


Abstract
Since the ban gene of bacteriophage P1 suppresses a number of conditionally lethal dnaB mutations in Escherichia coli, it was assumed that Ban protein is a DNA helicase (DnaB analogue) that can substitute for DnaB in the host replication machinery. We isolated and sequenced the ban gene, purified the product, and analysed the function of Ban protein in vitro and in vivo. Ban hydrolyses ATP, unwinds DNA and forms hexamers in the presence of ATP and magnesium ions. Since all existing conditionally lethal dnaB strains bear DnaB proteins that may interfere with the protein under study, we constructed a dnaB null strain by using a genetic set-up designed to provoke the conditional loss of the entire dnaB gene from E.coli cells. This novel tool was used to show that Ban restores the viability of cells that completely lack DnaB at 30°C, but not at 42°C. Surprisingly, growth was restored by the dnaB252 mutation at a temperature that is restrictive for ban and dnaB252 taken separately. This indicates that Ban and DnaB are able to interact in vivo. Complementary to these results, we demonstrate the formation of DnaB–Ban hetero-oligomers in vitro by ion exchange chromatography. We discuss the interaction of bacterial proteins and their phage-encoded analogues to fulfil functions that are essential to phage and host growth.
INTRODUCTION
DnaB of Escherichia coli is the paradigmatic member of the replicative helicases family of hexameric ring-shaped enzymes that use the energy of nucleotide hydrolysis to unwind duplex DNA at a replication fork (1). In addition, it has been recently suggested that DnaB could participate in DNA recombination, since DnaB drives DNA branch migration in vitro (2). DnaB is not only required for E.coli chromosome replication and cell growth, but also for the replication of most plasmids and bacteriophages that use this bacteria as a host. Yet, E.coli lysogens of particular phage systems were found to survive under conditions that are restrictive for dnaB function, thus suggesting the existence of bacteriophage-encoded functional homologues of DnaB (3–6). Particularly, bacteriophage P1bac mutants were shown to constitutively express a gene, ban (for DnaB analogue), that compensates for several dnaB-defective host mutations, notably insertions and unsuppressed amber mutations (5–8). Furthermore, converging genetic and biochemical approaches suggested that Ban and DnaB could interact and form hetero-hexamers in vivo (8,9). These data supported the hypothesis that Ban protein is not only able to substitute for DnaB in E.coli cells, but also to form functional hetero-oligomeric structures. However, because the ban gene was never isolated, nor the enzymatic properties of its purified product biochemically characterised, these issues have remained as open questions for more than 20 years. Here we address these questions and further investigate the properties of the Ban protein. We have: (i) isolated the ban gene and expressed it in a strain that completely lacks DnaB; (ii) purified the Ban protein and tested its DNA unwinding and ATP hydrolysis activities; (iii) tested in vivo and in vitro the hypothesis that Ban and DnaB are able to form hetero-oligomers. Our data show that Ban is a bona fide DNA helicase, and that it can substitute for DnaB in E.coli cells that completely lack this protein. We also provide genetic and biochemical evidence that DnaB and Ban are able to interact.
MATERIALS AND METHODS
Bacterial strains, plasmids and growth conditions
Table 1 lists the E.coli strains, plasmids and bacteriophages used in this study. Mutation rpsL266 was transferred from LN2843 (a gift from F. Cornet, CNRS, Toulouse, France) to W3110 by transduction with P1vir to yield MLM329. Bacteriophage P1Cm (10) was the source for the ban gene, and single-stranded circular DNA was obtained from M13mp18. To overproduce and purify the Ban protein, strains were grown in media as described previously (11). For other purposes, cultures were grown with aeration in Luria–Bertani (LB) broth (12), supplemented with 1.5% agar for solid medium. Antibiotics were added when appropriate to the following concentrations: ampicillin (sodium salt), 100 µg/ml; chloramphenicol, 20 µg/ml; kanamycin sulfate, 50 µg/ml; streptomycin, 200 µg/ml; tetracycline, 2.5 µg/ml.
Table 1. Strains and plasmids used in this work.
| Strains/plasmids | Genotypes/plasmid properties | Reference/source |
|---|---|---|
| LN2843 | thi-1, leu, thyA, deoB or C, supE, rpsL2666 (SmR), lacI::Ω–ApR | 37 |
| RS162 | thr-1, leuB6(Am), fhuA21, lacY1, glnV44(AS), LAM-, rfbD1, thyA6, rpsL67(SmR), thi-1, zjb-504::Tn10, dnaB252(ts), deoC1 | 29; CGSC stock |
| SCS1 | recA1, endA1, gyrA96, thi-1, hsdR17(rK– mK+), supE44, relA1, | Stratagene |
| W3110 | l-, IN(rrnD-rrnE)1, rph-1 | 38 |
| WM0331 | dnaB165/70, arg-28, deo, gal-11, his-47, hsdS-K12, lac-11, mal, pro-19, rbs, rpsL56, sul-1, thyA59, trp-25 | W. Messer, Max-Planck-Institut, Berlin |
| MLM329 | W3110 rpsL2666 (SmR) | This work |
| MLM337 | MLM329 dnaB368::Ω-Tc/pPS562 | This work |
| MLM368 | MLM329 dnaB368::Ω-Tc/pMLM121 | This work |
| MLM369 | MLM329 dnaB368::Ω-Tc/pMLM115 | This work |
| MLM370 | MLM329 dnaB368::Ω-Tc/pMLM105 | This work |
| MLM371 | MLM329 dnaB368::Ω-Tc/pTR101 | This work |
| Plasmids | ||
| pACYC184 | repp15A, CmR | 17 |
| pHP45Ω–Tc | repcolD, ApR, Ω–Tc | 19 |
| pFUS2 | reppMB1, araC-ParaBAD, KmR | 13 |
| pLN135 | reppSC101 repAts, rpsL+, CmR | 18 |
| pMLM105 | pFUS2, araC-ParaBAD::dnaB | This work |
| pMLM115 | pFUS2, araC-ParaBAD::ban | This work |
| pMLM116 | pFUS2, araC-ParaBAD::dnaB252 | This work |
| pPS562 | reppUC18, dnaB, dnaC | N. Dixon |
| pMOM01 | repp15A, lacIq, ApR | This work |
| pMLM117 | pMOM01, PA1/O4/O3::ban | This work |
| pMLM121 | pLN135::dnaB | This work |
| pMLM122 | dnaB chromosomal fragment in pACYC184 | This work |
| pMLM123 | dnaB chromosomal fragment in pLN135 | This work |
| pMLM124 | pMLM123 dnaB368 | This work |
| pMLM125 | pMLM124 dnaB368::Ω-Tc | This work |
| pMS470Δ8 | reppMB1, Ptac, lacI, ApR | 16 |
| pTR101 | pMS470Δ8::ban | This work |
Construction of ban overexpressing plasmids
pMLM115. Vector plasmid pFUS2 (KmR) (13), is a pMB1-derived replicon designed to allow the expression of genes under the control of the arabinose regulated ParaBAD promoter. Plasmid pMLM115, in which the ban gene is placed under the control of ParaBAD, was constructed by inserting a PCR fragment containing the ban gene amplified with primers ban5 (5′-CAGTACGCATATGTCTGCATC CCCTCTT) and ban3 (5′-GCGGAAGCTTAATAATCCTC TTGCCAGT), and cleaved with NdeI and HindIII (recognition sites are underlined in the primer sequences) into pFUS2 opened with NdeI and HindIII.
pMLM117. A vector fragment generated by EcoRI and BamHI restriction cutting of plasmid pMLM96 (14) was either self-ligated, to yield the control vector pMOM01, or ligated to the ban gene isolated from pTR101 as a BamHI–HindIII fragment (1415 bp), to yield plasmid pMLM117. This places ban expression under the control of lacIq and the LacI-regulated pA1/04/03 promoter (15). When needed, HindIII, BamHI and EcoRI generated ends were repaired by using Klenow polymerase prior to ligation.
pTR101. To construct the ban overexpression plasmid pTR101, the ban gene was amplified by PCR using P1Cm DNA as template and a pair of primers, each containing the recognition sequences for NdeI and HindIII, which are underlined (5′-TAATATATCATatgTCTGCATCCCCTCT TGAATCC, 5′-TAATAAGCttaATAATCC-TCTTGCCAG TCAGC). The codons delimiting the ban reading frame are in lower case. The fragment was inserted into the NdeI–HindIII digested Ptac/lacIq regulated plasmid pMS470Δ8 (16) to result in pTR101.
Construction of dnaB overexpressing plasmids
pMLM105/pMLM116. The procedures to construct plasmids pMLM105 and pMLM116 were identical, except for the dnaB alleles that were used (dnaB and dnaB252, respectively). PCR fragments containing either the dnaB or the dnaB252 genes were amplified from total genomic DNA of strains W3110 and RS162, respectively (Table 1) with primers dnaB56 (5′-GGAATTCTCGTCAGGGTCTGCTTCAT) and dnaB32 (5′-CAAGGATCCAACAGTTGCCGCTTGCATT), cleaved with EcoRI and BamHI and inserted into the EcoRI and BglII sites of pFUS2. This places the dnaB alleles under the control of the ParaBAD promoter.
pMLM121. To transfer the ParaBAD::dnaB construct from pMLM105 into the low copy number and replication temperature-sensitive plasmid pLN135 (Table 1), the large AlwNI–HindIII fragment of pMLM105 was inserted between the BglII and HindIII sites in pLN135, yielding pMLM121.
DnaB substitution in the chromosome: construction steps and corroboration by PCR and immunodetection
Construction of the dnaB368:: Ω–Tc integration plasmid pMLM125. In the pMLM122 plasmid, the HpaI–BamHI chromosomal fragment (4034 bp) containing the dnaB gene and its flanking regions was inserted into the pACYC184 plasmid (17) cleaved with NruI and BamHI. A BamHI–Eco47III fragment from pMLM122 containing the entire 4034 bp dnaB region was ligated to a vector fragment obtained from cleavage with XhoI (followed by end-repair using Klenow polymerase) and BglII of the integration-excision vector pLN135 (18), to give pMLM123. To replace the wild-type dnaB gene in pMLM123 with the dnaB368 amber mutant allele, borne by plasmid pBRdnaB368 (M. Lemonnier and R. Díaz-Orejas, unpublished results), the dnaB-containing NdeI–BstXI fragment from pMLM123 was excised and replaced with the equivalent NdeI–BstXI fragment from pBRdnaB368, to give pMLM124. Next, the bulk of the dnaB gene was removed by cutting pMLM124 with ClaI and the remaining plasmid DNA was end-repaired using Klenow polymerase and ligated to a SmaI Ω fragment containing tet obtained from pHP45Ω–Tc (19), to give plasmid pMLM125.
Integration of the dnaB368::Ω–Tc construct into the chromosome. The multi-step procedure to substitute the dnaB368::Ω–Tc mutation present in pMLM125 for dnaB+ in the chromosome was performed as described by Cornet et al. (18). Briefly, pMLM125 was used to transform the streptomycin-resistant (SmR) strain MLM329. Integration of pMLM125 into the chromosome was selected by plating cells on chloramphenicol-containing medium at 42°C. Colonies obtained were cultured in liquid in the same medium and temperature conditions and were transformed with a pUC18 derivative carrying the dnaB and dnaC genes (pPS562) (Table 1). Transformants were then plated at 37°C on medium containing streptomycin, to select for the excision of dnaB from the chromosome (this event is tightly linked to the loss of the pMLM125 borne rpsL+ allele that confers sensitivity to streptomycin), and tetracycline, to select for the presence of dnaB368::Ω–Tc in the chromosome. This procedure yielded the MLM337 strain.
Construction of MLM368, the indicator strain for the complementation of the dnaB368::Ω–Tc null mutation. Strain MLM337 was transformed with plasmid pMLM121. Cells were then cured from the resident pPS562 plasmid by replica plating for several rounds at 30°C in solid medium without ampicillin. Chloramphenicol was included to maintain the pMLM121 plasmid. This yielded the MLM368 strain.
Amplification of the dnaB locus by PCR. Total DNA was extracted from MLM337 (and from its parental dnaB+ counterpart MLM329, as a control) and was used as template in polymerase chain reactions (PCR) with primers dnaB53 (5′-CTGCTGCTTCGGTGCCTAATC), dnaB33 (5′-CGGCGA GACGCCATAAAGAAT) in one series of reactions, and with dnaB53 and tet1 (5′-TAGGCGCCGCCCTATACCTTGTCT) in a second series. Identical conditions were used for all the reactions: 10 ng of chromosomal DNA as template, 200 pmol of each primer, DNA polymerase Pfu (Stratagene) and 25 cycles of 95°C for 45 s, 62°C for 1 min, 72°C for 7.5 min followed by 72°C for 10 min.
Immunoblot analysis using anti-DnaB serum. Cells were grown exponentially at 30°C in medium containing kanamycin. At an A600 of ∼0.3, cells were induced or not with arabinose (0.2% final concentration). After 15 min, samples were collected and resuspended in SDS–mercaptoethanol buffer (20) and subjected to electrophoresis in 15% SDS–polyacrylamide gels. Proteins were transferred to Sequi-Blot PVDF membranes (Biorad) in Tris–glycine buffer (25 mM Tris, 192 mM glycine, 0.1% SDS, 20% methanol) using a Trans-Blot Semi-dry transfer cell (Biorad) as recommended by the manufacturer. Membranes were treated with polyclonal anti-DnaB serum from sheep (1:2500 dilution) and bound antibodies were detected using an ECL Western Blotting detection kit (Amersham Pharmacia Biotech) as recommended by the manufacturer.
Purification of the Ban protein
A 4.8 l culture of SCS1(pTR101) was grown at 37°C with shaking. At an A600 of 0.5, isopropyl β-d-thiogalactopyranoside was added to 1 mM. Shaking was continued for 5 h. Cells were centrifuged at 4000 g for 10 min, resuspended in 1 mM spermidine tris(hydrochloride), 200 mM NaCl and 2 mM EDTA (pH 7.5) (1 g of wet cells in 5 ml) and frozen in liquid nitrogen. All the following steps were performed at 0–4°C. Frozen cells were thawed (15 g in 75 ml) and adjusted to 40 mM Tris–HCl pH 7.6, 4% sucrose, 0.13% Brij-58, 50 mM NaCl, 2.5 mM dithiothreitol (DTT) and 0.3 mg/ml lysozyme. Following incubation for 1 h, the highly viscous lysate was centrifuged at 100 000 g for 60 min. The pellet was washed with 1 M NaCl in buffer A (20 mM Tris–HCl pH 7.6, 2.5 mM DTT and 1 mM EDTA) by rigid homogenisation to remove most of the soluble proteins. Then the pellet was resuspended twice in buffer A containing 6 M urea and 1 M NaCl to dissolve Ban. The supernatants of both urea steps were combined, and the proteins were precipitated with ammonium sulphate at 55% saturation. The pellet was solubilised in buffer B [20 mM Tris–HCl pH 7.6, 1 mM DTT, 0.1 mM EDTA and 10% (wt/vol) glycerol] containing 6 M urea and 100 mM NaCl. Ban was renatured by dialysis against buffer B containing 100 mM NaCl (fraction I, 107 ml). Fraction I was applied at 80 ml/h to a DEAE Sephacel column (2.6 × 20 cm), equilibrated with buffer B containing 100 mM NaCl. The column was washed with 250 ml of buffer B 100 mM NaCl. Proteins were eluted with an 800 ml linear gradient of 0.1 to 0.8 M NaCl in buffer B. Ban eluted at 500 mM NaCl. Peak fractions were pooled (fraction II, 95 ml). A 20 ml portion of fraction II was adjusted to 5 mM MgCl2 and loaded at 10 ml/h onto an ATP agarose column (0.9 × 7 cm) equilibrated with buffer B containing 450 mM NaCl and 5 mM MgCl2. The column was washed first with 15 ml of the same buffer, and then with 15 ml 10 mM AMP and 10 mM MgCl2 in buffer B without glycerol. Ban was eluted by 10 mM sodium pyrophosphate and 5 mM MgCl2 in buffer B without glycerol. The protein was concentrated by dialysis against 20% (wt/vol) polyethylene glycol 20 000 and dialysed against 50% (wt/vol) glycerol in buffer B containing 5 mM MgCl2, and stored at –20°C for at least 2 years without loss of activity.
Ban protein was also purified from the dnaB null strain MLM371 under native conditions. Based on the procedure described above, the potential inclusion bodies were extensively extracted in the presence of 1 M NaCl that eluted part of the Ban protein. Phenyl-Sepharose was used instead of ATP-agarose to obtain nearly homogenous protein. The protein was eluted with a linear gradient of ethylene glycol (0–80%). The yield is ∼30% of that of the urea procedure.
Helicase and ATPase assays
A forked helicase substrate based on that of Crute et al. (21) was used. To viral M13mp18 DNA, a 5′-32P-labeled 53mer oligonucleotide was annealed, resulting in a double-stranded segment of 31 bp and 22 unpaired nucleotides at the 3′ end. Helicase reactions were performed essentially as described at 30°C for 20 min in 20 µl of buffer containing 20 mM Tris–HCl pH 7.6, 10 mM MgCl2, 1 mM DTT, 2 mM ATP and 50 µg/ml bovine serum albumin (BSA) (22). To study the pH dependency of the reaction, Tris–HCl was replaced by 40 mM MES–NaOH pH 5.5, or 40 mM HEPES–NaOH pH 9.0; 45 fmol of helicase substrate was used per assay. The products were separated by electrophoresis on 10% polyacrylamide gels in 89 mM Tris–borate pH 8.3, containing 1 mM EDTA at 8 V/cm. The radioactivity present in the substrate and the displaced oligonucleotide was visualised using phosphor storage technology and quantified with the ImageQuant software version 5.0 (Amersham Pharmacia Biotech). The values plotted are averaged from at least two independent experiments.
ATP hydrolysis reactions were performed for 15 min at 30°C as previously described (22) in 20 µl of helicase buffer (see above) containing 0.2 mM ATP, 100 nCi [γ-32P]ATP, 1 µg viral M13mp18 DNA and 50 µg/ml BSA. Products were separated by thin layer chromatography and quantified as described above.
DEAE-chromatography of Ban/DnaB mixtures
The chromatography was performed at 4°C. Equimolar amounts of purified Ban and DnaB were mixed in buffer B containing 100 mM NaCl, 5 mM ATP and 5 mM MgCl2 and incubated at 30°C for 2 h. Then the mixture was loaded onto a DEAE-Sephacel column (0.9 × 7 cm) equilibrated in buffer B containing 100 mM NaCl, 5 mM ATP and 5 mM MgCl2. The column was washed with the same buffer. Proteins were eluted with a linear gradient of 0.1 to 1 M NaCl in buffer B containing 1 mM ATP and 5 mM MgCl2. Fractions were collected and analysed on denaturing polyacrylamide gels. After staining the protein bands with Serva Blue R, the gel was scanned with a densitometer and the signal strength produced by Ban and by DnaB within each fraction was determined using the ImageQuant software version 5.0 (Amersham Pharmacia Biotech). The signal strength corresponds to the amount of protein present in a protein band. The ratio Ban/DnaB was determined by dividing the signal obtained for Ban by the signal for DnaB.
RESULTS
Nucleotide sequence of ban and the deduced primary structure of the protein
Both strands of ban were sequenced by cycle sequencing using appropriate primers. The complete ban sequence is published in the GenBank database under the accession number AJ011592. Ban encodes a product of 453 residues resulting in a calculated mass of 50.3 kDa and an isoelectric point of 4.83. The comparison of the Ban and the DnaB amino acid sequences deduced by the respective nucleotide sequences revealed 84% similarity and 78% identity. Even more highly conserved are the C-terminal thirds of both proteins, where identity reaches ∼85% (Fig. 1). Especially conserved are the five helicase motifs of the DnaB family of helicases. The high degree of conservation is followed by cross-reactivity of Ban with anti DnaB serum (23). The difference in length between DnaB and Ban (471 versus 453 residues) is mainly due to a stretch of 19 N-terminal residues of DnaB that are absent in Ban (Fig. 1). The genes are less conserved than the proteins (69% identity) and did not hybridise (A. Jakschik and E. Lanka, unpublished observations).
Figure 1.

Comparison of the Ban and DnaB primary structures. The amino acid sequences of Ban (upper) and DnaB (lower) were aligned by the programme GAP of the Wisconsin programme package version 10.2, Genetics Computer Group (GCG) Madison, WI. Identical residues are marked by dashes between the sequence lines, whereas colons and dots highlight evolutionary similar residues. The five helicase motifs conserved in the DnaB family are marked by bars. The position of the DnaB252 mutation (a Gly→Asp change at position 299) is denoted by an asterisk beneath the DnaB sequence.
Purified Ban is essentially free of DnaB and hexamerises
To purify the Ban protein, we wanted to ensure that the ban reading frame we had defined encoded an active protein. Therefore, the ban overexpression plasmid pTR101 (see Materials and Methods) was introduced into the dnaB70-ts strain WM0331 (24). Spotted on solid medium, WM0331 (pTR101) grew at 42°C, whereas WM0331 containing the vector plasmid was not viable (data not shown). This demonstrates that the encoded Ban protein suppressed the dnaB mutation and therefore is functionally active. Thus, pTR101 was used to overproduce and purify the protein (Materials and Methods; Fig. 2A). The N-terminus of purified Ban was determined to be SASPLESMP, demonstrating that the initial formyl-methionine is cleaved off post-translationally. Crucial in purification was to separate Ban from the accompanying DnaB, that might co-purify due to the high overall similarity of both proteins (see above), and therefore result in misleading data on Ban activity. The difference in molecular mass of 2 kDa between Ban and DnaB allowed for a distinction to be made in denaturing polyacrylamide gel electrophoresis (50.3 versus 52.3 kDa, corresponding to 453 and 471 residues). Since polyclonal anti DnaB serum crossreacts with Ban (23), the Ban preparation was analysed by immunoblotting for the presence of minute DnaB impurities that were not detectable by staining procedures. Two samples of purified Ban (35 and 350 ng) were analysed and compared to an extract of SCS1 cells that carried the vector plasmid. Even for the higher amount of Ban only a faint band of the electrophoretic mobility of DnaB reacted with the anti-DnaB serum (Fig. 2B). Most of the crossreacting proteins present in the Ban preparation were absent in the Ban-free cell extract (Fig. 2B, lanes a-c), indicating that our Ban preparation is essentially free of DnaB impurities.
Figure 2.

Purification of Ban protein. (A) Gel electrophoresis of Ban. Aliquots of Ban fractions (see Materials and Methods) were electrophoresed on a 15% polyacrylamide gel containing 0.1% SDS and stained with Serva Blue R after electrophoresis. Lane a, crude cell extract; lane b, fraction I; lane c, fraction II; lane d, fraction III; lane e, molecular mass standards. (B) Immunoblot analysis of purified Ban protein. After gel electrophoresis the proteins were electroblotted onto a nitrocellulose membrane, followed by reaction with anti DnaB sheep serum and dichlorotriazinylaminofluorescein-conjugated goat anti sheep IgGs. Lane a, 35 ng Ban; lane b, 350 ng Ban; lane c, 7.5 µl of crude extract of SCS1 cells without plasmid; lane d, molecular mass standards.
Ban probably formed inclusion bodies in the cell, which had to be dissolved in 6 M urea. Therefore we determined the oligomeric state of Ban and DnaB preparations via FPLC gel filtration (Fig. 3). Ban, DnaB and mixtures of both proteins chromatographed on the column with the molecular mass of 320 kDa. The data indicate that both proteins Ban and DnaB form hexamers in solution. The chromatographic behaviour was the same whether native or denatured/renatured material was used. As expected, treatment with 6 M urea and gel filtration in the presence of urea yielded monomeric molecules eluting at the size of BSA (data not shown).
Figure 3.

FPLC size exclusion chromatography of Ban, DnaB and a mixture of Ban and DnaB. Chromatography was performed at 4°C. 150 µg of each protein was loaded onto a Superdex 200 HR 10/30 gel filtration column (Amersham Biosciences) equilibrated with gel filtration buffer (20 mM Tris–HCl pH 7.6, 2% ethylene glycol, 20 mM NaCl, 2 mM MgCl2, 1 mM DTT and 1 mM ATP). The Ban–DnaB mixture was incubated at room temperature for 30 min prior to loading. Protein was eluted at a flow rate of 0.4 ml/min. Absorption was followed at 280 nm. To ensure that the absorption observed was due to protein, fractions were collected for analysis by gel electrophoresis. Size standards are: bovine thyroglobulin, 670 kDa; bovine γ-globulin, 158 kDa; chicken ovalbumin, 44 kDa; and horse myoglobin, 17 kDa. Lower panel: aliquots of peak fractions collected were analysed by electrophoresis on a 15% polyacrylamide gel containing 0.1% SDS. Following electrophoresis, the gel was stained with Serva Blue R. The leftmost lane shows the proteins loaded onto the column.
Ban is a DNA helicase
Since Ban was able to hydrolyse ATP (Fig. 4A), we also tested the protein for DNA unwinding. The helicase substrate was viral M13mp18 DNA with a double-stranded segment of 31 bp and 22 unpaired nucleotides at the 3′ end resembling a replication fork. In the presence of 5 pmol Ban (as monomers) in the reaction mixture corresponding to a concentration of 250 nM, >50% of the oligonucleotide was displaced from the substrate (Fig. 4B and C). At 500 nM Ban, unwinding reached a plateau. As expected, no activity was observed in the absence of nucleotide triphosphates. Ban exerts its highest activity at pH 7.6 (Fig. 4A and B). Reactions still took place at pH 9.0, at pH 5.5 the turnover was negligible. The ATPase activity of Ban was identical in the presence/absence of ssDNA indicating that there is no stimulation of the nucleotide hydrolysing activity. Helicase activity has been determined in the presence of each of the eight nucleotides. Ribonucleoside triphosphates proved to be the best low molecular weight substrates with ATP and GTP as the most efficient cofactors for unwinding. Hydrolysis of UTP and CTP was ∼2-fold lower. dNTPs were not suitable to fuel Ban, since unwinding occurred at a negligible low rate (data not shown). The results obtained with Ban prepared from inclusion bodies solubilised in 6 M urea and with the soluble Ban fraction were almost identical. Since the denaturation and renaturation procedure applied for solubilisation of the inclusion bodies yielded an enzymatically active protein, the functional Ban conformation was recovered reasonably well in the renaturation process.
Figure 4.

Enzymatic activities of Ban as a function of protein concentration and pH. Reactions were performed as outlined in Materials and Methods. (A) ATPase activity in the absence of ssDNA. Addition of ssDNA resulted in identical values for hydrolysis. (B) Helicase activity. In both (A) and (B) circles represent the amount of product obtained at pH 7.6, whereas squares and triangles give the values for pH 9.0 and 5.5, respectively. (C) Autoradiogram of a 10% polyacrylamide gel showing the substrate DNA and the oligonucleotide displaced that results from the unwinding activity at pH 7.6. Lanes 1–8 contain the pmol amounts of Ban given below the gel. Lane 8, heat-denatured helicase substrate. Reactions were performed as outlined in Materials and Methods.
Ban helicase substitutes for E.coli dnaB
Since existing strains still encode N-terminal DnaB remnants of more than 10 amino-acid (aa) residues that may have the ability to interact with the protein under study, we aimed to construct a strain in which DnaB would be completely absent, in order to demonstrate that Ban may substitute for DnaB in E.coli. Therefore, the dnaB gene was replaced by the dnaB368::Ω-Tc construct in the E.coli chromosome (Materials and Methods; Fig. 5A). In this construct, 690 bp of the dnaB sequence contained between the two ClaI sites were removed and replaced by an Ω fragment containing the tetracycline resistance gene, tet (19). This means that ∼50% of the dnaB coding capacity is missing, including the coding sequences for the H1 and H1a helicase motifs. In addition, we introduced the dnaB368 mutation that changes the 10th codon of the dnaB gene to an amber stop codon (M. Lemonnier and R. Díaz-Orejas, unpublished results). This additional precaution was taken to avoid the synthesis of an 81 aa N-terminal fragment of DnaB encoded by the remaining 5′ sequences of the dnaB open reading frame that could constitute a source of interference.
Figure 5.

Structure of the dnaB region on dnaB+ and dnaB368::Ω-Tc strains. (A) Maps of the dnaB and dnaB368::Ω-Tc chromosomes. The open box which disrupts the dnaB gene in dnaB368::Ω-Tc chromosome represents the Ω fragment containing tet (arrow denotes direction of transcription). The asterisk on the dnaB interrupted gene symbolises the dnaB368 amber mutation. Small open arrows represent the position and 5′/3′ orientation of primers used in PCR experiments (see B). Recognition sites for restriction enzymes used in the construction steps (Materials and Methods) are indicated: C, ClaI; H, HpaI; B, BamHI; Bs, BstXI; N, NdeI. (B) Reaction products obtained from PCR amplification of the dnaB region. Template DNAs were from MLM329 (dnaB+; lanes 1 and 2) or MLM337 (dnaB368::Ω-Tc; lanes 3 and 4). Oligonucleotide couples used were dnaB53/dnaB33 (lanes 1 and 3) or dnaB53/tet1 (lanes 2 and 4). Leftmost lane is a 1 kb marker.
The dnaB368::Ω-Tc construct was made on a plasmid and was crossed into the E.coli chromosome to yield the MLM337 strain (Materials and Methods). In this strain, the essential dnaB product was provided by a multicopy plasmid, pPS562. To confirm the structure of the dnaB368::Ω-Tc locus in the chromosome, total DNA was extracted from MLM337 and was used as template in a series of PCR reactions using oligonucleotides dnaB53, dnaB33 and tet1 (Materials and Methods; Fig. 5A). The PCR product obtained with the couple of primers dnaB53/dnaB33 was a 3866 bp fragment when MLM337 DNA was used as template, which clearly differed from the 2405 bp fragment obtained with DNA from the parental dnaB+ strain W3110 (Fig. 5B). With the couple of primers dnaB53/tet1, PCR made on MLM337 DNA yielded a 1023 bp fragment, while the same reaction on W3110 yielded no product, as expected because the tet1 oligonucleotide hybridises to DNA corresponding to the tet gene, absent in W3110. Note that none of the primers used here hybridises to sequences of the dnaB gene contained in plasmid pPS562 present in MLM337. Nucleotide sequences of the obtained PCR products were determined, which confirmed their identity (data not shown). The structure of the dnaB368::Ω-Tc mutation in MLM337 was also established in Southern blot experiments using a probe specific to dnaB (data not shown). These results confirmed the substitution of dnaB368::Ω-Tc for dnaB in MLM337.
Strain MLM337 was transformed with a replication temperature-sensitive plasmid, pMLM121, which bears the dnaB gene and the dominant rpsL+ allele that confers sensitivity to streptomycin, and was cured of the pPS562 plasmid, to yield strain MLM368 (Materials and Methods). This novel strain was designed to select directly for the capacity of any plasmid-borne gene to complement the absence of dnaB. This can be performed by (i) a transformation of the MLM368 strain with a candidate plasmid; and (ii) a selection for the loss of the resident pMLM121 plasmid, by plating on streptomycin-containing medium either at 42°C, a temperature that prohibits pMLM121 replication, or even at 30°C, because pMLM121 is unstable at this temperature due to its low copy number, and probably to leakage of the repA-ts mutation. Therefore, transformations of strain MLM368 were carried out with the multicopy expression vector pFUS2 (KmR) (13) or its derivatives carrying the dnaB gene (pMLM105) or the ban gene (pMLM115). Transformants were selected and purified on kanamycin-containing medium at 30°C. Colonies obtained were diluted in liquid medium and plated at 30°C on medium containing either kanamycin and streptomycin to select for cells which have lost the pMLM121 plasmid, or kanamycin alone to allow growth of all cells. Then, we measured the ratio between the number of colonies obtained in each medium, which reflects the frequency of accumulation of cells that lack the dnaB plasmid pMLM121, and thus also reflects the capacity of given cells to grow in the absence of dnaB (Fig. 6A). As expected, colonies containing the dnaB plasmid pMLM105 produced cells lacking the pMLM121 plasmid (MLM370 cells) at a given frequency: ∼50% of the cells which were plated did not contain pMLM121 (Fig. 6A, column 1), whereas colonies containing the pFUS2 vector produced streptomycin-resistant cells, if any, at a frequency below our detection level (<10–5; Fig. 6A, column 2). This indicates that dnaB368::Ω-Tc cells are not viable unless a dnaB copy is provided in trans. Moreover, growth rates of MLM370 and of the parental MLM329 (dnaB+ chromosome) cells were comparable both at 30°C (doubling times of 44 and 43.2 min, respectively) and 42°C (30 and 28.9 min; data not shown). Thus, normal growth of dnaB368::Ω-Tc cells depends solely on the presence of the pMLM105 dnaB copy. Hence, the dnaB368::Ω-Tc construct does not exert any polar interference on the expression of neighbouring chromosomal genes that could significantly contribute to growth inhibition.
Figure 6.
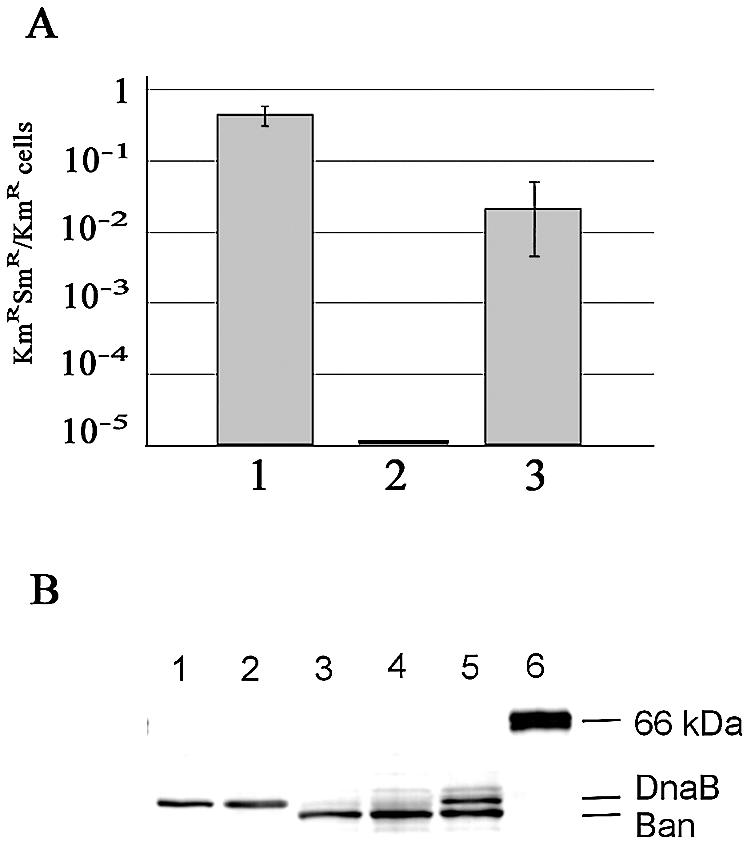
Loss of the dnaB gene in MLM369 (ban+) cells. (A) Frequency of appearance of cells that have lost dnaB. The number of colonies obtained on medium containing kanamycin and streptomycin over the number of colonies obtained on medium containing kanamycin alone was determined from cultures of MLM368 cells containing pMLM105 (dnaB+; column 1), pFUS2 (vector; column 2) or pMLM115 (ban; column 3) treated and plated as described in the Results. Error bars represent the standard deviation calculated from three independent experiments. (B) Western blot analysis of intracellular DnaB and Ban proteins following SDS–PAGE (10%). Samples were obtained as described in Materials and Methods. Lane 1, 10 ng of purified DnaB protein; lane 2, MLM329 (dnaB+) containing the pFUS2 vector; lane 3, MLM369 (dnaB–/ban+); lane 4, MLM369 (dnaB–/ban+) induced with 0.2% arabinose; lane 5, MLM329 (dnaB+) containing the pMLM115 (ban+) plasmid; lane 6, molecular mass standard BSA, 65 kDa.
The same analysis performed on colonies containing the ban plasmid pMLM115 showed that cells that had lost pMLM121 (MLM369 cells, genotype dnaB–/ban+) accumulated at a frequency of ∼10–2 (Fig. 6A, column 3). This difference, of about one order of magnitude compared to the pMLM105 situation (Fig. 6A, columns 1 and 3), can be attributed to the reduced growth rate of cells under the control of the Ban helicase (see next section), although the unlikely possibility that ban enhances the stability of the pMLM121 plasmid, and therefore delays its loss, cannot be completely ruled out. Nevertheless, these results indicated that the Ban helicase encoded by pMLM115 was able to support growth of E.coli cells in the absence of DnaB.
Loss of pMLM121 that carries the cat gene was confirmed by the sensitivity of MLM369 cells to chloramphenicol, and by purification and analysis of total cellular plasmid DNA. Additional evidence was provided by immunoblot experiments using anti-DnaB serum on crude protein extracts from MLM369 cells (Fig. 6B). Whereas DnaB protein was not detected (lanes 3–4), a lower molecular weight protein corresponding to Ban was observed. Moreover, the concentration of this protein increased in extracts of MLM369 in which ban expression from pMLM115 was induced by adding arabinose to the medium (compare lanes 3 and 4), confirming that this protein is Ban. Thus, the viability of the MLM369 strain demonstrates that, under the experimental conditions used, the Ban helicase is able to support growth of E.coli cells in the complete absence of the DnaB protein.
Ban and DnaB252 functionally interact in vivo
Next, we tested the old hypothesis that Ban and DnaB are able to interact and form mixed Ban:DnaB oligomers. Former experiments showed that P1bac mutants suppress the dnaB252-ts mutation (8,25). Furthermore, DnaB and Ban proteins were co-purified within a dnaB complementing fraction obtained from P1bac lysogens of dnaB mutants, among which was dnaB252 (9,25). This suggested that, at high temperatures, Ban could stabilise DnaB252 protomers, probably within a functional hetero-hexamer. Paradoxically, we found that growth of the MLM369 strain (dnaB–/ban+) was significantly reduced at 30°C (Fig. 7A) and dramatically impaired at 42°C (Fig. 7B), compared to its dnaB+ equivalent strain MLM370. Immediately after shifting cultures of MLM369 to 42°C increase in cell mass was blocked (Fig. 7B), and 4 h after the shift the viability had dropped by at least 1000-fold compared to MLM370 (data not shown). This result shows that Ban protein was either not functional or toxic at high temperatures in the absence of DnaB. Therefore, the reported observations of suppression of dnaB252 by ban could actually reflect the mutual stabilisation of both partner proteins through functional interaction. To test this hypothesis, dnaB252 and ban genes were placed under the control of distinct promoters and were expressed in dnaB368::Ω-Tc cells, alone or in combination. Cells that expressed dnaB252 or ban individually showed a marked decrease in viability (at least 1000-fold) at 42°C compared to cells expressing the wild-type dnaB gene (Fig. 8). In contrast, when both plasmids expressing dnaB252 and ban were present in the cells, viability was virtually restored. In the absence of IPTG, which induces ban expression, partial recovery of viability is observed (Fig. 8). This may reflect the production of a background level of Ban, due to promoter leakage, that is sufficient to form a few DnaB252:Ban functional mixed molecules. Our attempts to evaluate the Ban/DnaB252 molar ratio by immunodetection techniques were unsuccessful due to a fragment, presumably resulting from proteolytic cleavage of DnaB, that migrates at the very same position as Ban (see the faint band in Fig. 2B, lane C). Nevertheless, our experiments show that Ban and DnaB252 mutually compensate their deficiencies at high temperatures, and therefore functionally interact.
Figure 7.

Growth of the MLM369 (dnaB–/ban+) strain at 30 and 42°C. Single colonies of MLM369 (dnaB–/ban+, open circles) or MLM370 (dnaB+/ban–, filled circles) were picked and grown overnight at 30°C on LB medium with kanamycin. Cultures were then diluted in the same medium to an A600 of 0.02 to 0.04 and were split into two cultures; one was grown at 30°C (A) and the other at 42°C (B), both with the same shaking and aeration conditions. Samples were removed every hour to measure optical density at a wavelength of 600 nm. The graphics show a typical pattern, which was reproduced in three independent experiments.
Figure 8.

Reciprocal complementation of dnaB252 and ban. Cells were grown at 30°C (left panel) or 42°C (right panel) on LB medium containing kanamycin and ampicillin as described in Figure 7. IPTG (10 µM, final concentration) was present in all the cultures except d (see below). After 4 h of growth, 4 µl of serial 100-fold dilutions of the cultures were spotted on solid medium without IPTG and were incubated for 18 h at 30°C. All the strains were MLM329 dnaB368::Ω-Tc derivatives containing, in each case, the following pairs of compatible plasmids: (a) pMLM105 (pMB1 replicon, KmR, dnaB) + pMOM01 (p15A replicon, ApR vector); (b) pMLM117 (p15A replicon, ApR, ban) + pFUS2 (pMB1 replicon, KmR vector); (c) pMLM116 (pMB1 replicon, KmR, dnaB252) + pMOM01; (d) and (e) pMLM117 + pMLM116.
Ban and DnaB interaction in vitro
In an additional approach to test the hetero-oligomer hypothesis, we made use of the observation that Ban elutes at 500 mM NaCl from a DEAE column, whereas DnaB elutes at 450 mM (Fig. 9A). If both proteins are within the same oligomer, elution at an intermediate salt concentration is expected, e.g. the separated elution peaks of Ban and DnaB are expected to merge into a single elution maximum inbetween 450 and 500 mM NaCl caused by the mixed complex. Therefore, equimolar amounts of Ban and DnaB were mixed, incubated at 30°C for 2 h to allow for potential exchange of monomers, and then subjected to DEAE chromatography (see Materials and Methods). The elution profile of the Ban–DnaB mixture was similar to that of each single protein (Fig. 9B), and the ratio Ban/DnaB present in each fraction increased during elution, indicating that no prominent exchange of monomers between the Ban and DnaB oligomers occurred. Therefore, in a second attempt, the proteins of the equimolar mixture were denatured by adding urea, and renatured by dialysis. Then the mixture was treated as above. The elution profile of Ban and of DnaB was now identical with the bulk of protein eluting at 480 mM NaCl (Fig. 9C). This is in-between the values for each protein alone. The ratio Ban/DnaB during elution was constant at ∼1.1 (Fig. 9C), and the majority of protein eluted within a narrow range of salt concentration compared to the non-denatured mixture and each single protein. The results demonstrate that, in vitro, Ban and DnaB are able to interact and that within the complex the molar ratio of both polypeptides is ∼1.
Figure 9.

DEAE chromatography of Ban and DnaB. A mixture of equimolar amounts of Ban and DnaB was loaded onto a column of DEAE–sephacel equilibrated with buffer B containing 100 mM NaCl, 1 mM ATP and 5 mM MgCl2. Proteins were eluted with a linear gradient of 0.1–1 M NaCl and fractions were collected. Aliquots of each fraction were analysed by polyacrylamide gel electrophoresis. The relative amount of Ban and DnaB present in each fraction was determined and the ratio Ban/DnaB was calculated (see Materials and Methods). The relative amounts of proteins in the fractions were plotted against the NaCl concentration. (A) Individual chromatography of Ban and DnaB. (B) Ban and DnaB protein were mixed (see Materials and Methods) and then loaded onto the column. (C) Prior to loading, the protein mixture was denatured in 6 M urea and subsequently renatured by dialysis. The dashed black line and the black line represent the elution profiles of Ban and DnaB, respectively. The bold dashed line in (B) and (C) gives the ratio Ban/DnaB in the fractions collected.
DISCUSSION
The bacteriophage P1 ban gene product shares 84% similarity and 78% identity with the DnaB protein of E.coli. The purified Ban protein hydrolyses ATP, forms hexamers and unwinds duplex DNA in the presence of ATP and Mg2+ ions. Furthermore, Ban is able to sustain growth of an E.coli dnaB null strain. Altogether, these data demonstrate that Ban is a DNA helicase that substitutes for DnaB in the host replication machinery.
Only two further phage-encoded helicases, the Ban protein of phage P7 and the gene 12 protein of phage P22, have been shown to complement dnaB deficiencies in E.coli (3,4). While P1 and P7 Ban proteins share 98% identity (Malgorzata Lobocka, Polish Academy of Sciences, Warsaw, personal communication), P22 gene 12 protein exhibits <35% identity and 55% similarity with DnaB or Ban proteins (4; our unpublished observations). A similar situation is observed in the case of the IncI plasmid-encoded conjugative primases, which efficiently suppress E.coli dnaG mutations despite a low similarity in primary structure (26). Therefore, sequence conservation between Ban and DnaB might not be as crucial as the ability of Ban to properly interact with other replication proteins. With respect to this, the dnaB null strain MLM368 described in this work is an ideal tool to directly assay further bacterial replicative helicases, either phage or chromosome-encoded, for their ability to substitute for DnaB.
Our results show that Ban only partially complements the dnaB null mutation, as indicated by the decreased growth rate at 30°C and the complete growth block observed at 42°C. This suggests that Ban fits into the host replication machinery at low temperatures, but that the formation of Ban hexamers, or their interactions with host replication factors, are thermo-labile. On the other hand, the recently proposed involvement of DnaB in DNA recombination in vivo raises the possibility that Ban protein either could be deficient in recombination, or escape an essential regulatory control, thus leading to cell death under certain circumstances (2). Exposure of dnaB-ts mutants to non-permissive temperatures results in the formation of double-strand breaks (27). Furthermore, overexpression of dnaB leads to increased illegitimate recombination levels in E.coli, a phenomenon suggested to be linked to DnaB recombination functions (2,28). Whether the Ban-dependent death at high temperatures that is observed in the absence of DnaB is a reflection of deficient interaction with other replication proteins or a consequence of massive formation of double-strand breaks is an issue that remains to be addressed. Certainly, a future search for potential suppressors of MLM369 thermo-sensitive growth could lead to the isolation of mutations, either in the ban gene or in the host chromosome, which would enlighten the nature of Ban interactions in vivo.
Our results confirm the suppression of the dnaB252 thermo-sensitive mutation by Ban, reported earlier (8,25). In addition, we observed that the growth defect of Ban+/DnaB– cells at high temperatures is compensated by the overproduction of the DnaB252 mutant protein. The DnaB252 mutation, a G299E amino-acid substitution (Fig. 1), is unique in several features: (i) it is the only dnaB mutation described so far that affects initiation of replication and not elongation (25,29); (ii) the biochemical properties of purified DnaB252 protein, i.e. in vitro helicase, ATPase, ssDNA binding activities, are indistinguishable from those of wild-type DnaB, even at 42°C (24); and (iii) the dnaB252 mutation does not influence the effect of DnaB on UV-dependent illegitimate recombination (30). Moreover, overproduction of the E.coli helicase loader DnaC completely suppresses the dnaB252 mutation (31). Altogether, these data suggest that the DnaB252 protein is, from an enzymatical point of view, a fully proficient DnaB helicase. Therefore, the in vivo phenotype of dnaB252 could reflect altered DnaB–DnaC interactions and, as a consequence, deficient loading of the helicase into the replication initiation complex. Furthermore, Ban was shown to suppress several E.coli dnaC mutations (32). In light of these observations and of our results reported here, we propose that Ban and DnaB252 form functional hetero-hexamers in vivo in which Ban protomers provide an efficient interface of interaction with the DnaC loader. Further support to this hypothesis is supplied by: (i) Ban conserves the glycine residue that is changed in DnaB252; (ii) this residue is located in the C-terminal region, especially conserved between Ban and DnaB; (iii) this region corresponds to the C-terminal domain of DnaB, the likely interface of interaction with DnaC, as shown by cryo-electron microscopy studies (33).
Although our results are the first report of a mutual compensation of Ban and DnaB deficiencies in vivo, negative and positive functional interactions between Ban and several dnaB alleles have been reported (5–8; this work). Hence, there is no reason to assume that the Ban/DnaB252 interaction is allele specific. Associated wild-type DnaB and Ban proteins have been purified from P1 lysogen extracts (9). Furthermore, in this study we have shown that upon denaturation and renaturation of a mixture of both purified proteins, Ban and DnaB co-elute from a DEAE column at salt concentrations inbetween those observed for each protein alone, thus indicating formation of hetero-oligomers. Within these complexes, we found that the Ban/DnaB ratio was ∼1.1. What could be the nature of these hetero-oligomers? One possibility is that they form single hetero-hexamers. The observed Ban/DnaB ratio indicates an overall 1:1 ratio (3 Ban + 3 DnaB in the hexamer), although the whole range of possibilities leading to a 1:1 ratio might occur (Ban0–DnaB6, Ban1–DnaB5, …, Ban6–DnaB0). From our results we cannot conclude whether hetero-dimers are formed in a first step, which then assemble to hexamers, or whether hexamerisation takes place via other mechanisms. Alternatively, dimers of hexamers could be formed, i.e. dimers of one Ban homo-hexamer plus one DnaB homo-hexamer, or even dimers of hetero-hexamers, as long as the overall Ban/DnaB ratio is 1:1. However, the existence of such double-hexamers is highly unlikely, since in the size exclusion chromatography no material of a molecular mass higher than 320 kDa was observed. We assume that the steps of denaturation and renaturation of the mixture required to detect hetero-oligomer formation probably reflect the in vivo situation in so far that each monomer released from the ribosome has to assemble into an oligomer.
Concerning the rather puzzling issue of the biological significance of an exchange of subunits between Ban and DnaB, we need to consider the possibility that our experimental set-ups, and those of former studies, make use of artificial overproduction of Ban and DnaB proteins. Furthermore, the ban gene is normally repressed by the P1 phage C1 repressor in the prophage state, and therefore wild-type P1 lysogens are not able to compensate for dnaB deficiencies (5,34). P1 also encodes analogues to the E.coli SSB, UmuD′ and Tau proteins, but their precise biological role in the life cycle of the prophage is uncertain (35,36; Hansjörg Lehnherr, Ernst-Moritz-Arndt University, Greifswald, personal communication). However, recent studies on the P1 encoded single-stranded DNA-binding protein (SSB-P1), that shares 66% similarity with E.coli SSB, have provided some clues as to how P1 genes are expressed and interact with their E.coli functional homologues (35). These studies showed that: (i) like many plasmid-encoded SSB proteins, the phage-encoded SSB-P1 is able to substitute for the essential SSB protein of E.coli; (ii) P1 ssb gene is transcribed exclusively during stationary-phase growth in E.coli; and (iii) although P1 ssb is dispensable for phage growth during exponential-phase growth, it provides P1 with a selective advantage when exposed to stationary-phase host cells (35). Moreover, P1 ssb and ban genes are controlled by the same P1 regulator, Bof, suggesting that both genes could share a common regulatory pathway, albeit with distinct effects (Bof positively regulates ban and negatively regulates ssb; 34). Therefore, it is possible that under certain physiological conditions that could down-regulate dnaB expression and trigger ban expression, Ban could interact with the available DnaB molecules, and ultimately replace them to form functional hexamers for the mutual benefit of phage and host growth. Certainly, addressing this issue will require a new experimental focus on how dnaB and ban genes are expressed during the growth of bacterial P1 lysogens.
Acknowledgments
ACKNOWLEDGEMENTS
We thank F. Cornet (CNRS, Toulouse), J. Frey (University of Bern), N. Dixon (Australian National University, Canberra) and M. Berlyn (CGSC, New Haven, CT) for sending strains and plasmids, and C. Pardo-Abarrio and M. Schlicht for excellent technical assistance. This work was supported by grants from the European Commission grant QLRT-1999-30634 to R.D.-O. and E.L. R.D.-O. acknowledges support by the ‘Programa de Grupos Estrategicos’ (C.A.M.) 2000–2003 and grant BIO99-0859-CO1 from the Spanish MEC.
DDBJ/EMBL/GenBank accession no. AJ011592
REFERENCES
- 1.Lohman T.M. and Bjornson,K.P. (1996) Mechanisms of helicase-catalysed DNA unwinding. Annu. Rev. Biochem., 65, 169–214. [DOI] [PubMed] [Google Scholar]
- 2.Kaplan D.L. and O’Donnell,M. (2002) DnaB drives DNA branch migration and dislodges proteins while encircling two DNA strands. Mol. Cell, 10, 647–657. [DOI] [PubMed] [Google Scholar]
- 3.Selvaraj G. and Iyer,V.N. (1980) A dnaB analog function specified by bacteriophage P7 and its comparison to the similar function specified by bacteriophage P1. Mol. Gen. Genet., 178, 561–566. [DOI] [PubMed] [Google Scholar]
- 4.Backhaus H. and Petri,J.B. (1984) Sequence analysis of a region from the early right operon in phage P22 including the replication genes 18 and 12. Gene, 32, 289–303. [DOI] [PubMed] [Google Scholar]
- 5.D’Ari R., Jaffé-Brachet,A., Touati-Schwartz,D. and Yarmolinsky,M.B. (1975) A dnaB analog specified by bacteriophage P1. J. Mol. Biol., 94, 341–366. [DOI] [PubMed] [Google Scholar]
- 6.Ogawa T. (1975) Analysis of the dnaB function of Escherichia coli K12 and the dnaB-like function of P1 prophage. J. Mol. Biol., 94, 327–340. [DOI] [PubMed] [Google Scholar]
- 7.Sclafani R.A., Wechsler,J.A. and Schuster,H. (1981) The isolation and characterization of Escherichia coli dnaB::Tn10 insertion mutations. Mol. Gen. Genet., 182, 112–118. [DOI] [PubMed] [Google Scholar]
- 8.Touati-Schwartz D. (1979) A dnaB analog ban, specified by bacteriophage P1: genetic and physiological evidence for functional analogy and interactions between the two products. Mol. Gen. Genet., 174, 173–188. [DOI] [PubMed] [Google Scholar]
- 9.Lanka E., Mikolajczyk,M., Schlicht,M. and Schuster,H. (1978) Association of the prophage P1ban protein with the dnaB protein of Escherichia coli. J. Biol. Chem., 253, 4746–4753. [PubMed] [Google Scholar]
- 10.Kondo E. and Mitsuhashi,S. (1966) Drug resistance of enteric bacteria. VI. Introduction of bacteriophage P1CM into Salmonella typhi and formation of PldCM and F-CM elements. J. Bacteriol., 91, 1787–1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ziegelin G., Linderoth,N.A., Calendar,R. and Lanka,E. (1995). Domain structure of phage P4 α protein deduced by mutational analysis. J. Bacteriol., 177, 4333–4341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miller J.H. (1972) In: Experiments in Molecular Genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 352–355. [Google Scholar]
- 13.Lemonnier M., Bouet,J.-Y., Libante,V. and Lane,D. (2000) Disruption of the F plasmid partition complex in vivo by partition protein SopA. Mol. Microbiol., 38, 493–503. [DOI] [PubMed] [Google Scholar]
- 14.Potrykus K., Santos,S., Lemonnier,M., Díaz-Orejas,R. and Wegrzyn,G. (2002) Differential effects of Kid toxin on two modes of replication of lambdoid plasmids suggest that this toxin acts before, but not after, the assembly of the replication complex. Microbiology, 148, 2489–2495. [DOI] [PubMed] [Google Scholar]
- 15.Lanzer M. and Bujard,H. (1988) Promoters largely determine the efficiency of repressor action. Proc. Natl Acad. Sci. USA, 85, 8973–8977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Balzer D., Ziegelin,G., Pansegrau,W., Kruft,V. and Lanka,E. (1992) KorB protein of promiscuous plasmid RP4 recognizes inverted sequence repetitions in regions essential for conjugative plasmid transfer. Nucleic Acids Res., 20, 1851–1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chang A.C. and Cohen,S.N. (1978) Construction and characterization of amplifiable multicopy DNA cloning vehicles derived from the P15A cryptic miniplasmid. J. Bacteriol., 134, 1141–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cornet F., Louarn,J., Patte,J. and Louarn,J.M. (1996) Restriction of the activity of the recombination site dif to a small zone of the Escherichia coli chromosome. Genes Dev., 10, 1152–1161. [DOI] [PubMed] [Google Scholar]
- 19.Fellay R., Frey,J. and Krisch,H. (1987) Interposon mutagenesis of soil and water bacteria: a family of DNA fragments designed for in vitro insertional mutagenesis of gram-negative bacteria. Gene, 52, 147–154. [DOI] [PubMed] [Google Scholar]
- 20.Laemmli U.K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature, 227, 680–685. [DOI] [PubMed] [Google Scholar]
- 21.Crute J.J., Mocarski,E.S. and Lehman,I.R. (1988) A DNA helicase induced by herpes simplex virus type 1. Nucleic Acids Res., 16, 6585–6596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Scherzinger E., Ziegelin,G., Bárcena,M., Carazo,J.M., Lurz,R. and Lanka,E. (1997) The RepA protein of plasmid RSF1010 is a replicative DNA helicase. J. Biol. Chem., 272, 30228–30236. [DOI] [PubMed] [Google Scholar]
- 23.Edelbluth C., Lanka,E., von der Hude,W., Mikolajczyk,M. and Schuster,H. (1979) Association of the prophage P1ban protein with the dnaB protein of Escherichia coli. Overproduction of ban protein by a P1 bac crr mutant. Eur. J. Biochem., 96, 427–435. [DOI] [PubMed] [Google Scholar]
- 24.Saluja D. and Godson,G.N. (1995) Biochemical characterization of Escherichia coli temperature-sensitive dnaB mutants dnaB8, dnaB252, dnaB70, dnaB43, and dnaB454. J. Bacteriol., 177, 1104–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lanka E., Geschke,B. and Schuster,H. (1978) Escherichia coli dnaB mutant defective in DNA initiation: Isolation and properties of dnaB protein. Proc. Natl Acad. Sci. USA, 75, 799–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wilkins B.M. (1975) Partial suppression of the phenotype of Escherichia coli K-12 dnaG mutants by some I-like conjugative plasmids. J. Bacteriol., 122, 899–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Michel B., Ehrlich,S. and Uzest,M. (1997) DNA double-strand breaks caused by replication arrest. EMBO J., 16, 430–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yamashita T., Hanada,K., Iwasaki,M., Yamaguchi,H. and Ikeda,H. (1999) Illegitimate recombination induced by overproduction of DnaB helicase in Escherichia coli. J. Bacteriol., 181, 4549–4553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zyskind J.W. and Smith,D.W. (1977) Novel Escherichia coli dnaB mutant: direct involvement of the dnaB252 gene product in the synthesis of an origin-ribonucleic acid species during initiation of a round of deoxyribonucleic acid replication. J. Bacteriol., 129, 1476–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hanada K., Yamashita,T., Shobuike,Y. and Ikeda,H. (2001) Role of DnaB helicase in UV-induced illegitimate recombination in Escherichia coli. J. Bacteriol., 183, 4964–4969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sclafani R.A. and Wechsler,J.A. (1981) Deoxyribonucleic acid initiation mutation dnaB252 is suppressed by elevated dnaC+ gene dosage. J. Bacteriol., 146, 418–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sclafani R.A. and Wechsler,J.A. (1981) Suppression of dnaC alleles by the dnaB analog (ban protein) of bacteriophage P1. J. Bacteriol., 146, 321–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bárcena M., Ruiz,T., Donate,L.E., Brown,S.E., Dixon,N.E., Radermacher,M. and Carazo,J.M. (2001) The DnaB–DnaC complex: a structure based on dimers assembled around an occluded channel. EMBO J., 20, 1462–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schaefer T.S. and Hays,J.B. (1991) Bacteriophage P1 Bof protein is an indirect positive effector of transcription of the phage bac-1 ban gene in some circumstances and a direct negative effector in other circumstances. J. Bacteriol., 173, 6469–6474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bendsten J.D., Nilsson,A.S. and Lehnherr,H. (2002) Phylogenetic and functional analysis of the bacteriophage P1 single-stranded DNA-binding protein. J. Virol., 76, 9695–9701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McLenigan M.P., Kulaeva,O.I., Ennis,D.G., Levine,A.S. and Woodgate,R. (1999) The bacteriophage P1 HumD protein is a functional homolog of the prokaryotic UmuD′-like proteins and facilitates SOS mutagenesis in Escherichia coli. J. Bacteriol., 181, 7005–7013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cornet F., Mortier,I., Patte,J. and Louarn,J.M. (1994) Plasmid pSC101 harbors a recombination site, psi, which is able to resolve plasmid multimers and to substitute for the analogous chromosomal Escherichia coli site dif. J. Bacteriol., 176, 3188–3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bachmann B.J. (1972) Pedigrees of some mutant strains of Escherichia coli K-12. Bacteriol. Rev., 36, 525–557. [DOI] [PMC free article] [PubMed] [Google Scholar]