


Abstract
Several proteins involved in DNA synthesis are part of the so-called ‘replication factories’ that are anchored on non-chromatin nuclear structures. We report here that human kin17, a nuclear stress-activated protein, associates with both chromatin and non-chromatin nuclear structures in a cell cycle- and DNA damage-dependent manner. After l-mimosine block and withdrawal we observed that kin17 protein was recruited in the nucleus during re-entry and progression through S phase. These results are consistent with a role of kin17 protein in DNA replication. About 50% of the total amount of kin17 protein was detected on nuclear structures and could not be released by detergents. Furthermore, the amount of kin17 protein greatly increased in both G1/S and S phase-arrested cells in fractions containing proteins anchored to nuclear structures. The detection of kin17 protein showed for the first time its preferential assembly within non-chromatin nuclear structures in G1/S and S phase-arrested cells, while the association with these structures was found to be less stable in the G2/M phase, as judged by fractionation of human cells and immunostaining. In asynchronous growing cells, kin17 protein interacted with both chromatin DNA and non-chromatin nuclear structures, while in S phase-arrested cells it interacted mostly with non-chromatin nuclear structures, as judged by DNase I treatment and in vivo UV cross-linking. In the presence of DNA damage in S phase cells, the distribution of kin17 protein became mainly associated with chromosomal DNA, as judged by limited formaldehyde cross-linking of living cells. The physical interaction of kin17 protein with components of the nuclear matrix was confirmed and visualized by indirect immunuofluorescence and immunoelectron microscopy. Our results indicate that, during S phase, a fraction of the human kin17 protein preferentially associates with the nuclear matrix, a fundamentally non-chromatin higher order nuclear structure, and to chromatin DNA in the presence of DNA damage.
INTRODUCTION
DNA replication is a complex phenomenon and it is unclear how 3 × 109 bp of DNA per typical human cell are replicated with a high degree of fidelity in a relatively short time in a process that could involve thousands of origins of replication. Uncertainty still exists as to whether or not true origins of chromosomal DNA replication exist in mammals (1,2). Given this complexity, it is likely that nucleic acid metabolism is carried out on spatially organized domain structures in the cell nucleus. Evidence suggests that the nuclear matrix, a non-chromatin nuclear structure is a key player in organizing higher order chromatin/nuclear structures (3–5). In the case of DNA replication, fluorescence microscopic analyses revealed discrete granular sites of replication, i.e. replication sites or replication foci (6–8). Nascent DNA and several proteins involved in DNA synthesis have been found to attach to these non-chromatin nuclear structures, thereby forming replication foci (3,4,9). In addition, non-chromatin nuclear elements have also been implicated in the initiation step of DNA replication and putative origins of replication are often situated close to matrix attachment regions (10,11).
The kin17 protein was initially identified based on the cross-reacting property of antibodies raised against the stress-activated Escherichia coli RecA protein. The kin17 protein displays common epitopes with the RecA protein and shares 47% homology over a 40 residue stretch in the RecA C-terminal region (12). In RecA protein, this region is involved in the regulation of DNA binding and in the SOS response (13). Human kin17 is a 45 kDa nuclear protein which is remarkably conserved during evolution, ubiquitously expressed in mammals (14) and forms intranuclear foci in proliferating cells. To date, major features of the kin17 protein are its ability to: (i) bind to curved DNA found at the hot-spots of illegitimate recombination of human cells (15,16); (ii) complement the functions of the bacterial transcriptional factor H-NS, which binds to curved DNA and controls gene expression (17); (iii) be up-regulated after UV and ionizing radiation (18–23). We have reported that a fraction of kin17 protein is strongly and directly associated with chromosomal DNA in human cells. The amount of kin17 protein anchored to nuclear structures increases 2.6-fold after γ-irradiation and was found to be concentrated in large nuclear foci associated with replication protein A (RPA). Furthermore, clones expressing a human KIN17 antisense transcript elicited major disruptions in S phase progression as well as a significant decrease in clonogenic cell growth and proliferation (22). There is a relationship between kin17 protein, DNA replication and DNA damage, since these antisense clones displayed a 6-fold increase in their radiosensitivity in comparison with controls, suggesting an impaired tolerance to DNA damage (24). Blattner et al. have also highlighted a link between the presence of UV-induced DNA damage and the mouse KIN17 pathway in deficient Xeroderma pigmentosum complementation group A (XPA) mouse cells (20). Further more, the integrity of human global genome repair is crucial for the up-regulation of the human KIN17 gene. In particular, the presence of functional XPA and Xeroderma pigmentosum complementation group C (XPC) proteins is a prerequisite for the up-regulation of human KIN17 gene expression after UV-C (23). Interestingly, XPA, XPC and RPA proteins have been implicated in DNA damage recognition (25). Recently, we have reported a physical interaction between human kin17 protein and SV40 large T antigen. This interaction inhibited in vitro and in vivo DNA synthesis, suggesting a role of the kin17 protein in DNA replication (26). Taken together, these observations suggest that kin17 protein is a nuclear maintenance protein involved in DNA replication and in the cellular response to DNA damage (20–23,26).
The determination of the distribution of proteins implicated in DNA replication among subcellular fractions and their interactions with nuclear structures will cast light on how replication is initiated and otherwise regulated. Elucidation of the precise subnuclear compartmentalization of kin17 protein and its dynamic relocalization in human cells should help to uncover roles of this protein in chromosomal DNA replication. The aim of the present cell fractionation and immunostaining study was then to determine if the detergent-resistant nuclear kin17 protein could be associated with the nuclear matrix. We demonstrated for the first time that, in addition to its increased association with chromosomal DNA after DNA damage, kin17 protein is also detected in the nuclear matrix in all the phases of the cell cycle, becoming strongly associated with non-chromatin nuclear structures during S phase.
MATERIALS AND METHODS
Cell culture and synchronization
Human cervical carcinoma HeLa and human colorectal carcinoma RKO and HCT116 cell lines were grown in Dulbecco’s modified Eagle’s medium (DMEM) with 10% fetal calf serum and synchronized in early S phase with 0.5– 3 mM hydroxyurea (Sigma) or 15 µM aphidicolin (Sigma) treatment for 16–18 h, in G1/S with 50–400 µM l-mimosine (Sigma) treatment for 16–18 h or in G2/M phase with 0.2 µM nocodazole (Sigma) treatment for 16–18 h. Cells were released from their arrested state by adding fresh culture medium and their progression toward the cell cycle was analyzed 1, 3, 6, 9 and 24 h later by flow cytometry.
Purification and analysis of protein–protein and DNA–protein complexes from HeLa cells after in vivo cross-linking with formaldehyde
HeLa S3 cells were grown on 145 mm plastic dishes in DMEM with 5% fetal calf serum (Sigma). For DNA labeling, the medium of semi-confluent cultures was replaced by 20 ml of medium containing 15 µCi [methyl-3H]thymidine (25 Ci/mmol) and the cells were incubated for 24 h under normal culture conditions. Then cells were synchronized using a double thymidine block. Cells were exposed to 100 µM N-methyl-N-nitro-N-nitrosoguanidine (MNNG) (Sigma) for 3 h immediately after block release or to 200 µM MNNG 2 h 30 min after block release (30 min treatment) followed by two washes in phosphate-buffered saline (PBS). Formaldehyde cross-linking in vivo and DNA–protein complex preparation were performed exactly as described by Frouin et al. (27), based on the method of Gohring and Fackelmayer (28).
In vivo cross-linking of living cells with DSP
Cross-linking of living HeLa cells with dithiobis(succinimidylpropionate) (DSP) (Pierce) (29) was performed as described by Fujita et al. (30). Briefly, HeLa cells in 100 mm plates were washed three times with PBS at room temperature and mixed with 10 ml of cross-linking buffer (PBS, pH 8, 1 mM MgCl2, 100 mM sucrose, 0.01% Triton X-100) and DSP dissolved in dimethyl sulfoxide was added to a final concentration of 25–200 µg/ml. The plates were gently swirled for 10 min at room temperature and the reaction was stopped with 10 ml of TE buffer (10 mM Tris–HCl, pH 7.6, 1 mM EDTA) followed by two washes with ice-cold PBS. For immunofluorescence staining, HeLa cells on coverslips were fixed for 10 min with 3.7% formaldehyde in PBS and then permeabilized with methanol for 20 min at –20°C. When stained after DSP treatment and subsequent chromatin extraction, cells on coverlips were processed as described in ‘Cell fractionation’, before formaldehyde fixation.
Cell fractionation
To determine the association of the nuclear kin17 protein with non-ionic detergent-resistant nuclear structures in asynchronous and early S phase-arrested HCT116 and RKO cell lines, cells seeded at 5000 cells/cm2 3 days before trypsinization were lysed in buffer N (50 mM Tris–HCl, pH 7.9, 150 mM NaCl, 1% Igepal, 1 mM EDTA, protease inhibitor mixture) as described previously (22). The lysates were kept on ice for 30 min and soluble and insoluble protein fractions recovered after centrifugation (20 000 g for 15 min) were quantified by Bradford assay (Bio-Rad).
For experiments with DSP, DSP-treated and untreated HeLa cells were lysed for 5 min on ice with 1 ml of ice-cold cytoskeleton buffer with Triton X-100 (TX-CSK) (20 mM potassium phosphate, pH 6.8, 100 mM NaCl, 300 mM sucrose, 1 mM MgCl2, 1 mM EGTA, 0.1% Triton X-100, protease inhibitor cocktail, 200 µM Na3VO4, 20 mM NaF) (30). Cells were then subjected to centrifugation (3000 g for 5 min) to obtain Triton X-100-extractable fractions. The extracted nuclei were washed once more with the same volume of ice-cold TX-CSK buffer. The extracted nuclei were further extracted with buffer E (0.5 M NaCl, 1% Igepal, 10 mM Tris–HCl, pH 7.6) and centrifuged to obtain the salt-extractable fractions. The Triton X-100- and salt-extracted nuclei were digested with 1000 U/ml DNase I (10 U/µl) (Roche Molecular Biochemicals) in TX-CSK buffer containing 2 mM MgCl2 and protease inhibitor complete mixture at 25°C for 30 min and then NaCl was adjusted to a final concentration of 0.5 M. The samples were then centrifuged to obtain the solubilized chromatin fraction and the remaining non-chromatin nuclear structures.
To analyze the biochemical localization of kin17 protein without salt and DSP treatments, Triton X-100-extractable supernatants (TX-100 S) and extracted nuclear pellets (TX-100 P) were prepared from asynchronous HeLa cells as described above. The resuspended extracted nuclei from the second TX-CSK extraction were further treated with DNase I (1000 U/ml, 25°C, 30 min) to obtain DNase I-solubilized supernatants (DNase S) and remaining DNase-resistant nuclear pellets (DNase P). After incubation, insoluble and soluble materials were separated by centrifugation. All the samples were added to the same volumes of 2× SDS sample buffer (31) and boiled.
In vivo cross-linking of living cells with UV
Cross-linking of living HeLa cells with UV was performed as described (32) except that 1 M NaCl was used instead of 0.5 M NaCl. HeLa cells grown in 100 mm dishes (5 × 106 cells) were washed twice and 10 ml of PBS was added before irradiation with 254 nm UV (fluence rate 2.5 mJ/cm2) at the indicated doses in a UV chamber (XL-1000 UV Crosslinker; Spectronics Corp.). Dosimetry was performed with a CX-254 UV radiometer (Viber Lourmat, Marne la Vallée, France). The cells were then scraped with a rubber policeman, centrifuged and lysed in 500 µl of TX-CSK at 4°C for 10 min. The soluble and insoluble fractions were collected after centrifugation. Half of the washed insoluble fraction was further extracted with 1 M NaCl in the same buffer at 4°C for 30 min and centrifuged at 20 000 g for 20 min at 4°C to obtain the soluble and insoluble fractions.
Sample analyses
Immunoblotting and preparation and characterization of mouse monoclonal anti-kin17 protein antibodies were described previously (22,26). Purified IgG anti-kin17 protein Ig K36 and Ig K58 specifically recognize a single band of 45 kDa corresponding to the kin17 protein in SDS–PAGE (22,26). These antibodies were used at a final concentration of 25 ng/ml. Other mouse monoclonal antibodies used were anti-proliferating cell nuclear antigen (PCNA) (clone PC10, diluted to 50 mg/ml; Novo Castra), anti-RPA70 (diluted to 25 ng/ml; Oncogene Research Products) and anti-histone H1 (AE-4, 1/1000; Santa Cruz). Anti-histone H1 antibody binding was revealed by alkaline phosphatase-labeled anti-mouse monoclonal antibody (1/10 000; Sigma) and visualized using NBT/BCIP tablets (Roche). Peroxidase-labeled goat anti-mouse monoclonal antibody binding (1/10 000; Jackson Laboratories) was visualized using the ECL system (Amersham Pharmacia Biotech). Some samples were also subjected to 15% SDS–PAGE followed by Coomassie blue staining for detection of core histones.
Preparation of replication foci attached to the nuclear matrix
Replication foci attached to the nuclear matrix were prepared essentially as described (33). Briefly, asynchronous and early S phase-arrested HeLa cells seeded on glass coverslips were washed twice with Tris-buffered saline (TBS) (150 mM NaCl, 5 mM MgCl2, 10 mM Tris–HCl, pH 7.4) and permeabilized in situ with 0.1% Triton X-100 and 0.5 mM phenylmethylsulfonyl fluoride (PMSF) in TBS buffer at room temperature for 5 min, followed by a wash with TBS. The chromatin of the permeabilized cells was digested in situ with 40 U/ml DNase I in TBS at room temperature for 7 min and extracted twice with 20 mM Trizma, pH 7.4, containing 0.25 M ammonium sulfate and 0.2 mM MgCl2 at room temperature for 1 min. The samples were then washed twice with TBS. The cells were fixed with 4% paraformaldehyde in TBS at 4°C for 15 min, washed with TBS six times, permeabilized in TBS containing 0.5 mM CuSO4 and 0.5% Triton X-100 for 10 min at room temperature and rinsed four times in TBS buffer. Indirect immunofluorescence staining of fixed and permeabilized cells was performed as described below.
Indirect immunofluorescence staining
All staining procedures were carried out at room temperature, except for extraction where indicated. HeLa cells, plated at 5000 cells/cm2 on glass coverslips, were washed with PBS, fixed and permeabilized as indicated. The fixed and permeabilized cells were then incubated for 1 h with purified mouse monoclonal antibody IgG K36 (450 ng/ml) diluted in PBS containing 0.5% Tween 20, 12% bovine serum albumin (BSA) and 0.036% NaN3. Primary antibodies were incubated for 45 min with 2 µg/ml Cy3™-conjugated affinity-purified goat anti-mouse IgG (Jackson Laboratories Inc., USA). All washes after antibody incubation were done with PBS. Cells were counterstained with 4 µg/ml 4′,6-diamino-2,8-phenylindole (DAPI). Immunofluorescence staining was viewed using a Zeiss Axiophot 2 epifluorescence microscope coupled to a cooled Sensys 1400 camera (Photometrics, CA) monitored by the Zeiss KS300 3.0 program (22,26). Representative fields for each treatment are presented.
Nuclear matrix preparation and immunogold electron microscopy
Extraction treatments were carried out in situ on cells still attached to the plastic dishes as described by Puvion et al. (34). HeLa cells were rinsed in PBS and sequential extractions were performed as follows. Soluble proteins were removed by extraction in TMS buffer (0.25 M sucrose, 5 mM MgSO4, 50 mM Tris–HCl, pH 7.4, 1 mM PMSF) containing 1% Triton X-100 for 3 min at 4°C. After washing in TMS buffer, the cells were incubated for 30 min at 37°C in TMS buffer containing 50 µg/ml type IV pancreatic DNase I (Sigma). After three washes with LM buffer (10 mM Tris–HCl, pH 7.4, 0.2 mM MgSO4, 1 mM PMSF) at 4°C, the remaining structures were subjected to two consecutive 15 min treatments with HS buffer (2 M NaCl, 0.2 mM MgSO4, 10 mM Tris–HCl, pH 7.4, 1 mM PMSF) followed by a final wash with LM buffer for 5 min at 4°C. For electron microscopy the residual structures were fixed with 4% paraformaldehyde in PBS for 30 min at 4°C. The post-embedding immunogold procedure was carried out on sections of cells embedded in Lowicryl K4M and immunolabeling was performed using a mixture of anti-kin17 K36 and K58 monoclonal antibodies (1/10 in PBS containing 1% BSA) for 30 min at room temperature, followed by goat anti-mouse antibody conjugated to colloidal gold particles 5 nm in diameter (1/30 in PBS containing 1% BSA; Biocell Research Laboratories, Cardiff, UK). The grids were stained with uranyl acetate and observed with a Zeiss EM902 transmission electron microscope at 80 keV.
RESULTS
Association of kin17 protein with DNA synthesis
To determine a relationship of kin17 protein with the progression of the replication forks, we treated HeLa cells with l-mimosine (400 µM, 24 h) that induced a cell cycle arrest in late G1 phase close to the G1/S border, before the establishment of active DNA replication forks (35). As evidenced by flow cytometry analysis, treatment of asynchronously proliferating cells with l-mimosine for 24 h resulted in a population synchronized in late G1 phase and to a reduced percentage of cells in G2 phase (Fig. 1, compare 0 µM 24 h and 400 µM 24 h). After l-mimosine withdrawal, the arrested cells enter S phase rapidly and synchronously and their progression through the cell cycle was analyzed 1, 3, 6, 9 and 24 h later. As early as 1 h after l-mimosine removal we detected an increased kin17 protein level, probably associated with the re-initiation of DNA replication (compare lanes 2 and 3). This increase was maintained throughout the cell cycle (lanes 4–7). As control experiments, we detected p34cdc2 and cyclin B proteins in the same immunoblot. The p34cdc2/cyclin B complex is required for the G2 to M transition and these proteins are synthesized during late S and G2 phases (36). We found these proteins to be increased 6–9 h after drug withdrawal (lanes 5 and 6), corresponding to cells in late S and G2 phase. The recruitment of kin17 protein during re-initiation of DNA replication suggests a physiological role of this protein in this process.
Figure 1.

kin17 protein levels increased during the re-initiation of DNA synthesis and the progression into S phase. HeLa cells were treated with l-mimosine (400 µM) for 24 h. Cells were then released in fresh medium and recovered at the indicated times for cell cycle determination and immunoblot analysis of kin17, cyclin B and p34cdc2 protein contents.
Increased association of human kin17 protein with nuclear structures in G1/S and S phase-arrested cells
Since kin17 protein is recruited in synchronized cells released in S phase, we sought to determine whether kin17 protein remained bound to the sites of stalled replication forks. We then examined kin17 protein sublocalization in asynchronous and hydroxyurea- and l-mimosine-treated HeLa cells. Hydroxyurea and l-mimosine are agents known to block the elongation of DNA replication by reducing the dNTP pool. Hydroxyurea or l-mimosine treatment resulted in a population of cells predominantly stalled at the G1/S border, as determined by flow cytometry of bromodeoxyuridine (BrdU) pulse-labeled cells (data not shown).
We have suggested that kin17 protein participates in a cellular response to irradiation, thus helping to overcome the perturbation of DNA replication produced by unrepaired DNA lesions (22). Since in RKO cells ∼50% of kin17 protein is extracted with 1% non-ionic detergent (22), we asked whether the l-mimosine and hydroxyurea treatments leading to a G1/S and early S phase arrest, respectively, may change this distribution. Under the same experimental conditions, asynchronous and arrested RKO or HCT116 cells were lysed in buffer N to discriminate between ‘cytoplasmic and soluble nuclear proteins’ (detergent-extractable fraction) and ‘proteins anchored to nuclear structures’ (nuclei-bound fraction) (Fig. 2). Proteins extracted from each fraction were immunoblotted with anti-kin17 protein, anti-RPA or anti-PCNA antibodies and the relative amounts quantified. In asynchronous HCT116 cells, 30–50% of the kin17 protein, RPA or PCNA remained bound to nuclei, compared to only 10–20% in RKO cells, the remainder being present as a detergent-extractable form (compare lanes 1 and 10 from both detergent-extractable fraction and nuclei-bound fraction). In the nuclei-bound fraction of HCT116 cells, the maximal levels of kin17 protein were, respectively, 4- and 6-fold higher than in mock-treated cells after both l-mimosine and hydroxyurea treatments (the signals in lanes 5 and 9 over that in lane 1 in Fig. 2). In RKO cells, the highest levels were, respectively, 2.5- and 3-fold (lanes 13 and 17 over lane 10 in Fig. 2). A similar pattern was observed for RPA70 levels except for l-mimosine-treated RKO cells (lanes 11–14), whereas PCNA levels increased slightly in hydroxyurea-treated RKO cells (lanes 15–18). However, treatment of HCT116 and RKO cells with the lowest concentration of hydroxyurea used (0.5 mM, lanes 6 and 15) increased the level of detergent-resistant kin17 protein, PCNA and RPA70 to their maximum compared to mock-treated cells (lanes 1 and 10). In contrast, only minor modifications in kin17 protein or PCNA levels were noted in the detergent-extractable fractions of RKO cells after l-mimosine (lanes 11–14) or hydroxyurea (lanes 15–18) treatment compared to mock-treated cells (lane 10), whereas a modest 1.5- to 2-fold increase was noted in HCT116 cells after treatment (the signals in lanes 5 and 9 over lane 1). Interestingly, the increased RPA levels present in the nuclei-bound fractions of both cell lines were found to be linked to decreased levels of this protein in the corresponding detergent-extractable fractions. Similar results to hydroxyurea were obtained in cells arrested in S phase by aphidicolin treatment (data not shown). It is important to notice that l-mimosine, aphidicolin and hydroxyurea treatments stall DNA replication, induce DNA single-strand breaks and sister chromatid exchange and may cause oxidative cell stress. The possibility that the alterations of kin17 protein distribution by these chemicals may reflect a response to generic cell stress cannot be excluded. Nevertheless, l-mimosine treatment, which induces a cell cycle arrest in late G1 phase close to the G1/S border in HeLa cells (Fig. 1), has little effect on the level of kin17 protein. Although the whole content of kin17 protein was increased in S phase-arrested cells (by hydroxyurea), we constantly observed an increased association of this protein with nuclei-bound fractions in S phase cells (after l-mimosine withdrawal). It could then indicate that the main effect of these drugs observed on kin17 protein was not due to a response to generic cell stress.
Figure 2.

Increased association of human kin17 protein with nuclear structures in G1/S and S phase-arrested cells. Asynchronous (mock-treated, lanes 1 and 10), l-mimosine-treated (lanes 2–5 and 11–14) or hydroxyurea- treated (lanes 6–9 and 15–18) RKO and HCT116 cells were lysed in buffer N (containing 1% Igepal CA-630) as indicated in Materials and Methods. The detergent-extractable and nuclei-bound fractions were collected by centrifugation, immunoblotted with anti-kin17 protein, anti-RPA or anti-PCNA monoclonal antibodies and revealed by chemiluminescence. A representative pattern of three different experiments is shown.
RPA and kin17 proteins interact with both chromatin DNA and nuclear protein complexes
The increased association of kin17 protein with nuclear structures suggests that, in addition to DNA binding, a portion of kin17 protein could also interact with protein components organized into foci on non-chromatin structures. To test this hypothesis, we investigated the localization of kin17 protein in asynchronous HeLa cells using a fractionation method that includes a mild detergent extraction to produce extracted nuclear pellets, which were further extensively digested by DNase I to obtain solubilized supernatants and nuclear pellets resistant to nuclease. These fractions were immunoblotted with anti-kin17 protein, anti-RPA or anti-PCNA antibodies (Fig. 3A). In asynchronous HeLa cells, ∼40% of the total kin17 protein was present as a Triton X-100-extractable form, and the remainder was bound to nuclei (Fig. 3A, lanes 1 and 2). About 70% of the total PCNA was found in the detergent-extractable fraction, while ∼90% of the total RPA was sensitive to mild detergent extraction (Fig. 3A, lanes 1 and 2), as already described (37). Therefore, kin17 protein was strongly associated with nuclei compared to RPA and PCNA, two other nuclear proteins implicated in DNA transactions that are more easily solubilized by mild detergent extraction. To investigate whether the kin17 protein present in the detergent-resistant fraction was linked to chromatin or non-chromatin nuclear structures, the extracted nuclei were digested with DNase I (Fig. 3A). This drastic treatment (1000 U/ml DNase I at 25°C for 30 min) stripped ∼90% of core histones (Fig. 5B) and DNA (unpublished data). It is important to note that incubation at 25°C for 30 min of detergent-extracted nuclei without DNase I treatment revealed that ∼60–70% of the nucleus-bound kin17 protein, ∼90% of nucleus-bound RPA and ∼50% of nucleus-bound PCNA were released into the supernatant (Fig. 3A, lane 3 versus 4). Interestingly, DNase I digestion did not help to release further kin17 protein from the nuclear fraction (Fig. 3A, lane 6), as compared to samples processed under the same conditions but without nuclease digestion (Fig. 3A, lane 4), whereas bound RPA and bound PCNA were almost completely released into the supernatant (Fig. 3A, lane 5). For all three proteins, neither detectable proteolytic products nor a remarkable reduction in the total amount were found after incubation at 25°C for 30 min. This temperature-induced release has already been described for PCNA (38). Addition of ATP to the reaction mixture did not prevent the heat-induced dissociation of kin17, RPA or PCNA proteins bound to extracted nuclei (data not shown). Furthermore, the core histones were detected only in the nuclear pellet fractions, indicating that DNA was not solubilized at the incubation temperature (data not shown). We conclude that a fraction of kin17 protein interacts with non-chromatin nuclear structures, such as the nuclear matrix, while RPA and PCNA are mostly associated with nuclear structures sensitive to DNase I digestion.
Figure 3.


Endogenous kin17 protein associates with both DNA and nuclear structures as judged by selective extraction and immunostaining detection. (A) Triton X-100-extractable supernatants (TX-100 S, lane 1) and extracted nuclear pellets (TX-100 P, lane 2) were prepared from asynchronous HeLa cells. The extracted nuclear pellets were further treated with 1000 U/ml DNase I at 25°C for 30 min to obtain DNase I-solubilized supernatants [DNase(+) S, lane 5] and remaining DNase-resistant nuclear pellets [DNase(+) P, lane 6] or without DNase I at 25°C for 30 min [DNase(–) S, lane 3; DNase(–) P, lane 4]. These fractions were immunoblotted with anti-kin17 protein, anti-RPA or anti-PCNA monoclonal antibodies. (B) Indirect immunofluorescence detection of kin17 protein localization in in situ replication foci attached to the nuclear matrix prepared as described in Materials and Methods. Asynchronous (mock-treated) and hydroxyurea- or aphidicolin-arrested HeLa cells were fixed with paraformaldehyde (PAF) after Triton X-100 extraction (0.1% Triton-X100, panels a and e), DNase I treatment and ammonium sulfate extraction [0.1% Triton X-100 + (NH4)2SO4 + DNase I, panels b–d and f–h]. The extracted cells were then immunostained with anti-kin17 protein antibody (red). The samples were further treated with DAPI for DNA staining (blue). Digitized images of representative cells are shown at a magnification of 500×. Living asynchronous (C) or early S phase-arrested (D) HeLa cells on dishes were irradiated with UV at 50, 100, 150, 200 and 400 mJ/cm2 and subjected to successive extractions with Triton X-100 (Triton Ext., lanes 1–5) and 1 M NaCl (NaCl Ext., lanes 6–10) as described in Materials and Methods. Extracted kin17 and RPA proteins were detected by immunoblotting and revealed by chemiluminescence. A typical pattern is shown.
Figure 5.

Localization of kin17 protein in HeLa cell extracts after in vivo cross-linking with DSP. Triton X-100-extractable supernatants (TX-100 S, lane 1) and extracted nuclei (TX-100 P, lane 2) were prepared from asynchronous HeLa cells before and after treatment with 25–200 µg/ml DSP. The nuclei were further extracted with buffer containing 0.5 M NaCl and the salt extracts (0.5 M NaCl S, lane 3) and the nuclear pellets (0.5 M NaCl P, lane 4) were separated by centrifugation. The nuclei extracted with detergent and salt were then treated with DNase I followed by salt extraction to obtain solubilized chromatin fractions [DNase(+) S, lane 7] and insoluble non-chromatin nuclear pellets [DNase(+) P, lane 8] or without DNase I [DNase(–) S, lane 5; DNase(–) P, lane 6]. These fractions were immunoblotted with monoclonal anti-kin17 antibodies and revealed by chemiluminescence (A). The samples were also subjected to SDS–PAGE followed by Coomassie blue staining for detection of core histones (B).
To confirm the association of kin17 protein with non-chromatin nuclear structures, we prepared replication foci attached to the nuclear matrix in detergent-permeabilized HeLa cells after in situ DNase I digestion followed by ammonium sulfate extraction to eliminate loosely bound proteins and the digested DNA (33). After paraformaldehyde fixation, indirect immunofluorescence staining was performed with monoclonal anti-kin17 antibodies (Fig. 3B). Although the focal nuclear staining of kin17 protein was maintained, DNase I digestion and ammonium sulfate extraction slightly affect the structure of the nuclear foci. The foci seem fewer and fatter, suggesting that DNase I and ammonium sulfate treatment may induce the collapse of numerous small foci into each other. Furthermore, the intensity of kin17 protein detection was clearly increased in S phase-arrested cells (Fig. 3B, panels c and d) compared to asynchronous cells (Fig. 3B, panel b). These nuclear foci, which are still able to synthesize DNA in vitro (data not shown), remained associated with nuclear matrix structures despite the removal of >80% of the total nuclear DNA, as evidenced by DAPI staining of permeabilized cells before (Fig. 3B, panel e) and after chromatin extraction (Fig. 3B, panels f–h). Taken together, these results indicate that a fraction of kin17 protein is anchored to the nuclear matrix, a structure which is, as already described, well preserved during the isolation procedure (7,33).
The data described above, i.e. (i) the resistance to extensive nuclease treatment of bound kin17 proteins present in the detergent-extracted nuclei and (ii) the preservation of the staining pattern of kin17 protein in nuclear foci after in situ chromatin digestion, suggest that most of the nuclear fraction of kin17 protein (detergent-unextractable) is associated with non-chromatin nuclear structures. To check this hypothesis, we performed cross-linking experiments on living HeLa cells with increasing doses of UV (Fig. 3C and D). In this type of experiment only proteins that are directly associated with DNA may be cross-linked by UV and become high molecular weight complexes that cannot be extracted and detected by SDS–PAGE (39). The eventual association of kin17 protein with chromatin DNA in vivo will lead to a decreased amount of this protein in the fractions obtained after detergent and high salt extraction. Alternatively, if kin17 protein is bound to nuclear proteins by non-covalent interactions, it will be solubilized by detergent and salt extraction.
After irradiation with increasing UV doses, cells were subjected to in situ extractions with 0.1% Triton X-100 followed by 1 M NaCl. Only the soluble forms of kin17 protein from asynchronous (Fig. 3C) and early S phase cells (Fig. 3D) were detected by immunoblotting. In asynchronous cells, the amount of kin17 protein decreased with UV irradiation (Fig. 3C, lane 10), indicating that kin17 protein bound to nuclei after detergent extraction became unextractable upon UV irradiation. Interestingly, the kin17 protein present in the Triton-extractable fraction (cytoplasmic and nucleoplasmic proteins) seemed to be more efficiently cross-linked than the the Triton-unextractable nuclei-bound fraction. The UV dose required to make half of the kin17 protein covalently link to DNA was calculated to be between 100 and 200 mJ/cm2 (Fig. 3C, lanes 4 and 5). The situation was completely different in hydroxyurea-treated cells (Fig. 3D). Indeed, the nearly constant amounts of kin17 protein detected after both Triton extraction (Fig. 3D, lanes 1–5) and high salt extraction (Fig. 3D, lanes 6–10) indicated that this protein was not cross-linked to DNA in this UV range since salt extraction released almost all the kin17 protein. The RPA70 present in the Triton-extractable fraction showed a more efficient cross-linking to DNA in early S phase cells (Fig. 3D, lanes 1–5) than in asynchronous cells (Fig. 3C, lanes 1–5). RPA70 was scarcely cross-linked to DNA in the same UV range (Fig. 3C and D, lanes 6–10). Although the KIN17 gene is induced by UV, it should be noted that KIN17 mRNA starts to accumulate at 8 h after UVC irradiation and that the doses of UV used in the protein–DNA cross-linking experiments presented here were 50- to 250-fold higher than the UV dose used in stress response studies (20,23). From the UV cross-linking experiments, we suggest that, in early S phase cells, the nuclei-bound fraction (detergent-unextractable) of kin17 protein was predominantly linked to non-chromatin nuclear structures, such as the nuclear matrix, or with matrix-associated proteins and a small proportion bound directly to DNA, while RPA70 associated preferentially with chromatin DNA.
Relocalization of kin17 protein to chromatin after DNA damage in S phase HeLa cells
The results presented above show that in vivo only a part of kin17 protein is directly associated with chromosomal DNA in human cells, whereas the remainder is bound to non-chromatin structures. The amount of kin17 protein anchored to nuclear structures increased 2.6-fold after γ-irradiation and was found to be concentrated into large nuclear foci associated with RPA (22). These observations indicate that kin17 protein could be a nuclear maintenance protein involved in DNA replication and in the cellular response to DNA damage. We asked whether part of such a response involves changes in the subnuclear localization of the kin17 protein following other types of DNA damage. S phase synchronized HeLa cells were subjected to MNNG treatment before in vivo formaldehyde cross-linking. Next, protein–protein complexes (reflecting the fraction not associated with the chromatin) were separated from DNA–protein complexes (representing the proteins bound to chromatin) by density gradient fractionation as described elsewhere (22,27). Purified complexes were de-cross-linked by boiling in SDS sample buffer and analyzed for their protein components by SDS–PAGE. Immunoblot analysis of these fractions showed that kin17 protein in untreated cells was equally distributed in the unbound (free protein) versus bound fractions (data not shown). However, after DNA damage induced by 100 µM MNNG for 3 h (Fig. 4A) or 200 µM MNNG for 30 min (data not shown) a 5-fold increase in chromatin-bound kin17 protein was observed (compare lane 1 to 2), while the unbound fractions (free protein) remained unchanged (data not shown). As a control experiment, the fractions were probed with anti-histone H1 antibodies (Fig. 4B). The same level of chromatin-bound histone H1 was detected in untreated and MNNG-treated cells. These results indicate that the up-regulation of kin17 protein observed after DNA damage formation leads to an increased association of this protein with chromatin DNA.
Figure 4.
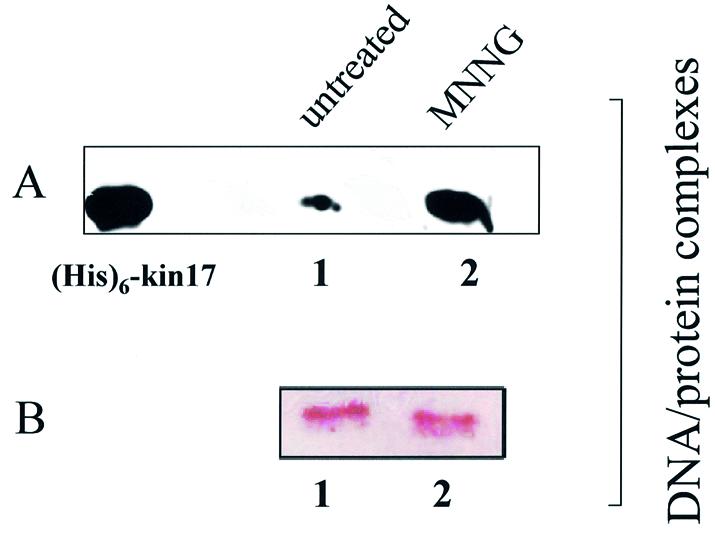
kin17 protein detection in purified protein–chromatin DNA and protein–protein complexes after in vivo cross-linking of living S phase HeLa cells with formaldehyde. After in vivo cross-linking with formaldehyde (1%, 4 min), the nuclei were isolated, lysed and both protein–chromatin DNA and protein–protein complexes were purified by equilibrium centrifugation in two consecutive cesium chloride gradients as described in Materials and Methods. After reversion of the cross-links by boiling, immunoblot analysis was performed on DNA–protein complexes obtained from untreated (A and B, lanes 1) and MNNG-treated (A and B, lanes 2) HeLa cells. (A) Treatment of cells for 3 h with 100 µM MNNG immediately after release from a double thymidine block. Detection of kin17 protein. (B) Detection of histone H1. (His)6-kin17, positive control [recombinant human (His)6-kin17 protein].
In vivo association of kin17 protein with non-chromatin nuclear structures in HeLa cells using protein–protein cross-linking
To investigate further the association of kin17 protein with non-chromatin nuclear structures, we cross-linked HeLa cells in vivo using a method described by Fujita et al. (30,38). Living cells are first cross-linked with the cell-permeable protein–protein (but not DNA–protein) cross-linker DSP (29) and then chromatin is extensively stripped by nuclease and salt treatments thus allowing exploration of the organization of nuclear proteins. If kin17 protein is associated with non-chromatin nuclear structures in vivo, they could be covalently cross-linked to these structures and, consequently, the bound protein would become resistant to chromatin stripping. If they bind to particular chromatin components but not to the structures, then they would be extracted together with chromatin.
We first performed cross-linking experiments and subsequent chromatin extraction with increasing DSP concentrations and detected kin17 protein by immunoblot in the fractions obtained (Fig. 5A). In asynchronous HeLa cells, ∼40% of kin17 protein was present in the fraction extracted with 0.1% Triton X-100 and the remainder was bound to nuclei (Fig. 3A, lanes 1 and 2, and Fig. 5A, lanes 1 and 2). This ratio remained unchanged when cells were treated with DSP even at the highest concentration tested (Fig. 5A). The constant distribution observed between extracted and remaining amounts of kin17 protein indicate that the treatment does not generate non-specific protein aggregation leading to artificial insolubility. Therefore, we used a DSP concentration of 200 µg/ml in the following experiments in agreement with a previous report (30).
In asynchronous cells, almost all of the Triton-insoluble kin17 protein and RPA were readily extracted with the buffer containing 0.5 M NaCl (Fig. 5A, lane 4 versus 3, and Fig. 6A, lane 3 versus 2). In contrast, in asynchronous DSP cross-linked cells the Triton X-100-insoluble kin17 protein became resistant to salt extraction and reached ∼80% (∼50% of total kin17 protein) at the highest concentration of DSP used, indicating that kin17 protein was covalently linked to other salt-insoluble nuclear protein(s) (Fig. 5A, lane 8).
Figure 6.


Subcellular localization of endogenous kin17 protein in HeLa cells during the cell cycle using in vivo cross-linking with DSP. Detection of kin17 protein, RPA or PCNA in asynchronous, early S phase- and G2/M-arrested cells, before and after treatment with DSP (200 µg/ml). Triton X-100-extractable supernatants (lanes 1 and 6) and extracted nuclei were prepared before (A) and after DSP cross-linking (B) of asynchronous or hydroxyurea-treated HeLa cells. The nuclei were further extracted with buffer containing 0.5 M NaCl and the salt extracts (lanes 2 and 7) and the nuclear pellets (lanes 3 and 8) were separated by centrifugation. The detergent- and salt-extracted nuclei were then treated with DNase I followed by salt extraction to obtain solubilized chromatin fractions (lanes 4 and 9) and insoluble non-chromatin nuclear pellets (lanes 5 and 10). (C) Triton X-100-extractable supernatants (0.1% TX-100 S) and extracted nuclei (0.1% TX-100 P) were prepared from nocodazole-treated cells with or without DSP. The nuclei were further extracted with buffer containing 0.5 M NaCl and the salt extracts and the nuclear pellets were separated by centrifugation. The detergent- and salt-extracted nuclei were then treated with DNase I followed by salt extraction to obtain solubilized chromatin fractions and insoluble non-chromatin nuclear pellets or without DNase I [DNase(–) S, DNase(–) P]. (D) Indirect immunofluorescence detection of kin17 protein localization in HeLa cells after DSP cross-linking and chromatin extraction. Asynchronous (left) or hydroxyurea-treated (early S phase, right) cells were fixed with formaldehyde directly (panels a and e), after extraction with Triton X-100 (panels b and f) or after chromatin extraction as described in Materials and Methods of cells treated with (panels c and g) or without DSP (w/o, panels d and h). The cells were then immunostained with anti-kin17 protein antibody (red) and treated with DAPI for DNA staining (blue). Digitized images of representative cells are shown at a magnification of 500×.
We asked further whether kin17 protein was linked to chromatin or to non-chromatin nuclear structures. After Triton and salt extraction, the DSP cross-linked nuclei were digested with DNase I followed by a second 0.5 M NaCl extraction. This treatment stripped ∼90% of core histones (Fig. 5B, lane 7 versus 8) and DNA (Fig. 6D, panels a and b versus c and d). Detergent-unextracted forms of both RPA and kin17 proteins were detected mostly in the resultant nuclear pellets (Fig. 6B, lane 5). In parallel, HeLa cells grown on coverslips were cross-linked with DSP and extracted with Triton X-100 and NaCl as described above but analyzed by immunocytochemistry. The observed nuclear staining revealed the characteristic dot distribution of kin17 protein in cells directly fixed with formaldehyde (Fig. 6D, panel a), after extraction with Triton X-100 (Fig. 6D, panel b) as well as after cross-linking with DSP (Fig. 6D, panel c). Only a slight residual fluorescence was detected in cells stripped without cross-linking (Fig. 6D, panel d), indicating that even after a drastic extraction, a small fraction of kin17 protein still remains tightly bound to the nucleus. We conclude that under physiological conditions in asynchronous cells, kin17 protein is completely extracted after nuclease digestion and salt extraction (Fig. 6A, lane 5), whereas in S phase-arrested cells this fraction became strongly bound to other nuclear proteins (Fig. 6A, lane 10). The protein–protein cross-linker DSP conserved these interactions and further confirmed that significant amounts of the bound fraction of kin17 protein and RPA are associated with non-chromatin nuclear structures in living HeLa cells.
Data for kin17 protein obtained with DSP-treated asynchronous HeLa cells (Fig. 5A and Fig. 6A and B, ‘asynchronous’) may mainly represent the situation of cells in G1 phase since more than 60% of cells were in this phase, as evidenced by flow cytometry of BrdU pulse-labeled cells (unpublished data). To check whether kin17 protein was still associated with non-chromatin nuclear structure in S phase, we analyzed hydroxyurea-treated HeLa cells arrested in early S phase with and without in vivo cross-linking with DSP. Without cross-linking, kin17 protein was completely extracted by salt treatment (Fig. 6A, lane 3), but in early S phase cells (Fig. 6A, HU) we detected a fraction of kin17 protein that became resistant to salt extraction (Fig. 6A, lane 8) and to DNase I digestion (Fig. 6A, lane 10). The major part of RPA in S phase was unextracted by detergent (Fig. 6A, lane 6) and became resistant to 0.5 M NaCl (Fig. 6A, lane 8) and nuclease extraction (Fig. 6A, lane 10). Taken together, the data shown in Figure 6A and B indicate that both kin17 protein and RPA were associated with non-chromatin nuclear structures in S phase. Immunofluorescence staining after fractionation showed that the cross-linked kin17 proteins are indeed in the nuclei of S phase HeLa cells (Fig. 6D, panel g), as they are in cells directly fixed with formaldehyde (Fig. 6D, panel e) or after extraction with Triton X-100 (Fig. 6D, panel f). Note that a weak staining could be observed after fractionation of cells in S phase without cross-linking (Fig. 6D, panel h).
As previously reported, kin17 protein also binds to chromatin DNA in HeLa cells synchronized in G2/M (22). We then analyzed the association of kin17 protein with non-chromatin nuclear structures in G2/M phase. Cell fractionation without cross-linking (Fig. 6C) suggested that the association of kin17 protein, RPA or PCNA with chromatin DNA and/or nuclease-resistant structures becomes weak in G2/M phase since these proteins were almost all efficiently extracted by detergent (Fig. 6C, 0.1% TX-100 S). On the other hand, kin17 protein, RPA or PCNA in G2/M phase were linked [Fig. 6C, DNase(+) P] to non-chromatin nuclear structures as efficiently as in asynchronous cells (Figs 5A and 6B), as shown after cross-linking with DSP.
We conclude that kin17 protein, RPA and PCNA have an increased association with non-chromatin nuclear structures in S phase of the cell cycle.
Immunoelectron microscopic localization of kin17 protein to the nuclear matrix prepared after in situ extraction of living HeLa cells
To avoid any eventual bias introduced by cross-linking treatments, we studied the interaction of kin17 protein with nuclear matrix prepared in living HeLa cells without cross-linking before extraction. We prepared residual structures corresponding to the nuclear matrix obtained by detergent, nuclease and salt extractions of living HeLa cells as described elsewhere (34). The protocol was applied to cells attached to plastic dishes, allowing preservation of the ultrastructure and more efficient extractions. By using this method, we obtained 100% completely preserved nuclear matrix, as revealed by electron microscopic observations. The nuclear matrix thus prepared contained less than 5% protected residual DNA (unpublished data). The successive extractions clearly reveal the three components of the nuclear matrix: the peripheral lamina (L), the internal or interchromatin matrix [with interchromatin granules (Ig) resistant to the treatment] and the residual nucleolus (NU) (Fig. 7A and C). The detection of kin17 protein with monoclonal antibodies revealed both scattered (Fig. 7A) and typical focal labeling restricted to the internal matrix (Fig. 7B–D). Considering the drastic treatment used to prepare nuclear matrix of living cells attached to plastic culture dishes (1% Triton X-100, extensive DNase I digestion and 2 M NaCl), few gold particles could be detected. Isolated gold particles were visualized on the fibers of the internal nuclear matrix but very rarely on interchromatin granules (Fig. 7A). It should be noted that foci of kin17 protein were preserved in 11% of the nuclear matrix observed, mostly close to the residual lamina (Fig. 7A–C), while the residual nucleoli were never labeled. The immunogold electron microscopy localization of kin17 protein demonstrated its association with the nuclear matrix.
Figure 7.

Immunogold electron microscopy localization of endogenous kin17 protein after in situ extraction of living HeLa cells. HeLa cells were extracted with Triton X-100, DNase I, and 2 M NaCl and embedded in Lowicryl. Immunolabeling was performed on sections using a mixture of anti-kin17 K36 and K58 monoclonal antibodies followed by goat anti-mouse antibody conjugated to colloidal gold particles 5 nm in diameter. Sections were stained with uranyl acetate. Gold particles were found either isolated along the fibers of the internal matrix (A and D) or in foci (arrows, A–D) close to the lamina (L). Interchromatin granules (Ig) and residual nucleoli (NU) were not labeled (A, C and D). Bars represent 0.5 µm.
DISCUSSION
The human kin17 protein is mainly localized in the nucleus of asynchronous cells. Several genotoxic agents trigger the nuclear accumulation of this protein, indicating its participation in a general cellular response (14,20–23). These reports have suggested the participation of kin17 protein in DNA metabolism, although its precise biological activity remains to be determined.
We show here that kin17 protein is present in two different forms in HeLa cells: a fraction containing nearly 40% of the total kin17 protein that is extractable by non-ionic detergent and a fraction tightly associated with the nucleus containing 60% of kin17 protein resistant to detergent, as judged by cell fractionation and immunocytochemical analysis. Under the same experimental conditions, RPA and PCNA are at least 70–90% extractable by mild detergent extraction. We have extended this type of analysis to drugs that arrest cells in different phases of the cell cycle. We showed that the levels of the nuclei-bound kin17 protein increased in S phase-arrested cells, suggesting that newly synthesized kin17 protein is in a nuclei-bound form. Furthermore, we show that in synchronized HeLa cells in G1/S phase the levels of total kin17 protein gradually increased during progression through S phase. Although the level of chromatin-bound kin17 protein fraction seems to be constant after G2/M arrest by nocodazole treatment (22), we show here that the association of kin17 protein with nuclear structures became less stable during mitosis, when resetting of the next replication cycle occurs, suggesting another possible role of kin17 protein in facilitating cell division.
In asynchronous cells, the fraction of kin17 protein extracted under mild detergent conditions mainly represents the chromatin-bound fraction, as judged by UV cross-linking experiments reported here. Interestingly, in S phase-arrested HeLa cells kin17 protein is barely bound to chromatin, suggesting a dynamic change in the association of this protein with other nuclear components during S phase. Nuclease treatment experiments further showed an interaction of kin17 protein with both nuclease-resistant nuclear structures and chromatin DNA, indicating that it does not associate simply with DNA. Our results also suggest that the detergent-extractable form of kin17 protein and/or the heat-sensitive portion of nuclear-bound kin17 protein (Fig. 3A) represents the part of kin17 protein that is effectively linked to chromatin DNA. In addition to this resistance to nuclease, a characteristic feature of nuclear-bound kin17 protein is its dissociation by salt wash within a wide concentration range (0.2–1 M NaCl), pointing to interactions with numerous partners and/or targets in nuclei (unpublished data). An association with nuclease-resistant nuclear structures using DNA as mediator may also be possible, but the data presented here are mostly in favor of an interaction with both chromatin DNA and one or more nuclease-resistant nuclear components.
We entertain the idea that the ‘nucleoplasmic pool’ of kin17 protein (extracted by mild detergent) could serve as a reservoir of kin17 protein that may later be associated with both chromatin and/or nuclear matrix during DNA replication. Indeed, during S phase kin17 protein interacts predominantly with non-chromatin nuclear structures where it could form a transient complex involved in DNA replication. Alternatively, the equilibrium of kin17 protein between the ‘nucleoplasmic pool’ versus the nuclei-bound form of kin17 protein (chromatin-bound plus non-chromatin-bound) would be required after generation of double-strand breaks induced by genotoxic agents like ionizing radiation (22). The simplest hypothesis would be that the nuclear accumulation of kin17 protein leads to an increase in the chromatin-bound fraction in order to facilitate the DNA replication process in spite of the presence of multiple damaged sites or other DNA lesions. This idea is reinforced by the data presented here showing that the methylating agent MNNG increases the amount of both kin17 protein and PCNA bound to chromatin DNA (data not shown for PCNA). Furthermore, it has been shown that PCNA interacts with other components of the high molecular weight replication complex and at the same time with chromatin (27). We hypothesize that after DNA lesion formation the complex containing kin17 and other proteins such as PCNA undergoes a dynamic rearrangement leading to the interaction of these proteins in or around DNA damage. Our results indicate that kin17 protein is one of several factors that equilibrate between the free nuclear pool and assembly into large structures (40,41), further confirming the notion that the nucleus is a highly dynamic organelle in which the assembly of compartments in response to metabolic requirements of the cell may be a general feature (41).
Nuclear morphology is strikingly deformed after the ectopic expression of kin17 protein in human cells, leading to the inhibition of DNA synthesis and to a drastic drop in clonogenic growth (42,43). Furthermore, the inhibition of in vitro and in vivo DNA replication triggers by a direct interaction between human kin17 protein and SV40 large T antigen (26), together with the results presented here, point to a role of kin17 protein in the regulation of DNA replication. DNA replication takes place in specialized compartments, such as the nuclear matrix, in which specific factors assemble in highly organized structures (4). The nuclear matrix is a non-chromatin substructure of the nucleus upon which chromatin is organized, DNA and RNA are processed and differential gene expression may be managed. The nuclear matrix is experimentally seen after removal of the nucleoplasm and chromatin by detergent extraction, nuclease digestion and high salt extraction (33). Proteins that remain within the nuclear structure after these treatments are defined as being ‘nuclear matrix associated’. However, some proteins may coagulate under high salt conditions and thereby be artificially detected as matrix elements. Alternatively, some proteins associated with the matrix in vivo may be stripped by salt extraction. Furthermore, exposure of cells to non-physiological buffers alters the native nuclear architecture and could also result in non-specific aggregation of matrix-related proteins. These limitations were circumvented by using the in vivo cross-linking method with the permeable chemical protein–protein (but not DNA–protein) cross-linker DSP, as described (30). We demonstrated that kin17 protein covalently linked to the nuclear structures became resistant to chromatin stripping and then associated with these non-chromatin nuclear structures in vivo during all the phases of the cell cycle.
The average amount of kin17 protein in several proliferating human cell lines is ∼2 × 104–1 × 105 molecules/cell nucleus (26). This amount of kin17 protein is similar to that of other nuclear proteins involved in DNA metabolism, such as RPA, AP endonuclease 1 (APE1), DNA ligase 1 and DNA polymerase β (37,44). Several of these nuclear proteins localize either partly or completely in distinct ‘bodies’ or subnuclear compartments that produce a punctate staining pattern when analyzed by indirect immunofluorescence. Immunocytochemical analyses detect kin17 as a protein mainly localized in nuclei with a staining pattern that resembles those of other proteins involved in DNA replication (22,26,42). The observed intranuclear foci are substructures arising as a consequence of activities such as DNA replication and may reflect the assembly of dedicated ‘factories’ at specific sites in the nucleus (45). Evidence for multiprotein DNA replication complexes has been accumulating in recent years (27,46–49). Recently, a model of DNA replication was proposed to represent the multiprotein DNA replication complex termed the ‘DNA synthesome’. The synthesome ‘core’ comprised proteins involved in the elongation phase of DNA replication (DNA polymerases α and δ, primase, replication factor C, DNA helicase, ligase I and topoisomerase II), whereas the proteins involved in the initiation phase of DNA replication (PCNA, RPA and topoisomerase I) are more loosely associated with it (47). We have recently co-purified from HeLa cells the endogenous kin17 protein as a component of a high molecular weight protein complex with components of the ‘DNA synthesome’ such as PCNA, RPA, DNA polymerase and ATPase activities (manuscript in preparation). The implication of kin17 protein in DNA replication is reinforced by the results presented here together with recent findings, i.e. (i) the inhibition of in vitro and in vivo DNA synthesis by a physical interaction between human kin17 protein and SV40 large T antigen, (ii) the increased expression of kin17 protein in cells entering S phase, (iii) the blockage in early-to-mid S phase of cells expressing a KIN17 antisense transcript, (iv) the preferential association of kin17 protein with the nuclear matrix in S phase and (v) the co-purification of kin17 protein with the DNA synthesome. The molecular characterization of this matrix-associated protein and the determination of its biological activity will help to shed some light on replication regulation and on the role of the nuclear matrix in the activity of kin17 protein.
Acknowledgments
ACKNOWLEDGEMENTS
B. Dutrillaux is gratefully acknowledged for his continuous support. We particularly thank E. Puvion for stimulating discussions and advice on nuclear structure, E. Pichard for electron microscopy, C. Créminon, Y. Frobert, J. Grassi and all the staff of the Service de Pharmacologie et d’Immunologie for monoclonal antibodies against human kin17 protein and M.-F. Poupon for cell lines. This work was supported by Electricité de France contract 8702 and by the CNR Target Project on Biotechnology and CNR Agenzia 2000.
REFERENCES
- 1.Boulikas T. (1996) Common structural features of replication origins in all life forms. J. Cell. Biochem., 60, 297–388. [DOI] [PubMed] [Google Scholar]
- 2.Boulikas T. (1995) Chromatin domains and the prediction of MAR sequences. Int. Rev. Cytol., 162A, 279–388. [DOI] [PubMed] [Google Scholar]
- 3.Cook P.R. (1988) The nucleoskeleton: artefact, passive framework or active site? J. Cell Sci., 90, 1–6. [DOI] [PubMed] [Google Scholar]
- 4.Berezney R., Mortillaro,M.J., Ma,H., Wei,X. and Samarabandu,J. (1995) The nuclear matrix: a structural milieu for genomic function. Int. Rev. Cytol., 162A, 1–65. [DOI] [PubMed] [Google Scholar]
- 5.Nickerson J.A. (2001) Experimental observations of a nuclear matrix. J. Cell Sci., 114, 463–474. [DOI] [PubMed] [Google Scholar]
- 6.Nakamura H., Morita,T. and Sato,C. (1986) Structural organizations of replicon domains during DNA synthetic phase in the mammalian nucleus. Exp. Cell Res., 165, 291–297. [DOI] [PubMed] [Google Scholar]
- 7.Hozak P., Hassan,A.B., Jackson,D.A. and Cook,P.R. (1993) Visualization of replication factories attached to nucleoskeleton. Cell, 73, 361–373. [DOI] [PubMed] [Google Scholar]
- 8.Ma H., Samarabandu,J., Devdhar,R.S., Acharya,R., Cheng,P.C., Meng,C. and Berezney,R. (1998) Spatial and temporal dynamics of DNA replication sites in mammalian cells. J. Cell Biol., 143, 1415–1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cook P.R. (1999) The organization of replication and transcription. Science, 284, 1790–1795. [DOI] [PubMed] [Google Scholar]
- 10.Dijkwel P.A. and Hamlin,J.L. (1988) Matrix attachment regions are positioned near replication initiation sites, genes and an interamplicon junction in the amplified dihydrofolate reductase domain of Chinese hamster ovary cells. Mol. Cell. Biol., 8, 5398–5409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dijkwel P.A. and Hamlin,J.L. (1995) Origins of replication and the nuclear matrix: the DHFR domain as a paradigm. Int. Rev. Cytol., 162A, 455–484. [DOI] [PubMed] [Google Scholar]
- 12.Angulo J.F., Rouer,E., Mazin,A., Mattei,M.G., Tissier,A., Horellou,P., Benarous,R. and Devoret,R. (1991) Identification and expression of the cDNA of KIN17, a zinc-finger gene located on mouse chromosome 2, encoding a new DNA-binding protein. Nucleic Acids Res., 19, 5117–5123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kurumizaka H., Aihara,H., Ikawa,S., Kashima,T., Bazemore,L.R., Kawasaki,K., Sarai,A., Radding,C.M. and Shibata,T. (1996) A possible role of the C-terminal domain of the RecA protein. A gateway model for double-stranded DNA binding. J. Biol. Chem., 271, 33515–33524. [DOI] [PubMed] [Google Scholar]
- 14.Kannouche P., Mauffrey,P., Pinon-Lataillade,G., Mattei,M.G., Sarasin,A., Daya-Grosjean,L. and Angulo,J.F. (2000) Molecular cloning and characterisation of the human Kin17 cDNA encoding a component of the UVC-response that is conserved among metazoans. Carcinogenesis, 21, 1701–1710. [DOI] [PubMed] [Google Scholar]
- 15.Mazin A., Milot,E., Devoret,R. and Chartrand,P. (1994) KIN17, a mouse nuclear protein, binds to bent DNA fragments that are found at illegitimate recombination junctions in mammalian cells. Mol. Gen. Genet., 244, 435–438. [DOI] [PubMed] [Google Scholar]
- 16.Mazin A., Timchenko,T., Menissier-de Murcia,J., Schreiber,V., Angulo,J.F., de Murcia,G. and Devoret,R. (1994) Kin17, a mouse nuclear zinc finger protein that binds preferentially to curved DNA. Nucleic Acids Res., 22, 4335–4341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Timchenko T., Bailone,A. and Devoret,R. (1996) Btcd, a mouse protein that binds to curved DNA, can substitute in Escherichia coli for H-NS, a bacterial nucleoid protein. EMBO J., 15, 3986–3992. [PMC free article] [PubMed] [Google Scholar]
- 18.Biard D.S., Saintigny,Y., Maratrat,M., Paris,F., Martin,M. and Angulo,J.F. (1997) Enhanced expression of the Kin17 protein immediately after low doses of ionizing radiation. Radiat. Res., 147, 442–450. [PubMed] [Google Scholar]
- 19.Kannouche P., Pinon-Lataillade,G., Tissier,A., Chevalier-Lagente,O., Sarasin,A., Mezzina,M. and Angulo,J.F. (1998) The nuclear concentration of kin17, a mouse protein that binds to curved DNA, increases during cell proliferation and after UV irradiation. Carcinogenesis, 19, 781–789. [DOI] [PubMed] [Google Scholar]
- 20.Blattner C., Kannouche,P., Litfin,M., Bender,K., Rahmsdorf,H.J., Angulo,J.F. and Herrlich,P. (2000) UV-induced stabilization of c-fos and other short-lived mRNAs. Mol. Cell. Biol., 20, 3616–3625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Masson C., Menaa,F., Pinon-Lataillade,G., Frobert,Y., Radicella,J.P. and Angulo,J.F. (2001) Identification of KIN (KIN17), a human gene encoding a nuclear DNA-binding protein, as a novel component of the TP53-independent response to ionizing radiation. Radiat. Res., 156, 535–544. [DOI] [PubMed] [Google Scholar]
- 22.Biard D.S.F., Miccoli,L., Despras,E., Frobert,Y., Créminon,C.C. and Angulo,J.F. (2002) Ionizing radiation triggers chromatin-bound HSAkin17 complex in human cells. J. Biol. Chem., 277, 19156–19165. [DOI] [PubMed] [Google Scholar]
- 23.Masson C., Menaa,F., Pinon-Lataillade,G., Frobert,Y., Chevillard,S., Radicella,J.P., Sarasin,A. and Angulo,J.F. (2003) Global genome repair is required to activate KIN17, a UVC-responsive gene involved in DNA replication. Proc. Natl Acad. Sci. USA, 100, 616–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Despras E., Miccoli,L., Créminon,C., Rouillard,D., Angulo,J.F. and Biard,D.S.F. (2003) Depletion of kin17, a human DNA-replication protein, increases the radiosensitivity of RKO cells. Radiat. Res., 159, 748–758. [DOI] [PubMed] [Google Scholar]
- 25.Wakasugi M. and Sancar,A. (1999) Order of assembly of human DNA repair excision nuclease. J. Biol. Chem., 274, 18759–18768. [DOI] [PubMed] [Google Scholar]
- 26.Miccoli L., Biard,D.S.F., Créminon,C. and Angulo,J.F. (2002) Human kin17 protein directly interacts with the SV40 large T antigen and inhibits DNA replication. Cancer Res., 62, 5425–5435. [PubMed] [Google Scholar]
- 27.Frouin I., Montecucco,A., Biamonti,G., Hubscher,U., Spadari,S. and Maga,G. (2002) Cell cycle-dependent dynamic association of cyclin/Cdk complexes with human DNA replication proteins. EMBO J., 21, 2485–2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gohring F. and Fackelmayer,F.O. (1997) The scaffold/matrix attachment region binding protein hnRNP-U (SAF-A) is directly bound to chromosomal DNA in vivo: a chemical cross-linking study. Biochemistry, 36, 8276–8283. [DOI] [PubMed] [Google Scholar]
- 29.Baumert H.G. and Fasold,H. (1989) Cross-linking techniques. Methods Enzymol., 172, 584–609. [DOI] [PubMed] [Google Scholar]
- 30.Fujita M., Ishimi,Y., Nakamura,H., Kiyono,T. and Tsurumi,T. (2002) Nuclear organization of DNA replication initiation proteins in mammalian cells. J. Biol. Chem., 277, 10354–10361. [DOI] [PubMed] [Google Scholar]
- 31.King J. and Laemmli,U.K. (1973) Bacteriophage T4 tail assembly: structural proteins and their genetic identification. J. Mol. Biol., 75, 315–337. [DOI] [PubMed] [Google Scholar]
- 32.Tatsumi Y., Tsurimoto,T., Shirahige,K., Yoshikawa,H. and Obuse,C. (2000) Association of human origin recognition complex 1 with chromatin DNA and nuclease-resistant nuclear structures. J. Biol. Chem., 275, 5904–5910. [DOI] [PubMed] [Google Scholar]
- 33.Nakayasu H. and Berezney,R. (1989) Mapping replicational sites in the eucaryotic cell nucleus. J. Cell Biol., 108, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Puvion E., Duthu,A., Harper,F., Ehrhart,J.C., Viron,A. and May,P. (1988) Intranuclear distribution of SV40 large T-antigen and transformation-related protein p53 in abortively infected cells. Exp. Cell Res., 177, 73–89. [DOI] [PubMed] [Google Scholar]
- 35.Krude T. (1999) Mimosine arrests proliferating human cells before onset of DNA replication in a dose-dependent manner. Exp. Cell Res., 247, 148–159. [DOI] [PubMed] [Google Scholar]
- 36.Pines J. and Hunter,T. (1989) Isolation of a human cyclin cDNA: evidence for cyclin mRNA and protein regulation in the cell cycle and for interaction with p34cdc2. Cell, 58, 833–846. [DOI] [PubMed] [Google Scholar]
- 37.Treuner K., Eckerich,C. and Knippers,R. (1998) Chromatin association of replication protein A. J. Biol. Chem., 273, 31744–31750. [DOI] [PubMed] [Google Scholar]
- 38.Fujita M., Kiyono,T., Hayashi,Y. and Ishibashi,M. (1997) In vivo interaction of human MCM heterohexameric complexes with chromatin. Possible involvement of ATP. J. Biol. Chem., 272, 10928–10935. [DOI] [PubMed] [Google Scholar]
- 39.Carr A. and Biggin,M.D. (1999) A comparison of in vivo and in vitro DNA-binding specificities suggests a new model for homeoprotein DNA binding in Drosophila embryos. EMBO J., 18, 1598–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lamond A.I. and Earnshaw,W.C. (1998) Structure and function in the nucleus. Science, 280, 547–553. [DOI] [PubMed] [Google Scholar]
- 41.Leonhardt H., Rahn,H.P. and Cardoso,M.C. (1999) Functional links between nuclear structure, gene expression, DNA replication and methylation. Crit. Rev. Eukaryot. Gene Expr., 9, 345–351. [DOI] [PubMed] [Google Scholar]
- 42.Kannouche P. and Angulo,J.F. (1999) Overexpression of kin17 protein disrupts nuclear morphology and inhibits the growth of mammalian cells. J. Cell Sci., 112, 3215–3224. [DOI] [PubMed] [Google Scholar]
- 43.Biard D.S., Kannouche,P., Lannuzel-Drogou,C., Mauffrey,P., Apiou,F. and Angulo,J.F. (1999) Ectopic expression of (Mm)Kin17 protein inhibits cell proliferation of human tumor-derived cells. Exp. Cell Res., 250, 499–509. [DOI] [PubMed] [Google Scholar]
- 44.Sokhansanj B.A., Rodrigue,G.R., Fitch,J.P. and Wilson,D.M.R. (2002) A quantitative model of human DNA base excision repair. I. Mechanistic insights. Nucleic Acids Res., 30, 1817–1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Leonhardt H., Rahn,H.P., Weinzierl,P., Sporbert,A., Cremer,T., Zink,D. and Cardoso,M.C. (2000) Dynamics of DNA replication factories in living cells. J. Cell Biol., 149, 271–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hickey R.J. and Malkas,L.H. (1997) Mammalian cell DNA replication. Crit. Rev. Eukaryot. Gene Expr., 7, 125–157. [DOI] [PubMed] [Google Scholar]
- 47.Malkas L.H. (1998) DNA replication machinery of the mammalian cell. J. Cell. Biochem., 30/31, 18–29. [PubMed] [Google Scholar]
- 48.Simbulan-Rosenthal C.M., Rosenthal,D.S., Hilz,H., Hickey,R., Malkas,L., Applegren,N., Wu,Y., Bers,G. and Smulson,M.E. (1996) The expression of poly(ADP-ribose) polymerase during differentiation-linked DNA replication reveals that it is a component of the multiprotein DNA replication complex. Biochemistry, 35, 11622–11633. [DOI] [PubMed] [Google Scholar]
- 49.Lebel M., Spillare,E.A., Harris,C.C. and Leder,P. (1999) The Werner syndrome gene product co-purifies with the DNA replication complex and interacts with PCNA and topoisomerase I. J. Biol. Chem., 274, 37795–37799. [DOI] [PubMed] [Google Scholar]