codon for Leu167 is always base-paired with the conserved U493, the first nucleotide of the codon for F165 or Y165. This base is fully conserved since it plays two different roles: one as a part of the amino acid code and the other as a loop-keeper in the respective RNA stem–loop structure.
Indication of overlapping messages
The question as to whether highly conserved genetic codes tend to overlap, or whether this overlapping of genetic codes creates highly conserved sequences, has yet to be answered. It seems that highly conserved genetic codes tend to be overlapped. The RNA secondary structure detected in the nef gene sequence is located in the β3/β4 regions of very conserved amino acid sequences. A possible explanation for this phenomenon is that the high level of conservation of one biological signal dictates the conservation of another pattern. Another possibility is that mutual conservation is advantageous from the point of view of minimizing the targets for damage. That is, if the region is important and each mutation is critical, then the appearance of another important code in this region, superimposed on the first, is of evolutionary preference, since it reduces the number of vulnerable sites in this sequence. Thus, one would except to find more hidden codes within the sequences of highly conserved genes. Two separate equally vulnerable sites would have a larger overall target size and a lower rate of survival.
Acknowledgments
ACKNOWLEDGEMENTS
We thank the members of the Genome Diversity Center for discussions and critical comments on the paper, Drs Irit Or, Marilyn Safran, Shifra Ben Dor and Vered Halifa-Caspi from the Bioinformatics and Biological Services at the Weizmann Institute of Science for their helpful support, Ms Na’ava Rubinstein and Ms Robin Permut for the editing.
REFERENCES
- 1.Trifonov E.N. (1989) The multiple codes of nucleotide sequences. Bull. Math. Biol., 51, 417–432. [DOI] [PubMed] [Google Scholar]
- 2.Trifonov E.N. (1990) Making sense of the human genome. In Sarma,R.H. and Sarma,M.H. (eds), Structure and Methods. Adenine Press, Albany, NY, Vol. 1, pp. 69–77. [Google Scholar]
- 3.Trifonov E.N. (1996) Interfering contexts of regulatory sequence elements. CABIOS, 12, 423–429. [DOI] [PubMed] [Google Scholar]
- 4.Dayton E., Powell,D. and Dayton,A. (1989) Functional analysis of CAR, the target sequence for the Rev protein of HIV-1. Science, 246, 1625–1629. [DOI] [PubMed] [Google Scholar]
- 5.Malim M., Hauber,J., Le,S.-Y., Maizel,J. and Cullen,B. (1989) The HIV-1 rev trans-activator acts through a structured target sequence to activate nuclear export of unspliced viral mRNA. Nature, 338, 254–257. [DOI] [PubMed] [Google Scholar]
- 6.Konings D.A.M. (1992) Coexistence of multiple codes in messenger RNA molecules. Comp. Chem., 16, 153–163. [Google Scholar]
- 7.Geyer M., Munte,C.E., Schorr,J., Kellner,R. and Kalbitzer,H.R. (1999) Structure of the anchor-domain of myristoylated and non-myristoylated HIV-1 Nef protein. J. Mol. Biol., 289, 123–138. [DOI] [PubMed] [Google Scholar]
- 8.Lama J., Mangasarian,A. and Trono,D. (1999) Cell-surface expression of CD4 reduces HIV-1 infectivity by blocking Env incorporation in a Nef- and Vpu inhibitable manner. Curr. Biol., 9, 622–631. [DOI] [PubMed] [Google Scholar]
- 9.Ross T.M., Oran,E.A. and Cullen,B.R. (1999) Inhibition of HIV-1 progeny virion release by cell-surface CD4 is relieved by expression of the viral Nef protein. Curr. Biol., 9, 613–621. [DOI] [PubMed] [Google Scholar]
- 10.Geyer M., Fackler,O.T. and Peterlin,B.M. (2001) Structure–function relationships in HIV-1 Nef. EMBO Rep., 2, 580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Echarri A., Gonzalez,M.E. and Carrasco,L. (1996) Human immunodeficiency virus (HIV) Nef is an RNA binding protein in cell-free systems. J. Mol. Biol., 262, 640–651. [DOI] [PubMed] [Google Scholar]
- 12.Geyer M. and Peterlin,B.M. (2001) Domain assembly, surface accessibility and sequence conservation in full length HIV-1 Nef. FEBS Lett., 496, 91–95. [DOI] [PubMed] [Google Scholar]
- 13.Ahmad N. and Venkatesan,S. (1988) Nef protein of HIV-1 is a transcriptional repressor of HIV-1 LTR. Science, 241, 1481–1485. [DOI] [PubMed] [Google Scholar]
- 14.Kaminchik J., Bashan,N., Pinchasi,D., Amit,B., Sarver,N., Johnston,M.I., Fischer,M., Yavin,Z., Gorecki,M. and Panet,A. (1990) Expression and biochemical characterization of human immunodeficiency virus type 1 nef gene product. J. Virol., 64, 3447–3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Littman D.R. (1994) Immunodeficiency viruses. Not enough sans Nef. Curr. Biol., 4, 618–620. [DOI] [PubMed] [Google Scholar]
- 16.Grate L. (1998) Potential SECIS elements in HIV-1 strain HXB2. J. AIDS Hum. Retrovirol., 17, 398–403. [DOI] [PubMed] [Google Scholar]
- 17.Feng S. and Holland,E. (1988) HIV-1 tat trans-activation requires the loop sequence within TAR. Nature, 334, 165–167. [DOI] [PubMed] [Google Scholar]
- 18.Le S.-Y., Chen,J. and Maizel,J. (1991) Detection of unusual RNA folding regions in HIV and SIV sequences. CABIOS, 7, 51–55. [DOI] [PubMed] [Google Scholar]
- 19.Le S.-Y., Malim,M., Cullen,B. and Maizel,J. (1990) A highly conserved RNA folding region coincident with the Rev response element of primate immunodeficiency viruses. Nucleic Acids Res., 18, 1613–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peleg O., Brunak,S., Trifonov,E.N., Nevo,E. and Bolshoy,A. (2002) RNA secondary structure and sequence conservation in C1 region of human immunodeficiency virus type 1 env gene. AIDS Res. Hum. Retroviruses, 18, 867–878. [DOI] [PubMed] [Google Scholar]
- 21.Huynen M., Perelson,A., Vieira,W. and Stadler,P. (1996) Base pairing probabilities in a complete HIV-1 RNA. J. Comput. Biol., 3, 253–274. [DOI] [PubMed] [Google Scholar]
- 22.Fontana W., Konings,D.A.M., Stadler,P.F. and Schuster,P. (1993) Statistics of RNA secondary structures. Biopolymers, 33, 1389–1404. [DOI] [PubMed] [Google Scholar]
- 23.Schuster P. (1993) RNA based evolutionary optimization. Orig. Life Evol. Biosphere, 23, 373–391. [DOI] [PubMed] [Google Scholar]
- 24.Schuster P. (1996) The role of neutral mutations in the evolution of RNA molecules. In Suhai,S. (ed.), Theoretical and Computational Methods in Genome Research. Springer-Verlag, Heidelberg, pp. 287–304. [Google Scholar]
- 25.Eddy S.R. and Durbin,R. (1994) RNA sequence analysis using covariance models. Nucleic Acids Res., 22, 2079–2088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Trifonov E.N. and Bolshoi,G. (1983) Open and closed ribosomal RNA, the only two universal structures encoded in the nucleotide sequences. J. Mol. Biol., 169, 1–13. [DOI] [PubMed] [Google Scholar]
- 27.Browner M.F. and Lawrence,C.B. (1985) Comparative sequence analysis as a tool for studying the secondary structure of mRNAs. Nucleic Acids Res., 13, 8645–8660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jaeger J., Turner,D. and Zuker,M. (1990) Predicting optimal and suboptimal secondary structure for RNA. Methods Enzymol., 183, 281–306. [DOI] [PubMed] [Google Scholar]
- 29.Chan L., Zuker,M. and Jacobson,A. (1991) A computer method for finding common base paired helices in aligned sequences: application to the analysis of random sequences. Nucleic Acids Res., 19, 353–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hofacker I., Fontana,W., Stadler,P., Bonherffer,L., Tacker,M. and Schuster,P. (1994) Fast folding and comparison of RNA secondary structures. Monatsh. Chem., 125, 167–188. [Google Scholar]
- 31.Hofacker I.L., Fekete,M., Flamm,C., Huynen,M.A., Rauscher,S., Stolorz,P. and Stadler,P.F. (1998) Automatic detection of conserved RNA structure elements in complete RNA virus genomes. Nucleic Acids Res., 26, 3825–3836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Juan V. and Wilson,C. (1999) RNA secondary structure prediction based on free energy and phylogenetic analysis. J. Mol. Biol., 289, 935–947. [DOI] [PubMed] [Google Scholar]
- 33.Hofacker I.L., Fekete,M. and Stadler,P.F. (2002) Secondary structure prediction for aligned RNA sequences. J. Mol. Biol., 319, 1059–1066. [DOI] [PubMed] [Google Scholar]
- 34.Lück R., Steger,G. and Riesner,D. (1996) Thermodynamic prediction of conserved secondary structure: application to the RRE element of HIV, the tRNA-like element of CMV and the mRNA of prion protein. J. Mol. Biol., 258, 813–836. [DOI] [PubMed] [Google Scholar]
- 35.Grillo G., Attimonelli,M., Liuni,S. and Pesole,G. (1996) CLEANUP: a fast computer program for removing redundancies from nucleotide sequence databases. CABIOS, 12, 1–8. [DOI] [PubMed] [Google Scholar]
- 36.Kullback S. and Leibler,R.A. (1951) On information and sufficiency. Ann. Math. Stat., 22, 79–86. [Google Scholar]
- 37.Cover T. and Thomas,J.A. (1991) Elements of Information Theory. John Wiley and Sons, Inc., New York. [Google Scholar]
- 38.Schneider T.D., Stormo,G.D., Gold,L. and Ehrenfeucht,A. (1986) Information content of binding sites on nucleotide sequences. J. Mol. Biol., 188, 415–431. [DOI] [PubMed] [Google Scholar]
- 39.Schneider T.D. and Stephens,R.M. (1990) Sequence logos: a new way to display consensus sequences. Nucleic Acids Res., 18, 6097–6100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gorodkin J., Heyer,L.J., Brunak,S. and Stormo,G.D. (1997) Displaying the information contents of structural RNA alignments: the structure logos. CABIOS, 13, 583–586. [DOI] [PubMed] [Google Scholar]
- 41.Higgins D., Thompson,J. and Gibson,T. (1996) Using CLUSTAL for multiple sequence alignments. Methods Enzymol., 266, 383–402. [DOI] [PubMed] [Google Scholar]
- 42.Thompson J., Higgins,D. and Gibson,T. (1994) CLUSTALW: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res., 22, 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zuker M. and Stiegler,P. (1981) Optimal computer folding of large RNA sequences using thermodynamic and auxiliary information. Nucleic Acids Res., 9, 133–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zuker M. (1989) On finding all suboptimal foldings of an RNA molecule. Science, 244, 48–52. [DOI] [PubMed] [Google Scholar]
- 45.Zuker M. (1994) Prediction of RNA secondary structure by energy minimization. Methods Mol. Biol., 25, 267–294. [DOI] [PubMed] [Google Scholar]
- 46.Zuker M. (2000) Calculating nucleic acid secondary structure. Curr. Opin. Struct. Biol., 10, 303–310. [DOI] [PubMed] [Google Scholar]
- 47.Mathews D.H., Sabina,J., Zuker,M. and Turner,D.H. (1999) Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure. J. Mol. Biol., 288, 911–940. [DOI] [PubMed] [Google Scholar]
- 48.Hertz G.Z. and Stormo,G.D. (1994) Identification of consensus patterns in unaligned DNA and protein sequences: a large deviation statistical basis for penalizing gaps. In Lim,H.A. and Cantor,C.M. (eds), Proceedings of the Third International Conference on Bioinformatics and Genome Research. World Scientific Publishing Co. Ltd, Singapore, pp. 201–216. [Google Scholar]
- 49.Woese C.R., Gutell,R., Gupta,R. and Noller,H.F. (1983) Detailed analysis of the higher-order structure of 16S-like ribosomal ribonucleic acids. Microbiol. Rev., 47, 621–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gutell R.R., Weiser,B., Woese,C.R. and Noller,H.F. (1985) Comparative anatomy of 16-S-like ribosomal RNA. Prog. Nucleic Acid Res. Mol. Biol., 32, 155–216. [DOI] [PubMed] [Google Scholar]
- 51.Gutell R.R., Noller,H.F. and Woese,C.R. (1986) Higher order structure in ribosomal RNA. EMBO J., 5, 1111–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tuplin A., Wood,J., Evans,D.J., Patel,A.H. and Simmonds,P. (2002) Thermodynamic and phylogenetic prediction of RNA secondary structures in the coding region of hepatitis C virus. RNA, 8, 824–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Simmonds P. and Smith,D.B. (1999) Structural constraints on RNA virus evolution. J. Virol., 73, 5787–5794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Arimondo P.B., Gelus,N., Hamy,F., Payet,D., Travers,A. and Bailly,C. (2000) The chromosomal protein bind to the TAR and RBE RNA of HIV-1. FEBS Lett., 485, 47–52. [DOI] [PubMed] [Google Scholar]
- 55.Lacey S.F. and Larder,B.A. (1994) Mutagenic study of codons 74 and 215 of the human immunodeficiency virus type 1 reverse transcriptase, which are significant in nucleoside analog resistance. J. Virol., 68, 3421–3424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rey D., Hughes,M., Pi,J.T., Winters,M., Merigan,T.C. and Katzenstein,D.A. (1998) HIV-1 reverse transcriptase codon 215 mutation in plasma RNA: immunologic and virologic responses to zidovudine. The AIDS Clinical Trials Group Study 175 Virology Team. J. AIDS Hum. Retrovirol., 17, 203–208. [DOI] [PubMed] [Google Scholar]
- 57.Tisdale M., Alnadaf,T. and Cousens,D. (1997) Combination of mutations in human immunodeficiency virus type 1 reverse transcriptase required for resistance to the carbocyclic nucleoside 1592U8. Antimicrob. Agents Chemother., 41, 1094–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Harris D., Yadav,P.N. and Pandey,V.N. (1998) Loss of polymerase activity due to Tyr to Phe substitution in the YMDD motif of human immunodeficiency virus type-1 reverse transcriptase is compensated by Met to Val substitution within the same motif. Biochemistry, 37, 9630–9640. [DOI] [PubMed] [Google Scholar]
- 59.West J.T., Weldon,S.K., Wyss,S., Lin,X., Yu,Q., Thali,M. and Hunter,E. (2002) Mutation of the dominant endocytosis motif in human immunodeficiency virus type 1 gp41 can complement matrix mutations without increasing Env incorporation. J. Virol., 76, 3338–3349. [DOI] [PMC free article] [PubMed] [Google Scholar]