


Abstract
Triple helix-forming oligonucleotides covalently linked to topoisomerase I inhibitors, in particular the antitumor agent camptothecin, trigger topoisomerase I-mediated DNA cleavage selectively in the proximity of the binding site of the oligonucleotide vector. In the present study, we have performed a systematic analysis of the DNA cleavage efficiency as a function of the positioning of the camptothecin derivative, either on the 3′ or the 5′ side of the triplex, and the location of the cleavage site. A previously identified cleavage site was inserted at different positions within two triplex site-containing 59 bp duplexes. Sequence-specific DNA cleavage by topoisomerase I occurs only with triplex conjugates bearing the inhibitor at the 3′-end of the oligonucleotide and on the oligopyrimidine strand of the duplex. The lack of targeted cleavage on the 5′ side is attributed to the structural differences of the 3′ and 5′ duplex–triplex DNA junctions. The changes induced in the double helix by the triple-helical structure interfere with the action of the enzyme according to a preferred spatial organization. Camptothecin conjugates of oligonucleotides provide efficient tools to probe the organization of the topoisomerase I–DNA complex and will be useful to understand the functioning of topoisomerase I in living cells.
INTRODUCTION
Human topoisomerase I (Topo I) is a ubiquitous nuclear enzyme involved in the control of DNA topology and thus in DNA metabolism and chromosome segregation (1). The reaction between double-stranded DNA and Topo I produces a covalent 3′-phosphorotyrosyl adduct, usually referred to as the cleavage complex. Under physiological conditions, the covalent intermediate is barely detectable, because a fast religation step occurs after relaxation of the DNA constraints. A number of drugs, such as the antitumor alkaloid camptothecin (CPT), can convert Topo I into a cell poison by blocking the religation step, thereby enhancing the formation of persistent DNA breaks responsible for cell death (2,3).
The crystal structure of a short DNA fragment complexed with Topo I as well as molecular modeling and binding studies using various DNA sequences and Topo I mutants all indicate that, in the covalent complex, the enzyme encircles DNA and makes tight contacts with the sequence upstream of the cleavage site (4–6). Very recently, the three-dimensional structure of the ternary complex formed by DNA, a truncated Topo I and the CPT-like inhibitor topotecan has been solved by X-ray diffraction (7). Interestingly, topotecan specifically binds to the DNA–Topo I interface by intercalation in the DNA double helix at the cleavage site between the upstream (–1) and downstream (+1) base pairs. This new molecular model, whereby one particular conformation of the ternary complex is captured in the crystal packing, greatly helps to better understand the positioning of the CPT inhibitor within the covalent complex, but it does not provide information on the dynamics of the system. In fact, the ternary complex can probably adopt several conformations, all playing a role in the inhibition of the nucleophilic attack on the tyrosylphophodiester bond by the cleaved 5′-OH DNA strand during the DNA religation step of the catalytic cycle (4,7–11). Over the past 5 years, we and others (10,12–14) have developed an original strategy to direct the activity of the DNA–Topo I–CPT ternary complex via the design of oligonucleotide–drug conjugates capable of recruiting the enzyme at specific sites within the target DNA sequence. Triplex-forming oligonucleotides (TFO) conjugated to CPT (or other Topo I poisons such as rebeccamycin derivatives) direct DNA cleavage by Topo I at specific sites. In the present study, we have used this strategy to probe the geometry of the cleavage complex, by rational design of novel TFO–drug conjugates and different target DNA sequences. A systematic analysis of the position of the potential Topo I cleavage site with respect to the location of the CPT inhibitor at the 3′ or 5′ extremity of the TFO enabled us to delineate some of the molecular constraints which govern the geometry of the ternary complex and orient the DNA cleavage. This dual approach, whereby the ligand or the bioreceptor was tuned, allowed us to dissect the various molecular combinations susceptible to recruit Topo I cleavage at specific sites. It revealed that only a subset of TFO–CPT/DNA combinations lead to DNA cleavage. The panoply of tools presented here with their corresponding DNA molecular constructions allowed us to probe the geometry of the DNA–Topo I–CPT ternary complex.
MATERIALS AND METHODS
Drugs
All drugs and conjugates were dissolved in dimethylsulfoxide and then diluted further with water. The final dimethylsulfoxide concentration never exceeded 0.3% (w/v) in all assays. The CPT inhibitor was attached to the 3′- or 5′-end of the TFO. 7-Ethyl-10-hydroxycamptothecin acetic acid (2) was derived, as shown in Figure 1, from 7-ethyl-10-hydroxycamptothecin (SN-38), synthesized according to the procedures described in Sawada et al. (15) without major modifications.
Figure 1.

Synthetic route used for the preparation of 7-ethyl-10-hydroxycamptothecin acetic acid (2).
Synthesis of 7-ethyl-10-hydroxycamptothecin acetic acid (2). Chloroacetic acid (57.2 mg, 0.60 mmol) was added to the CPT derivative SN-38 (7-ethyl-10-hydroxycamptothecin, 100.7 mg, 0.256 mmol), followed by addition of an aqueous solution of 6.4 M NaOH (0.28 ml, 1.80 mmol). The reaction mixture was concentrated to dryness under vacuum and then irradiated in a domestic microwave oven (900 W) three times for 4 min each. The solid was dissolved in a minimum volume of water (21.5 ml) and, after acidification to pH 3 by a solution of 1 N hydrochloric acid, it was stirred at room temperature for 1 h. After filtration through Celite, the residue was purified by flash chromatography (CH2Cl2/MeOH 95/5, then 9/1, then 85/15, then CH3CN/water 95/5) to afford 7-ethyl-10-hydroxycamptothecin acetic acid (2) as a pale yellow powder: yield 99.5 mg (86.5%); silica gel TLC Rf = 0.33 (85/15 CH3CN/H2O); melting point 255°C (decomposition); [α]D20: +15 (c 0.5, MeOH); IR (KBr, cm–1) ν: 3423 (COOH); 1747 (CO); 1658 (CO amide); 1596, 1510 (C=C); 1385 (CH3); 1330 (C-O lactone); 1H NMR (300 MHz, CD3OD) δ: 1.01 (t, 3H, J = 7.3 Hz, CH3-18); 1.39 (t, 3H, J = 7.5 Hz, 7-CH2CH3); 1.95 (q, 2H, J = 7.3 Hz, CH2-19); 3.18 (q, 2H, J = 7.5 Hz, 7-CH2CH3); 4.59 (s, 2H, OCH2CO2H); 5.22 (s, 2H, CH2N-5); 5.37 and 5.57 (AB, 2H, JAB = 16.2 Hz, CH2O-17); 7.37 (d, 1H, Jmeta = 2.5 Hz, H-9); 7.55 (dd, 1H, Jortho = 9 Hz, Jmeta = 2.5 Hz, H-11); 7.59 (s, 1H, H-14); 8.02 (d, 1H, Jortho = 9 Hz, H-12); 13C NMR (75 MHz, CD3OD) δ: 8.2 (CH3-18); 13.8 (7-CH2CH3); 23.9 (7-CH2CH3); 32.1 (CH2-19); 50.8 (CH2N-5); 66.7 (CH2O-17); 68.7 (OCH2CO2H); 74.3 (C20); 99.0 (C14-H); 104.1 (C9-H); 119.6 (C16); 124.5 (C11-H); 129.1–129.6 (C6,C8); 131.8 (C12-H); 146.0–146.2–148.1–150.4–152.8 (C2,C3,C7,C13,C15); 159.2–159.5 (C10, CO16a); 175.0 (CO21); MS (C.I./NH3): m/z: 451 [M + H]+, 407 [M – CO2 + H]+; MS (ES–) m/z: 449 [M – H]–; calculated C24H22N2O7, 450.
Oligonucleotides and DNA fragment
Oligonucleotides were purchased from Eurogentec (Belgium) and purified using Micro Bio Spin® 6 Chromatography columns (Bio-Rad, USA). Concentrations were determined spectrophotometrically at 25°C using molar extinction coefficients at 260 nm calculated from a nearest neighbor model (16).
Synthesis of CPT conjugates. CPT derivative 2 bearing an acid function was conjugated to the terminal amino group of different linker arms at the 3′- or 5′-end of the oligonucleotide. The different synthetic procedures have been previously described (10,17). The linker arms were attached by reaction of the corresponding diamine to the 3′ or 5′ phosphorylated oligonucleotide activated by treatment with 4-dimethylaminopyridine, dipyridyl disulfide and triphenylphosphine as reported (18). The conjugates were purified by reverse phase HPLC using a linear acetonitrile/buffer gradient [0–80% CH3CN in 0.2 M aqueous (NH4)OAc] and characterized by UV spectroscopy, denaturing gel electrophoresis and mass spectrometry (Q-star I). TFO-L3-CPT MS (ES–) m/z: 5629 [M+H]+ (calculated 5632); CPT-L3-TFO MS (ES–) m/z: 5630 [M+H]+ (calculated 5632); CPT-L6-TFO MS (ES–) m/z: 5672 [M+H]+ (calculated 5674); CPT-L10-TFO MS (ES–) m/z: 5730 [M+H]+ (calculated 5730); CPT-LO8-TFO MS (ES–) m/z: 5708 [M+H]+ (calculated 5706); CPT-LO13-TFO MS (ES–) m/z: 5788 [M+H]+ (calculated 5778); MGB-CPT 1436 [M+H]+ (calculated 1437).
Preparation of the DNA targets. The plasmid pBSK(+/–) was bought from Promega (USA) and the 77 bp target duplex was inserted between the BamHI and EcoRI sites. The detailed procedures for isolation, purification and labeling of the 324mer DNA fragment, radiolabeled at the 3′-end on the oligopyrimidine-containing strand, have been described previously (13). The two radiolabeled 59 bp target duplexes were obtained by labeling one strand at the 3′-end by terminal transferase (Ozyme, UK) and [α-32P]ddATP (Amersham, USA), followed by incubation with the complementary non-labeled strand for 5 min at 90°C and hybridization upon slow cooling to room temperature. The radiolabeled fragments were then purified by gel electrophoresis. The nomenclature of the strands is as follows: R stands for the oligopurine-containing strand and Y for the oligopyrimidine-containing strand.
Topoisomerase I cleavage assays
The radiolabeled target duplexes (50 nM) were incubated for 1 h at 30°C, in 50 mM Tris–HCl, pH 7.5, containing 60 mM KCl, 10 mM MgCl2, 0.5 mM DTT, 0.1 mM EDTA and 30 µg/µl BSA, in the presence of either the ligand or/and the drugs at the indicated concentrations (total reaction volume 10 µl). Ten units of enzyme (Invitrogen) were added to the duplex, preincubated as described above, followed by incubation for 20 min at 30°C. DNA–Topo I cleavage complexes were dissociated by addition of SDS (final concentration 0.25%). After ethanol precipitation, all samples were resuspended in 6 µl of formamide, heated at 90°C for 4 min and then chilled on ice for 4 min, before being loaded onto a denaturing 8 or 10% polyacrylamide gel (19:1 acrylamide: bisacrylamide), for the long and short targets, respectively, containing 7.5 M urea in 1× TBE buffer (50 mM Tris base, 55 mM boric acid, 1 mM EDTA). Gels were scanned with a Molecular Dynamics 445SI PhosphorImager and a standard normalization relative to total loading was performed for the determination of cleavage levels.
RESULTS
The synthesis of 7-ethyl-10-hydroxycamptothecin acetic acid (2), a new CPT derivative used for the preparation of the TFO–CPT conjugates, attached through position 10 of the drug, was carried out as outlined in Figure 1. The starting material was compound SN-38, the active metabolite of the clinically used drug irinotecan. Compound 2 was obtained in 86.5% overall yield by reaction of the phenol function of SN-38 with chloroacetic acid under basic conditions with microwave irradiation. The procedure was adapted from a published work (19) describing the reaction for various substituted phenols and 1- and 2-naphtols. Helpfully, with this solvent-free procedure there is no need to use a protecting group for chloroacetic acid, so a deprotection step after coupling is no longer required.
CPT was attached to the 3′- or 5′-end of the oligonucleotide through a diaminopropyl linker, following the synthetic procedures detailed in Grimm et al. (18) (TFO-L3-CPT and CPT-L3-TFO in Fig. 2B). The choice of linker size was guided by a model previously built for the DNA/Topo I/TFO conjugate complex, optimizing the length and nature of the linker for efficient cleavage (10,14).
Figure 2.

(A) Sequence of the TFOs and the 77 bp DNA target used in this study. The TFO binds in the major groove parallel to the oligopurine strand of the duplex. The orientation of the triple helix is defined by the orientation of the oligopurine strand of the duplex. The triplex site is in bold. The 77 bp duplex target sequence was inserted between the BamHI and EcoRI sites of plasmid pbSK(+/–). M, 5-methyl-2′-deoxycytidine; P, 5-propynyl-2′-deoxyuridine. (B) Structure of the camptothecin conjugates used in this study. 7-Ethyl-10-hydroxycamptothecin acetic acid was attached through N-succinimide activation to the terminal amino group of the linker attached to the extremity of the TFO or MGB as described previously (18). L3, L6 and L10, propyldiamine, hexyldiamine and decyldiamine; LO8, 1,8-diaminodioxaoctane; LO13, 4,7,10-trioxa-1,13-tridecandiamine.
Targeting topoisomerase I-mediated DNA cleavage: 3′ side versus 5′ side
DNA cleavage by Topo I is induced at a specific site upon covalent linkage of a CPT poison to a TFO (10,12). In previous studies, cleavage was always targeted to the 3′ side of the triplex site (conventionally, the orientation of the triple helix is defined by reference to the 5′→3′ polarity of the oligopurine strand of the duplex). Here we investigated whether targeting was also possible on the 5′ side of the triplex. We used a previously described target duplex (13) (Fig. 2A), containing a triplex site (in bold) and several CPT-sensitive sites (called sites a, b and c), and a 16 nt TFO.
The cleavage properties of the conjugates were studied with a 324 bp target duplex, radiolabeled on the oligopyrimidine strand (Fig. 3). As expected from previous work (10,13), conjugation of the CPT derivative 2 to the 3′-end of the TFO (conjugate TFO-L3-CPT) specifically directed Topo I-mediated cleavage only to site b on the 3′ side of the triple helix (Fig. 3, lane 4). Cleavage was abolished at the other sites sensitive to CPT (sites a and c, lanes 3 and 10). In sharp contrast, no cleavage was observed when the CPT ligand was attached to the 5′ end of the TFO (lane 5). At this stage, two reasons could be invoked: either the orientation of the CPT residue was not suitable for promoting DNA cleavage or site a, located 8 bp from the 5′ end of the triplex, was too far from the triple helix and could not be reached by the inhibitor. To address this issue, we increased the length and flexibility of the linker arm. Compound 2 was attached to the 5′ end of the TFO through 1,6-diaminohexane (CPT-L6-TFO), 1,10 diaminodecane (CPT-L10-TFO), 1,8-diaminodioxaoctane (CPT-LO8-TFO) or 4,7,10-trioxa,1,13-tridecandiamine (CPT-LO13-TFO) (Fig. 2B). Analysis of the cleavage products of Topo I upon binding of these conjugates to the target clearly showed that, in all cases, no cleavage was induced on the 5′ side of the triple helix (Fig. 3, lanes 6–9). The TFO had no effect alone (lane 11) and only partially protected site b from cleavage in the presence of the free CPT derivative 2 (lane 12).
Figure 3.
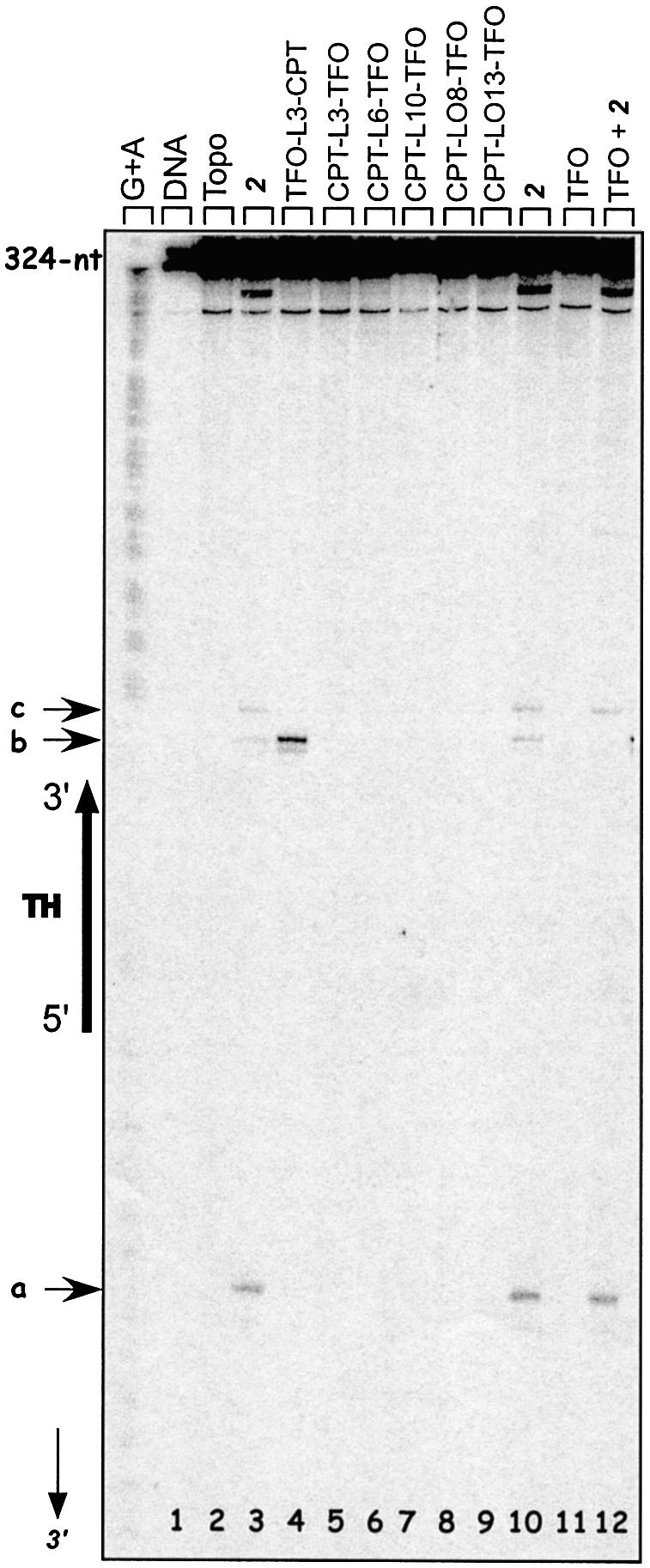
Sequence analysis of the Topo I-mediated cleavage products of the 324 bp target duplex (50 nM) 3′-end radiolabeled on the oligopyrimidine-containing strand. Adenine/guanine-specific Maxam–Gilbert chemical cleavage reactions were used as markers. The positions of the cleavage sites are indicated (sites a–c), as well as the triple helix region and the nucleotide positions along the radiolabeled oligopyrimidine strand (arrow). Lane 1, duplex; lane 2, duplex incubated with Topo I; lanes 3–12, duplex incubated with Topo I in the presence of 5 µM 2 (lane 3 and 10), 0.5 µM TFO-L3-CPT (lane 4), 0.5 µM CPT-L3-TFO (lane 5), 0.5 µM CPT-L6-TFO (lane 6), 0.5 µM CPT-L10-TFO (lane 7), 0.5 µM CPT-LO8-TFO (lane 8), 0.5 µM CPT-LO13-TFO (lane 9), 5 µM TFO (lane 11) or 5 µM TFO + 5 µM 2 (lane 12).
The lack of cleavage on the 5′ side could not be related to a lower stability of the complexes formed by these conjugates. Attachment of the CPT derivative to the oligonucleotide extremity increased triplex formation at least 4-fold and all conjugates were equally potent in terms of triplex stability (Table 1). Cleavage was also analyzed on the other strand of the duplex. Again, no cleavage was observed in the presence of the TFO conjugates either 3′ or 5′ substituted (data not shown); however, no sensitive CPT site was present near the triplex site on the oligopurine-containing strand.
Table 1. Stability of the triple helix.
| Conjugate | Kd (µM)a |
|---|---|
| TFO | 1.2 ± 0.5 |
| TFO-L3-CPT | 0.25 ± 0.05 |
| CPT-L3-TFO | 0.24 ± 0.06 |
| CPT-L10-TFO | 0.31 ± 0.06 |
| CPT-L010-TFO | 0.27 ± 0.06 |
| CPT-L014-TFO | 0.22 ± 0.06 |
aKd values were measured as described in Arimondo et al. (33), after incubation for 24 h at 37°C in 50 mM Tris–HCl, pH 7.5, 60 mM KCl, 10 mM MgCl2, 10% sucrose and 0.5 µg/µl tRNA.
Briefly, the results indicate that Topo I can be controlled to induce site-specific DNA cleavage at the 3′ end of the triplex but not at the opposite 5′ end. In these experiments, the DNA target was fixed and the location of the CPT inhibitor was changed depending on its attachment to one or the other extremity of the TFO. However, no CPT-inducible site was present on the target close to the 5′-end of the triplex, other than site a, located at 7 bp on the oligopyrimidine-containing strand. Next, we designed a reverse system, in which the ligand was fixed, whereas the DNA target was manipulated to place the potential cleavage site at different specific locations with respect to the DNA ligand.
Moving the position of the cleavage site
As a potential cleavage site, we chose site b, detected on the 324 bp DNA fragment, and its adjacent base pairs, 6 bp in total (Fig. 2A). We designed two 59 bp duplexes containing the triplex site in the middle and the above-described cleavage site b in different positions (in red and underlined), as illustrated in Figure 4. Short duplexes were chosen in order to facilitate the detection of weak cleavage sites. To design the systems, we referred to our previous model, based on X-ray structures, indicating that Topo I binds asymmetrically to DNA and that there is a potential ‘cavity’ towards the 3′ side of the cleavage site where the triple helical structure fits (14). Furthermore, we considered the literature data indicating that Topo I cleaves only one strand and that the DNA region located 5′ (upstream) to the covalently linked nucleotide in the cleavage complex is essential for binding, while the 3′ region (downstream) is covered by the enzyme, but it is not essential (5) and can thus allow TFO binding. Based on these data, the two configurations designated TRY and TRYi were built (Fig. 4). In the TRY system, site b was situated on the 5′ side of the triplex site on the oligopurine strand of the duplex, called site bR5′, and also on the oligopyrimidine strand to the 3′ side of the triplex, site bY3′ (Fig. 4A). Of note, the latter is identical to the original site b on the long DNA target. This configuration should be the most favorable for Topo I-mediated cleavage. System TRYi was designed in the opposite configuration, i.e. site b was now located on the 5′ side of the triplex on the oligopyrimidine strand of the duplex, called site bY5′, and on the 3′ side of the oligopurine strand, site bR3′ (Fig. 4B). A priori we expected no cleavage in this configuration because the presence of the triplex should hinder Topo I binding.
Figure 4.

Model 59 bp duplexes used in this study. (A) System TRY and (B) System TRYi. Sequence of the duplexes. The triple helix site is in bold and the same cleavage site b is indicated in red and underlined. The nomenclature of site b is as follows. The strand of the duplex, R or Y, is indicated as superscript followed by the side of the triple helix, 5′ or 3′, on which the site is located. For example, bR5′ stands for a site b positioned on the R strand of the duplex and on the 5′ side of the triplex end. Numbers indicate the other cleavage sites.
Figure 5 compares Topo I-mediated cleavage on each strand of the two target systems, TRY and TRYi, in the presence of CPT derivative 2 (lane 3), 5′ conjugate CPT-L3-TFO (lane 4), 3′ conjugate TFO-L3-CPT, (lane 5) or TFO alone (lane 6) and in the presence of free derivative 2 (lane 7). CPT-stimulated cleavage occurred at the predicted sites bR5′, bY3′, bR3′ and bY5′ (lane 3), as well as at other sites, which are numbered. The positions of all sites are mentioned in Figure 4. Interestingly, on all targets the presence of the triple helical structure, formed by either the oligonucleotide alone (lane 6) or in the presence of CPT (lane 7), appeared to stimulate Topo I-mediated cleavage at precise sites (bY3′, 3 and 2 on TY, 4′ on TYi, bR5′ on TR and 1′ on TRi), the efficacy depending on the DNA sequence. As expected, on the oligopyrimidine strand of duplex TRY (far left) the 3′ conjugate TFO-L3-CPT (lane 5) induced potent and highly specific cleavage at site bY3′, as observed on the 324 bp target duplex (Fig. 3). Unexpectedly, the same 3′ conjugate stimulated cleavage specifically at the weak site 4′ on the oligopyrimidine strand of the reverse system TRYi, situated at 4 bp from the 3′ triplex end, equivalent to site bY3′ on TY. Only the 3′ TFO-L3-CPT conjugate induced specific cleavage on the 3′ side of the triple helix on the oligopyrimidine strand (Y) of the duplex. No targeted cleavage was detected with the 5′ conjugate (lane 4). The cleavage observed with all triplexes was rather stimulated by formation of the triple-helical structure, whether the free or conjugated TFO was used, and independently of the site of attachment (5′ or 3′) and of the presence of free CPT. This triplex-induced cleavage was also clearly present on the oligopurine strand of the two duplexes (right) at site bR5′ on TR and 1′ on TRi. We studied the organization of this cleavage in more detail.
Figure 5.

Positioning of Topo I in the presence of the triple-helical structure. Sequence analysis of the Topo I-mediated cleavage products on each 3′-end radiolabeled (50 nM) strand of the duplexes. Adenine/guanine-specific Maxam–Gilbert chemical cleavage reactions were used as markers. The positions of the cleavage sites are indicated on each strand according to the nomenclature defined in Figure 4. The orientation of the triple helix region is indicated, as well as the duplex system analyzed; radiolabeled strands of the duplexes are indicated by asterisks for each system. Lane 1, target duplex; lane 2, duplex incubated with Topo I; lanes 3–7, duplex incubated with Topo I and in the presence of 5 µM 2 (lane 3), 0.5 µM CPT-L3-TFO (lane 4), 0.5 µM TFO-L3-CPT (lane 5), 5 µM TFO (lane 6) or 5 µM TFO + 5 µM 2 (lane 7).
Spatial organization of the cleavage
The geometry of the cleavage induced in the presence of the triple helix seems well defined. This is illustrated in Figure 6, which reports, on a logarithmic scale, the efficacy of cleavage in the presence of the different triple helices [formed by 3′ conjugate TFO-L3-CPT (right hatched bars), 5′ conjugate CPT-L3-TFO (left hatched bars), TFO (white bars) and TFO + CPT (black bars)], compared to CPT alone, for each site on the two duplexes. In this representation, a ratio of intensity of cleavage for the triplex over the intensity for CPT alone higher than 1 indicates that the triplex induced stronger cleavage than the free Topo I poison. Conversely, a ratio less than 1 refers to a higher cleavage intensity with free CPT than with the triple helix. The position of the cleavage sites with respect to the triplex site agreed perfectly with our model: on the 5′ side of the triplex, cleavage was observed only on the oligopurine strand of the duplex, whether these sites were the built-in site b (site bR5′; Fig. 6A, top) or an unpredicted site (such as site 1′ in the top panel of Fig. 6B); whereas on the 3′ side of the triple-helical structure cleavage was observed only on the oligopyrimidine strand of the duplex, at sites bY3′, 3 and 2 on TY (Fig. 6A, bottom) and 4′ and 3′ on TYi (Fig. 6B, bottom).
Figure 6.


Comparison of the cleavage positions and intensities on each target duplex strand. The ratios between the intensity of cleavage in the presence of the triple helices and the intensity in the presence of CPT alone are presented, on a logarithmic scale, for duplexes TRY (A) and TRYi (B) at each cleavage site according to the position along the target. Data were compiled from eight independent experiments. TFO-L3-CPT, right-hatched bars; CPT-L3-TFO, left-hatched bars; TFO, white bars; TFO + CPT, black bars.
In order to confirm that no triplex-directed CPT targeting is possible on the 5′ side of the triple helix, we tested all 5′ conjugates (CPT-L3-TFO, CPT-L6-TFO, CPT-L10-TFO, CPT-LO8-TFO and CPT-LO13-TFO; Fig. 2B), differing by the length and nature of the linker, on the short DNA targets. Even at the optimized site bR5′, no cleavage was specifically induced by the conjugates (data not shown). Only triplex structure-stimulated cleavage was observed according to the spatial arrangement described above.
DISCUSSION
We have performed a systematic analysis of the control of Topo I-mediated DNA cleavage by TFO–CPT conjugates. The aim of the work was to study whether triplex-directed Topo I-mediated DNA cleavage could be induced on the 5′ side of the triple-helical structure, as it was previously demonstrated on the 3′ side (10,12–14). To answer this question, we developed two complementary strategies: the first one consisted of designing a TFO conjugate linked to CPT at its 5′-end, instead of the 3′-end; the second was to use two short DNA targets containing a fixed triple helix site in the middle and a potential cleavage site on either side with respect to the triplex on each strand of a 59 bp duplex (systems TRY and TRYi, described in Fig. 4). The 6 bp sequence of the disposable cleavage site was identified from a study of the cleavage sites on a long 324 bp DNA fragment. This two-front strategy, whereby the ligand or the bioreceptor were modulated, allowed us to dissect the various molecular combinations capable of recruiting Topo I cleavage at specific sites.
Both approaches reveal two major points. First, the presence of the triple-helical structure masks defined Topo I sites, in agreement with previous findings suggesting that the region upstream (5′ side) of the cleavage site is essential for Topo I binding (4–6,20–22). Therefore, the presence of the triple helix in this region inhibited cleavage, while its presence in the region downstream (3′ side) stimulated cleavage. Second, the local conformation and the DNA sequence modulate the extent of stimulated DNA cleavage at the sites which are not masked by the triple helix.
The first point is schematized in Figure 7, summarizing all presented data and illustrating the specific spatial organization of the cleavage complex necessary for DNA cleavage in the presence of the triple-helical structure. Whether the triple helix is formed by the unlinked TFO or by the conjugates, on either the 3′ or the 5′ side, DNA cleavage can only occur: (i) on the oligopyrimidine-containing strand of the duplex on the 3′ side of the triplex, and this site can be efficiently targeted specifically by 3′ TFO–CPT conjugates; (ii) on the oligopurine-containing strand of the duplex on the 5′ side of the duplex, but in this latter case no CPT targeting is possible by TFO conjugates. Accordingly, in the case of the TRYi duplex in the presence of the triple-helical structures, cleavage was not observed at the built-in CPT-sensitive Topo I sites bR3′ and bY5′, because the triple helical structure hindered Topo I, which could only fit upstream of the triple helix and therefore cleave at sites 1′ and 4′. These results further confirm the model where the triple helix structure is positioned in a ‘hole’ present on the bottom side of the enzyme bound to DNA (14). This spatial arrangement is in good agreement with previous findings reporting that single lesions in DNA stimulate cleavage when located downstream of the cleavage site and inhibit topoisomerase binding when located upstream (reviewed in 23). Interestingly, similar cleavage profiles have been obtained with hydrocarbon diol epoxide adducts lying in the minor groove of DNA (24) and, in this study, with the minor groove conjugate MGB–CPT (see Supplementary Material).
Figure 7.

Schematic representation of the interaction between Topo I and DNA in the presence of the triple helices of the two systems studied. (A) TRY; (B) TRYi.
The second point, supported by the fact that Topo I is an enzyme highly sensitive to DNA conformation, is illustrated by several considerations. We did not detect targeted DNA cleavage when the CPT derivative was attached to the 5′-end of the TFO with either strategy used in this study. Neither a lack of stability of the triplex nor an insufficient length of the linker nor the absence of a CPT-sensitive site can be invoked. The conjugates formed stable triplexes as verified by PAGE (Table 1) and DNase I footprinting experiments performed under the same experimental conditions as the Topo I cleavage assays (data not shown). The length and nature of the aminoalkyl linker between the oligonucleotide and the inhibitor has been varied (Fig. 2B) and in all cases no cleavage occurred on the 5′ side (Fig. 3). In our second approach, a CPT-sensitive site was built on the 5′ side of the triple helix on both DNA strands (sites bR5′ and bY5′; Fig. 4). Again, no cleavage was observed with all the 5′ conjugates. At first sight, the absence of DNA cutting on the 5′ side may appear surprising, but a posteriori we realized that this was probably due to the significantly different structural behavior of the triplex junctions for the 5′ and 3′ sides. Several studies have clearly identified different structures for the 5′ duplex–triplex junction as opposed to the opposite 3′ junction (25,26). When CPT is linked to the 3′-end of the TFO, DNA becomes hyper-reactive to Topo I, because the cleavage site, situated at 4 or 8 bp from the triplex end, is easily accessible to the enzyme. The 3′ junction displays the CPT moiety in a favorable conformation for Topo I inhibition. In contrast, the 5′ junction is not apt to accommodate the CPT moiety, which likely cannot be positioned correctly by the linker in the cleavage site. Structural analyses of these junctions by nuclear magnetic resonance spectroscopy illustrate this issue (27,28).
An unexpected result was the enhanced Topo I-mediated DNA cleavage by the triple-helical structure alone, according to a precise spatial arrangement. Several scenarios can be envisaged to account for this stimulation of DNA cleavage in the presence of a triple helix. Formation of the triplex induces specific structural modifications of the DNA target, thereby facilitating binding of Topo I to the target sequence. Furthermore, it is known that triple helix induces structural changes in the double helix even at a distance from the triplex site. In particular, triplex formation generally induces a bend at the duplex junction (26). Topo I is highly sensitive to the DNA conformation and displays a higher affinity for bent DNA as opposed to linear DNA (29). The role of DNA bending in Topo I poisoning is not yet well understood but drugs that induce DNA bending, such as the antitumor agent nogalamycin, generally stimulate Topo I-mediated DNA cleavage (30). Similarly, the presence of a bend in DNA, induced by a poly(dA)·poly(dT) tract in the region 3′ to the cleavage site, stimulates Topo I binding and cleavage (22,31). In fact, a curvature in DNA will change the orientation of the phosphate charges and thus influence Topo I binding to DNA (4). Alternatively, both covalent and non-covalent modifications of DNA have been shown to be effective mechanisms for the stimulation of Topo I-mediated DNA cleavage. Abasic sites, base mismatches, nicks, modified base-containing DNA and alkylated adducts can induce cleavage in the vicinity of a locally modified DNA structure (23,24,32), suggesting a role for Topo I in DNA damage procession. The local structural torsion induced by the triplex may be specifically recognized by Topo I and viewed as a damage-like modification to be eliminated. Finally, it is also plausible that the enzyme considers the triplex structure as an obstacle in its progression, functioning as a processive/scanning enzyme. At this hurdle, the enzyme would make a pause, and cleave at its sitting point.
An interesting new model of the mechanism of action of Topo I hypothesizes that the enzyme primarily maintains an inactive conformation and once bound to DNA shifts to the active state (32). In this case, all distortions in the DNA duplex, as for example the presence of a triple helix, will have a structural impact on the extent of Topo I–DNA recognition. Subsequently, depending on the nature of the DNA conformation at the binding site, the enzyme will be efficiently or loosely converted to the active state.
In conclusion, we have shown here that triplex-directed Topo I-mediated DNA cleavage requires a specific organization of the ternary complex with an optimal positioning of the inhibitor with respect to the DNA target site for the enzyme. The study provides a better understanding of the molecular architecture necessary to design drug–TFO conjugates acting as site-specific Topo I-dependent nucleases. Such rationally designed small molecules could prove to be useful antitumor drugs directed selectively against genes bearing the targeted triplex-binding site and, in the future, this may be further exploited to improve the efficacy of chemotherapeutic cancer treatments. It also illustrates the utility of such TFO–drug conjugates as potent and tunable molecular tools to probe the structure and organization of protein–DNA complexes. TFO–CPT conjugates will be useful to understand the functioning of Topo I in living cells. The study of this aspect of the research is now under way in our laboratories.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at NAR Online.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Lionel Dubost and Dr Jean-Paul Brouard of the Mass Spectroscopy Service of the MNHN for the mass spectra analysis and Prof. Sidney Hecht for helpful discussions. This work was supported by grants from the Ligue Nationale Contre le Cancer (to P.B.A.), from the Ligue Nationale Contre le Cancer (Equipe Labellisée La Ligue) (to C.B.) and from the European Community (grant INTAS 01-0638 to A.B.).
REFERENCES
- 1.Champoux J.J. (2001) DNA topoisomerases: structure, function and mechanism. Annu. Rev. Biochem., 70, 369–413. [DOI] [PubMed] [Google Scholar]
- 2.Wang J.C. (1996) DNA topoisomerases. Annu. Rev. Biochem., 65, 635–692. [DOI] [PubMed] [Google Scholar]
- 3.Pommier Y., Pourquier,P., Fan,Y. and Strumberg,D. (1998) Mechanism of action of eukaryotic DNA topoisomerase I and drugs targeted to the enzyme. Biochim. Biophys. Acta, 1400, 83–105. [DOI] [PubMed] [Google Scholar]
- 4.Redinbo M.R., Stewart,L., Kuhn,P., Champoux,J.J. and Hol,W.G. (1998) Crystal structures of human topoisomerase I in covalent and noncovalent complexes with DNA. Science, 279, 1504–1513. [DOI] [PubMed] [Google Scholar]
- 5.Stewart L., Redinbo,M.R., Qiu,X., Hol,W.G.J. and Champoux,J.J. (1998) A model for the mechanism of human topoisomerase I. Science, 279, 1534–1541. [DOI] [PubMed] [Google Scholar]
- 6.Redinbo M.R., Champoux,J.J. and Hol,W.G. (2000) Novel insights into catalytic mechanism from a crystal structure of human topoisomerase I in complex with DNA. Biochemistry, 39, 6832–6840. [DOI] [PubMed] [Google Scholar]
- 7.Staker B.L., Hjerrild,K., Feese,M.D., Behnke,C.A., Burgin,A.B.,Jr and Stewart,L. (2002) The mechanism of topoisomerase I poisoning by a camptothecin analog. Proc. Natl Acad. Sci. USA, 99, 15387–15392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fan Y., Weinstein,J.N., Kohn,K.W., Shi,L.M. and Pommier,Y. (1998) Molecular modeling studies of the DNA-topoisomerase I ternary cleavable complex with camptothecin. J. Med. Chem., 41, 2216–2226. [DOI] [PubMed] [Google Scholar]
- 9.Kerrigan J.E. and Pilch,D.S. (2001) A structural model for the ternary cleavable complex formed between human topoisomerase I, DNA and camptothecin. Biochemistry, 40, 9792–9798. [DOI] [PubMed] [Google Scholar]
- 10.Arimondo P.B., Boutorine,A., Baldeyrou,B., Bailly,C., Kuwahara,M., Hecht,S.M., Sun,J.S., Garestier,T. and Hélène,C. (2002) Design and optimization of camptothecin conjugates of triple helix-forming oligonucleotides for sequence-specific DNA cleavage by topoisomerase I. J. Biol. Chem., 277, 3132–3140. [DOI] [PubMed] [Google Scholar]
- 11.Laco G.S., Collins,J.R., Luke,B.T., Kroth,H., Sayer,J.M., Jerina,D.M. and Pommier,Y. (2002) Human topoisomerase I inhibition: docking camptothecin and derivatives into a structure-based active site model. Biochemistry, 41, 1428–1435. [DOI] [PubMed] [Google Scholar]
- 12.Matteucci M., Lin,K.-Y., Huang,T., Wagner,R., Sternbach,D.D., Mehrotra,M. and Besterman,J.M. (1997) Sequence-specific targeting of duplex DNA using a camptothecin-triple helix forming oligonucleotide conjugate and topoisomerase I. J. Am. Chem. Soc., 119, 6939–6940. [Google Scholar]
- 13.Arimondo P.B., Bailly,C., Boutorine,A., Sun,J.S., Garestier,T. and Hélène,C. (1999) Targeting topoisomerase I cleavage to specific sequences of DNA by triple helix-forming oligonucleotide conjugates. A comparison between a rebeccamycin derivative and camptothecin. C. R. Acad. Sci. III, 322, 785–790. [DOI] [PubMed] [Google Scholar]
- 14.Arimondo P.B., Bailly,C., Boutorine,A., Moreau,P., Prudhomme,M., Sun,J.S., Garestier,T. and Hélène,C. (2001) Triple helix-forming oligonucleotides conjugated to indolocarbazole poisons direct topoisomerase I-mediated DNA cleavage to a specific site. Bioconjugate Chem., 12, 501–509. [DOI] [PubMed] [Google Scholar]
- 15.Sawada S., Okajima,S., Aiyama,R., Nokata,K., Furuta,T., Yokokura,T., Sugino,E., Yamaguchi,K. and Miyasaka,T. (1991) Synthesis and antitumor activity of 20(S)-camptothecin derivatives: carbamate-linked, water-soluble derivatives of 7-ethyl-10-hydroxycamptothecin. Chem. Pharm. Bull. (Tokyo), 39, 1446–1450. [DOI] [PubMed] [Google Scholar]
- 16.Cantor C.R., Warshaw,M.M. and Shapiro,H. (1970) Oligonucleotides interactions. III. Circular dichroism studies of the conformation of deoxyoligonucleotides. Biopolymers, 9, 1059–1077. [DOI] [PubMed] [Google Scholar]
- 17.Arimondo P.B., Bailly,C., Boutorine,A., Ryabinin,V., Syniakov,A., Sun,J.S., Garestier,T. and Hélène,C. (2001) Directing topoisomerase I-mediated DNA cleavage to specific sites by camptothecin tethered to minor and major groove ligands. Angew. Chem. Int. Ed., 40, 3045–3048. [DOI] [PubMed] [Google Scholar]
- 18.Grimm G., Boutorine,A. and Hélène,C. (2000) Rapid routes of synthesis of oligonucleotide conjugates from non-protected oligonucleotides and ligands possessing different nucleophilic or electrophilic functional groups. Nucleosides Nucleotides Nucleic Acids, 19, 1943–1965. [DOI] [PubMed] [Google Scholar]
- 19.Villemin D. and Hammadi,M. (1996) Environmentally desirable synthesis without use of organic solvent. Synthesis of aryloxyacetic acids. Synthetic Commun., 26, 4337–4341. [Google Scholar]
- 20.Stevnsner T., Mortensen,U.H., Westergaard,O. and Bonven,B.J. (1989) Interactions between eukaryotic DNA topoisomerase I and a specific binding sequence. J. Biol. Chem., 264, 10110–10113. [PubMed] [Google Scholar]
- 21.Svejstrup J.Q., Christiansen,K., Andersen,A.H., Lund,K. and Westergaard,O. (1990) Minimal DNA duplex requirements for topoisomerase I-mediated cleavage in vitro. J. Biol. Chem., 265, 12529–12535. [PubMed] [Google Scholar]
- 22.Krogh S., Mortensen,U.H., Westergaard,O. and Bonven,B.J. (1991) Eukaryotic topoisomerase I–DNA interaction is stabilized by helix curvature. Nucleic Acids Res., 19, 1235–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pourquier P. and Pommier,Y. (2001) Topoisomerase I-mediated DNA damage. Adv. Cancer Res., 80, 189–216. [DOI] [PubMed] [Google Scholar]
- 24.Pommier Y., Kohlhagen,G., Laco,G.S., Kroth,H., Sayer,J.M. and Jerina,D.M. (2002) Different effects on human topoisomerase I by minor groove and intercalated deoxyguanosine adducts derived from two polycyclic aromatic hydrocarbon diol epoxides at or near a normal cleavage site. J. Biol. Chem., 277, 13666–13672. [DOI] [PubMed] [Google Scholar]
- 25.Collier D.A., Mergny,J.L., Thuong,N.T. and Hélène,C. (1991) Site-specific intercalation at the triplex-duplex junction induces a conformational change which is detectable by hypersensitivity to diethylpyrocarbonate. Nucleic Acids Res., 19, 4219–4224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chomilier J., Sun,J.S., Collier,D.A., Garestier,T., Hélène,C. and Lavery,R. (1992) A computational and experimental study of the bending induced at a double-triple helix junction. Biophys. Chem., 45, 143–152. [DOI] [PubMed] [Google Scholar]
- 27.Asensio J.L., Dosanjh,H.S., Jenkins,T.C. and Lane,A.N. (1998) Thermodynamic, kinetic and conformational properties of a parallel intermolecular DNA triplex containing 5′ and 3′ junctions. Biochemistry, 37, 15188–15198. [DOI] [PubMed] [Google Scholar]
- 28.Asensio J.L., Brown,T. and Lane,A.N. (1999) Solution conformation of a parallel DNA triple helix with 5′ and 3′ triplex-duplex junctions. Struct. Fold Des., 7, 1–11. [DOI] [PubMed] [Google Scholar]
- 29.Perini R., Caserta,M. and Di Mauro,E. (1993) DNA tridimensional context affects the reactivity of eukaryotic DNA topoisomerase I. J. Mol. Biol., 231, 634–645. [DOI] [PubMed] [Google Scholar]
- 30.Sim S.P., Pilch,D.S. and Liu,L.F. (2000) Site-specific topoisomerase I-mediated DNA cleavage induced by nogalamycin: a potential role of ligand-induced DNA bending at a distal site. Biochemistry, 39, 9928–9934. [DOI] [PubMed] [Google Scholar]
- 31.Shen C.C. and Shen,C.K. (1990) Specificity and flexibility of the recognition of DNA helical structure by eukaryotic topoisomerase I. J. Mol. Biol., 212, 67–78. [DOI] [PubMed] [Google Scholar]
- 32.Lesher D.T., Pommier,Y., Stewart,L. and Redinbo,M.R. (2002) 8-Oxoguanine rearranges the active site of human topoisomerase I. Proc. Natl Acad. Sci. USA, 99, 12102–12107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Arimondo P.B., Barcelo,F., Sun,J.S., Maurizot,J.C., Garestier,T. and Hélène,C. (1998) Triple helix formation by (G,A)-containing oligonucleotides: asymmetric sequence effect. Biochemistry, 37, 16627–16635. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.