

Abstract
Clostridium perfringens enterotoxin (CPE), a 35-kDa polypeptide, induces cytotoxic effects in the enterocyte-like CaCo-2 cell culture model. To identify the mammalian cell death pathway(s) mediating CPE-induced cell death, CaCo-2 cultures were treated with either 1 or 10 μg of CPE per ml. Both CPE doses were found to induce morphological damage and DNA cleavage in CaCo-2 cells. The oncosis inhibitor glycine, but not a broad-spectrum caspase inhibitor, was able to transiently block both of those pathological effects in CaCo-2 cells treated with the higher, but not the lower, CPE dose. Conversely, a caspase 3/7 inhibitor (but not glycine or a caspase 1 inhibitor) blocked morphological damage and DNA cleavage in CaCo-2 cells treated with the lower, but not the higher, CPE dose. Collectively, these results indicate that lower CPE doses cause caspase 3/7-dependent apoptosis, while higher CPE doses induce oncosis. Apoptosis caused by the lower CPE dose was shown to proceed via a classical pathway involving mitochondrial membrane depolarization and cytochrome c release. As the CPE concentrations used in this study for demonstrating apoptosis and oncosis have pathophysiologic relevance, these results suggest that both oncosis and apoptosis may occur in the intestines during CPE-associated gastrointestinal disease.
Approximately 5% of Clostridium perfringens isolates produce a 35-kDa single polypeptide named C. perfringens enterotoxin (CPE). CPE is responsible for most (if not all) gastrointestinal (GI) symptoms of C. perfringens type A food poisoning (44), the third most commonly identified food-borne disease in developed countries (24, 26). Enterotoxin-producing C. perfringens type A isolates also cause up to 5 to 20% of all cases of non-food-borne human GI disease (e.g., antibiotic-associated diarrhea) and certain veterinary diarrheas (26, 48).
CPE has a potent but complicated cytotoxic action that begins with binding of this toxin to protein receptors (31, 32, 46, 47, 54, 55). Those receptors, which possibly include certain members of the claudin family of tight-junction proteins (14, 15, 49), are present on the apical surface of enterocytes or polarized enterocyte-like cell lines such as CaCo-2 cells. Once bound, CPE localizes in an ∼155-kDa, sodium dodecyl sulfate (SDS)-resistant plasma membrane complex whose formation rapidly permeabilizes the CPE-treated cell to molecules of <∼300 Da (27, 46, 47). That effect disrupts the normal cellular colloid-osmotic equilibrium (25, 29). It also induces morphological damage (22, 28) that permits other (still unbound) CPE molecules access to the basolateral surface of epithelial cells, where the toxin then interacts with occludin (another tight-junction protein) to form an ∼200-kDa plasma membrane complex (46, 47). Formation of this second, larger SDS-resistant CPE complex triggers the internalization of occludin, possibly along with CPE and certain claudins (46, 47), an effect causing paracellular permeability changes that may contribute to CPE-induced diarrhea (43, 49).
The cytotoxic action of CPE produces substantial histopathologic damage in the small intestines of rabbit models (33-37, 45). This damage, which includes epithelial desquamation and villus shortening developing within 30 min of higher-dose CPE treatment, appears to play a key role in initiating CPE-induced intestinal fluid and electrolyte transport changes. For example, a close temporal correlation has been demonstrated between the development of histopathologic damage and the onset of intestinal fluid and electrolyte transport effects in the CPE-treated rabbit ileum (45). Additionally, only those CPE doses causing intestinal damage can induce fluid and electrolyte transport alterations in the rabbit ileum (36).
The cell death pathway(s) by which CPE kills enterocytes to produce intestinal damage and subsequent GI symptoms has not yet been evaluated. The literature now recognizes two major categories of mammalian cell death (18, 39), apoptosis and oncosis (often incorrectly referred to as necrosis [18, 19, 39]). Apoptosis, or programmed cell death, typically involves (i) activation of specific proteases, (ii) membrane bleb formation, (iii) nuclear and chromatin condensation, and (iv) cleavage of DNA into fragments of 200-bp increments (11, 50). Increasingly appreciated as a diverse process, apoptosis sometimes involves mitochondrial release of cytochrome c to activate the caspase family of cysteine proteases (17, 40). Apoptotic cells are easily phagocytosed and thus generate minimal inflammation (20, 50).
In contrast, oncosis is an “accidental” death process occurring when cells are exposed to extreme insults (3, 5, 20, 50). Oncosis results in a chaotic breakdown of cellular integrity and is typically accompanied by (i) random DNA degradation; (ii) significant plasma membrane damage, which can be transiently inhibited by extracellular glycine; and (iii) organelle and cell swelling, sometimes with membrane blebbing (21, 23). Although caspase activation is not traditionally considered a component of oncosis, a caspase 1-dependent oncosis form referred to as pyrotosis has recently been identified (5, 6). In contrast to apoptosis, oncosis is a proinflammatory process (5, 50).
To better understand the pathophysiologic effects of CPE, we now report studies evaluating the death pathway(s) by which this enterotoxin kills the polarized human enterocyte-like CaCo-2 cell line, which is a commonly used in vitro model for studying CPE action (46, 47).
MATERIALS AND METHODS
Materials.
CPE was purified to homogeneity from C. perfringens strain NCTC8239 as previously described (38). Aliquots (2 mg) of this purified native CPE were radiolabeled by using lactoperoxidase glucose oxidase (Bio-Rad) and 2 mCi of Na125I (specific activity, 17 mCi/mg; ICN Radiochemicals) (31). By utilizing their vector-encoded His6 sequences, recombinant CPE (rCPE) and the G49D rCPE mutant (which is receptor binding capable but noncytotoxic) were enriched by metal affinity chromatography (16).
The cell-permeative broad-spectrum caspase inhibitor Z-VAD-FMK, cell-permeative caspase 3/7 inhibitor DEVD-CHO, cell-permeative caspase 1 inhibitor YVAD-CHO, caspase 1 colorimetric substrate Ac-YVAD-pNA, and caspase 3/7 colorimetric substrate Ac-DEVD-pNA were purchased from Biomol. Other chemicals were purchased from Sigma, unless otherwise noted.
Cell culture.
CaCo-2 cells were routinely maintained as previously described (46, 47) and, unless otherwise noted, inoculated at 700,000 cells/60-mm2 petri dish (Falcon) for experiments. Those cells were then grown to confluency (∼5 to 6 days).
Pretreatment of CaCo-2 cell cultures with apoptosis or oncosis inhibitors.
For pretreatment with a caspase or oncosis inhibitor, confluent CaCo-2 cultures were washed twice with warm Hanks balanced salts solution containing 1.7 mM Ca2+ and 1.8 mM Mg2+ (HBSS). Those washed cultures were then incubated for 2 h at 37°C in HBSS that contained or did not contain a 50 μM final concentration of a specified caspase inhibitor (i.e., Z-VAD-FMK, DEVD-CHO, or YVAD-CHO) or a 5 mM final concentration of the oncosis inhibitor glycine. These concentrations of caspase and oncosis inhibitors are proven effective dosages for inhibiting apoptosis and oncosis, respectively (5, 7, 40).
CPE treatment of CaCo-2 cultures.
CaCo-2 cultures that had or had not been subjected to inhibitor pretreatment (as described above) were washed twice with warm HBSS and then treated for 30 or 60 min at 37°C in 2 ml of HBSS containing either 1 or 10 μg of CPE per ml. For cultures that had been previously pretreated with a caspase or oncosis inhibitor, the same concentration of inhibitor present during pretreatment was also included in the CPE treatment buffer.
Cells remaining adherent after CPE treatment were detached with Versene for 10 min at room temperature (RT). The detached cells were combined with any nonadherent cells spontaneously released from the same petri dish during the treatment period or collected during subsequent washings. The collected cells were then quantified with a hemocytometer.
Analysis of cellular and nuclear morphology.
Adherent and released cells from CPE-treated CaCo-2 cultures were collected separately and examined by phase-contrast photomicroscopy with a Nikon Diaphot microscope. To examine nuclear morphology, adherent and released cells from each CPE-treated petri dish were separately washed with HBSS and then fixed with 3% paraformaldehyde for 20 min before permeabilization for 15 min with 0.2% Triton X-100. The permeabilized cells were washed with phosphate-buffered saline containing 0.5 mM Ca2+ and 0.5 mM Mg2+ (PBS) and treated with 0.5 μg of 4,6-diamino-2-phenylindole (DAPI) per ml for 1 h to visualize nuclei with a Nikon Diaphot fluorescence microscope and a DAPI filter.
DNA cleavage analysis.
CaCo-2 cells collected from each CPE-treated (as described above) petri dish were centrifuged, and the resultant pellet was washed once with HBSS and once with PBS. An aliquot (360 μl) of lysis buffer (10 mM Tris-Cl [pH 7.4], 150 mM NaCl, 10 mM EDTA, 0.4 37 SDS), along with 40 μl of proteinase K (final concentration, 1 μg/μl; Gibco), was added to each cell pellet. The mixtures were incubated at 65°C for 15 min, followed by overnight incubation at 37°C. The resultant lysates were microcentrifuged for 5 min, and each supernatant was then treated with RNase A (final concentration, 0.25 μg/μl; Roche) for 90 min at 4°C. DNA was extracted from each lysate by using phenol-chloroform (1:1) and then precipitated with 3 M sodium acetate and ethanol. Each precipitated DNA was washed once with 80% ethanol and air dried. These dried DNAs were individually dissolved in 100 μl of TE (10 mM Tris-Cl, 1 mM EDTA [pH 8.0]) and then quantified with a spectrophotometer (Smartspec; Bio-Rad). An aliquot (20 μg) of each DNA preparation was electrophoresed in a 2% agarose gel at 30 V for 7 h. After being stained with ethidium bromide, the gel was viewed by using the Bio-Rad Fluro-S Gel documentation system.
In specified DNA cleavage experiments, CaCo-2 cultures pretreated with a caspase or oncosis inhibitor were washed twice with warm HBSS before treatment for 8 h at 37°C with either 2 μM staurosporine or 5 μM ionomycin. These treatments were performed in the presence of the same concentration of the caspase or oncosis inhibitor (if any) used to pretreat those samples.
Analysis of CPE large-complex formation.
Formation of CPE-containing large complexes was evaluated as previously described (46, 47). Briefly, CPE-treated CaCo-2 cells collected by centrifugation were washed once with PBS. An aliquot (2 × 106 cells) of the washed cells was then resuspended in 5 μl of Benzonase (25 U/μl; Novagen)-45 μl of PBS, and then 50 μl of 2× SDS sample buffer (2% SDS, 10% glycerol, 0.02% bromophenol blue, 60 mM Tris [pH 6.8], without β-mercaptoethanol) was added to the mixture. After 10 min at RT, the mixture was electrophoresed (without sample boiling) overnight on a 4% acrylamide gel containing SDS. Separated proteins were electrotransfered onto a nitrocellulose membrane (Bio-Rad) and subjected to Western immunoblotting with anti-CPE rabbit polyclonal antibody and goat anti-rabbit horseradish peroxidase-conjugated secondary antibody. Immunoreactivity was detected by using WestPico chemiluminescent substrate (Pierce).
Evaluation of 125I-CPE-specific binding to CaCo-2 cultures.
CaCo-2 cultures that had or had not been pretreated with a caspase or oncosis inhibitor (as described above) were washed twice with warm HBSS and then treated with 2 ml of HBSS containing 1 μg of 125I-CPE that did (to determine nonspecific binding) or did not (to determine total binding) also contain a 200-fold excess of native CPE. This binding buffer also contained the same concentration of caspase or oncosis inhibitor present (if any) during sample pretreatment. After 15 min of incubation at 37°C, the 125I-CPE-treated cultures were washed twice with warm HBSS to remove unbound CPE, and adherent cells were detached for 10 min at RT with Versene solution. The detached cells were combined with any nonadherent cells spontaneously released from the same petri dish during CPE treatment or subsequent washings. The combined cells from each petri dish were washed once with PBS and then resuspended in 50 μl of PBS. The number of cells and amount of radioactivity present in each sample were determined with a hemocytometer and a Packard gamma counter, respectively. The counts per minute detected in each sample were converted to nanograms of 125I-CPE for calculating nanograms of specifically bound 125I-CPE (31, 46, 47), which equals nanograms of 125I-CPE in the total binding sample − nanograms of 125I-CPE in the matching nonspecific binding sample.
Assay for CPE-induced membrane permeability alterations in CaCo-2 cells.
To assess CPE-induced membrane permeability changes in CaCo-2 cells, 86Rb release from radiolabeled CaCo-2 cultures was measured as previously described (46, 47). Briefly, confluent CaCo-2 cultures were radiolabeled for 2 h at 37°C in complete minimal essential medium containing 4 μCi of 86RbCl (specific activity, 10.4 mCi/mg; NEN) per well that contained or did not contain either a 50 μM concentration of a caspase inhibitor (Z-VAD-FMK, DEVD-CHO, or YVAD-CHO) or a 5 mM concentration of the oncosis inhibitor glycine. After two washings with warm HBSS, radiolabeled cultures were incubated at 37°C for 15 min in 2 ml of HBSS containing either 1 or 10 μg of CPE per ml along with the same concentration of the caspase or oncosis inhibitor present (if any) during pretreatment.
After CPE treatment, the supernatant from each culture well was collected and the radioactivity present in that sample was determined with a gamma counter. The counts per minute obtained were used to calculate, as previously described (31, 46, 47), the percentage of 86Rb release specifically induced by CPE, which equals 100 × [(86Rb release in a CPE-treated well − spontaneous 86Rb release)/(maximal 86Rb release − spontaneous 86Rb release)]. Spontaneous release represents radiolabel released during the 15-min treatment period from cultures receiving only HBSS (no CPE) and averaged ∼103 cpm/culture. Maximal release corresponds to the total amount of cytoplasmic 86Rb radioactivity present in each radiolabeled culture at the start of the experiment and was determined to be ∼0.8 × 104 to 1 × 104 cpm/culture. As previously described (31, 46, 47), maximal release values were obtained by lysing 86Rb-labeled cultures with a 1:1 mixture of 0.1 M citric acid and saponin buffer (0.3 M sucrose, 1 mM MgCl2 · 6H2O, 1 mM KH2PO4, and 0.5% saponin [pH 6.8]). The presence of caspase or oncosis inhibitors during pretreatment and treatment did not affect either spontaneous or maximal release values (data not shown).
Mitochondrial membrane perturbation analysis.
The fluorescent dye JC-1 (5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolylcarbo-cyanine iodide), a lipophilic cation that selectively enters mitochondria (42, 53), is commonly used for mitochondrial membrane perturbation studies (42, 53). This dye normally exists in a monomeric form that emits at a λ of 527 nm after excitation at a λ of 490 nm. However, in the presence of mitochondrial membrane potential changes, JC-1 dye forms aggregates that exhibit a large shift in emission to a λ of 590 nm, which can be readily detected with a flow cytometer.
To evaluate CPE effects on mitochondrial membrane perturbation, confluent CaCo-2 cultures that had or had not been pretreated with a caspase or oncosis inhibitor (as described above) were washed twice with warm HBSS and then treated for 15 min at 37°C with HBSS containing either 1 or 10 μg of CPE per ml, along with the same concentration of caspase or oncosis inhibitor (if any) that had been present during pretreatment. After this CPE treatment, cultures were washed twice with warm HBSS and adherent cells were detached for 10 min at RT with Versene. The detached cells were combined with any nonadherent cells spontaneously released from the same culture during treatment or subsequent washings. The collected cells were then washed once with PBS, resuspended in PBS, and quantified with a hemocytometer. A 0.5-ml aliquot (containing 0.5 × 106 cells) of the cell suspension was incubated for 15 min at RT in the dark with 1 μg of JC-1 dye (Molecular Probes) per ml dissolved in PBS. After that incubation, the cells were washed twice with warm PBS and then resuspended in 0.5 ml of PBS. Changes in mitochondrial membrane potential in those samples were then determined by using a FACSCalibur flow cytometer (Becton Dickinson) equipped with a 488-nm argon laser. A minimum of 50,000 cells per sample were acquired in list mode and analyzed by using the FlowJo (Tree Star Inc.) program.
Evaluation of the presence of cytochrome c in mitochondrial versus cytosolic fractions.
CaCo-2 cells were inoculated into 150-mm2 petri dishes (Sarstedt) at a density of 3 × 106 cells/petri dish, and those cultures were then grown to confluency (∼5 to 6 days). After two washes with warm HBSS, the confluent cultures were treated for 30 min at 37°C with 20 ml of HBSS containing either 1 or 10 μg of CPE per ml. After this CPE treatment, the cultures were washed twice with HBSS and both adherent and released cells were collected by centrifugation as described above. The resultant cell pellet was washed once with PBS and resuspended in 5 ml of PBS, and the cells were counted with a hemocytometer. After centrifugation, an aliquot containing 108 cells from each sample was brought to 100 μl with mitochondrial isolation buffer (MIB) (51) (0.3 M sucrose, 10 mM MOPS [morpholinepropanesulfonic acid], 1 mM EDTA, 4 mM KH2PO4 [pH 7.4]) and that suspension was incubated on ice for 20 min. After homogenization with 120 strokes in a Dounce homogenizer, the resultant homogenate was transferred to a microcentrifuge tube and centrifuged at 650 × g for 15 min. The supernatant was removed and kept on ice, while the pellet was resuspended in 100 μl of MIB and then homogenized again. The homogenate was transferred to a microcentrifuge tube and centrifuged (10,000 × g) at 4°C for 15 min. The supernatant was removed and kept on ice, while the pellet was resuspended in 50 μl of MIB and used as the mitochondrial fraction. The supernatants from both centrifugations described above were combined and centrifuged (10,000 × g) at 4°C for 1 h to remove cellular organelles. The resultant supernatant was then used as the cytosolic fraction.
The presence of cytochrome c in the prepared cytosolic and mitochondrial fractions extracted from 2 × 107 cells was evaluated by separately boiling and electrophoresing those fractions on a 12% acrylamide gel containing SDS. The separated proteins were then electrotransfered onto a nitrocellulose membrane (Bio-Rad) and subjected to Western immunoblotting with the anti-cytochrome c monoclonal antibody 7H8.2C12 (PharMingen) and goat anti-mouse-horseradish peroxidase conjugate as the secondary antibody. The blot was detected with WestFemto chemiluminescent substrate (Pierce).
Caspase activity analysis.
CPE-treated CaCo-2 cells (as described above) were collected by centrifugation, and 2 × 107 cells per ml were resuspended in lysis buffer {50 mM HEPES [pH 7.4], 0.1% 3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate [CHAPS], 1 mM dithiothreitol, 0.1 mM EDTA}. After 5 min on ice, each lysate was centrifuged (10,000 × g) for 10 min at 4°C and the resultant supernatants were used to measure caspase activity. In these caspase activity assays, a caspase 1 or caspase 3/7 substrate (see Materials and Materials) was dissolved in assay buffer (50 mM HEPES [pH 7.4], 100 mM NaCl, 0.1% CHAPS, 10 mM dithiothreitol, 1 mM EDTA, 10% glycerol) at a concentration of 2 mM. Cell extract (10 μl, prepared as described above), one dissolved caspase substrate (10 μl), and assay buffer were then combined in a total volume of 100 μl, and the mixtures were incubated at 37°C for 4 h. Cleavage of these caspase substrates results in release of p-nitroaniline, which was detected by measuring absorption at 405 nm with an enzyme-linked immunosorbent assay plate reader. The amount of substrate cleaved was then calculated according to the manufacturer's instructions.
In a few specified experiments, confluent CaCo-2 cultures were treated for 8 h at 37°C with either a 2 μM concentration of the known (4) caspase 3/7 inducer staurosporine or a 100-U/ml concentration of the known (41) caspase 1 inducer recombinant human alpha interferon (IFN-α) (specific activity, 2.8 × 108 U/mg of protein; Calbiochem).
RESULTS
CPE-induced morphological changes in CaCo-2 cells.
As previously reported for CPE-treated Vero cells (28), CaCo-2 cultures exhibited monolayer disruption, cell rounding, cell detachment from the underlying matrix, and membrane bleb formation following treatment with HBSS containing 1 μg of CPE per ml (Fig. 1, top row). Under this CPE treatment condition, morphological damage was fully apparent within 60 min, by which time >95% of cells were no longer able to exclude the viability dye trypan blue (data not shown).
FIG. 1.

Morphological damage in CaCo-2 cells treated with 1 μg of CPE per ml. Confluent CaCo-2 cells were pretreated for 2 h at 37°C with HBSS that contained or did not contain a specified caspase inhibitor (50 μM final concentration) or the oncosis inhibitor glycine (5 mM final concentration), as indicated on the left Those cultures were then treated for 60 min in HBSS with or without 1 μg of CPE per ml, along with the same inhibitor (if any) used during pretreatment. Following that treatment, the culture supernatant was gently removed and photomicrographs were taken of each culture dish. CaCo-2 cells detaching from each petri dish during the treatment period were placed on microscope slides for photomicrography. Culture supernatants not shown contained no detectable detached CaCo-2 cells.
To evaluate whether procaspase activation might be involved in the cytotoxic effects shown in the top row of Fig. 1, CaCo-2 cells were incubated with a specified caspase inhibitor or the oncosis inhibitor glycine prior to and during HBSS treatment with or without 1 μg of CPE per ml (Fig. 1, bottom four rows). Those studies revealed that the concentrations of caspase and oncosis inhibitors used in this experiment were not themselves toxic to CaCo-2 cells in the absence of CPE. Furthermore, they demonstrated that neither the caspase 1 inhibitor YVAD-CHO nor glycine was able to prevent development of the typical morphological changes induced by 1 μg of CPE per ml. However, morphological changes did not develop when CaCo-2 cells were incubated with either the broad-spectrum caspase inhibitor Z-VAD-FMK or the caspase 3/7 inhibitor DEVD-CHO prior to and during 60 min of treatment with 1 μg of CPE per ml.
The ability of Z-VAD-FMK and DEVD-CHO to inhibit the morphological damage caused by 60 min of treatment with 1 μg of CPE per ml (Fig. 1) suggested that low CPE doses might induce caspase-mediated apoptosis in CaCo-2 cells. Therefore, the CPE-treated CaCo-2 cultures were also examined for nuclear chromatin condensation, another apoptosis hallmark. In CaCo-2 cells treated for 60 min with 1 μg of CPE per ml, but not in control cells treated only with HBSS, nuclear chromatin condensation was readily apparent (data not shown). That CPE-induced nuclear chromatin condensation was inhibited by incubating CaCo-2 cells with Z-VAD-FMK or DEVD-CHO, but not with YVAD-CHO or glycine, prior to and during 60 min of treatment with 1 μg of CPE per ml (data not shown).
To investigate whether cell death mechanisms might be dependent upon CPE dosage, confluent CaCo-2 cultures were also treated with 10 μg of CPE per ml. As shown in Fig. 2, CaCo-2 cultures receiving this higher CPE dose developed extensive morphological damage (including cell rounding, cell detachment, and membrane bleb formation) within 30 min of treatment, by which time >95% of cells no longer excluded trypan blue (data not shown). Most DAPI-stained CaCo-2 cells treated with 10 μg of CPE per ml did not exhibit nuclear condensation (data not shown).
FIG. 2.

Morphological damage in CaCo-2 cells treated with 10 μg of CPE per ml. Confluent CaCo-2 cells were pretreated for 2 h at 37°C with HBSS that contained or did not contain the broad-spectrum caspase inhibitor Z-VAD-FMK (50 μM final concentration) or oncosis inhibitor glycine (5 mM final concentration), as indicated. Those cultures were then treated for 30 or 60 min (as indicated) in HBSS with or without 10 μg of CPE per ml, along with the same inhibitor (if any) used during pretreatment. After treatment, photomicrographs were taken of each culture dish.
In contrast to the experiments with the results shown in Fig. 1, CaCo-2 cells incubated with Z-VAD-FMK prior to and during their 30 or 60 min of treatment with 10 μg of CPE per ml still developed morphological damage (Fig. 2). Similarly, the morphological damage induced by this higher CPE dose was not blocked by incubation with YVAD-CHO or DEVD-CHO prior to and during this toxin treatment (data not shown). However, the morphological changes caused by 10 μg of CPE per ml were inhibited for at least 30 min when CaCo-2 cultures were incubated with a 5 mM concentration of the oncosis inhibitor glycine prior to and during this higher-dose toxin treatment (Fig. 2).
CPE-induced DNA cleavage in CaCo-2 cells.
Apoptosis typically involves cleavage of nucleosomal DNA into fragments with a ladder-like pattern corresponding to 180- to 200-bp multiples (11, 50). Oncosis also induces DNA cleavage, which can result in either nondistinct DNA smearing or cleavage into distinct DNA fragments similar to those observed during apoptosis (7, 9).
Therefore, to test further the morphology results suggesting that CPE causes apoptosis at lower doses but oncosis at higher doses (Fig. 1 and 2), experiments were performed to determine whether CPE induces DNA cleavage in CaCo-2 cells and, if so, whether that cleavage can be prevented by a specific caspase- or oncosis-associated inhibitor(s). Initially, control experiments were performed to verify our ability to distinguish between apoptosis- versus oncosis-mediated DNA cleavage. As shown in Fig. 3A, these positive control experiments demonstrated a distinct ladder pattern of nucleosomal DNA cleavage fragments in CaCo-2 cells treated with either 2 μM staurosporine (which is known to induce caspase-mediated apoptosis [4]) or 5 μM ionomycin (which is known to induce oncosis [7]). As expected, the staurosporine-induced DNA fragmentation could be blocked by 50 μM Z-VAD-FMK but not by 5 mM glycine. In contrast, the ionomycin-induced DNA fragmentation could be blocked by 5 mM glycine but not by 50 μM Z-VAD-FMK.
FIG. 3.

CPE-induced DNA cleavage in CaCo-2 cells treated with 1 μg of CPE per ml. (A) Confluent CaCo-2 cells were pretreated for 2 h at 37°C with HBSS that contained or did not contain either the broad-spectrum caspase inhibitor Z-VAD-FMK or the oncosis inhibitor glycine. Those cultures were then treated for 60 min at 37°C in HBSS with or without 1 μg of CPE per ml, along with the same inhibitor (if any) used during pretreatment. After CPE treatment, DNA was extracted from each sample and run on a 2% agarose gel, followed by staining with ethidium bromide. CaCo-2 cells treated for 8 h at 37°C with either 2 μM staurosporine (Staurosp.) or 5 μM ionomycin (IONO) served as positive controls for apoptosis and oncosis, respectively. Control,CaCo-2 cells treated only with HBSS (no CPE). The arrows indicate the migration of DNA size markers. (B) Confluent CaCo-2 cells were pretreated for 2 h at 37°C with HBSS that contained or did not contain a specified caspase inhibitor and then treated for 60 min at 37°C in HBSS with or without 1 μg of CPE per ml, along with the same inhibitor (if any) used during pretreatment. After CPE treatment, DNA was extracted from each sample and run on a 2% agarose gel, followed by staining with ethidium bromide. Control, depicts CaCo-2 cells treated only with HBSS (no CPE). The arrows indicate the migration of DNA size markers.
When similar DNA cleavage analyses were performed with CaCo-2 cells treated for 1 h with 1 μg of CPE per ml, a distinct ladder pattern of nucleosomal DNA cleavage fragments was also observed (Fig. 3A). That lower CPE dose-induced DNA cleavage was blocked by a 50 μM concentration of the broad-spectrum caspase inhibitor Z-VAD-FMK but not by a 5 mM concentration of the oncosis inhibitor glycine (Fig. 3A). Furthermore, experiments performed to help identify the specific caspases involved in this low-CPE-dose-induced DNA fragmentation did not detect DNA fragmentation in CaCo-2 cultures incubated with Z-VAD-FMK or the caspase 3/7-specific inhibitor DEVD-CHO prior to and during treatment with 1 μg of CPE per ml (Fig. 3B). Similar incubation with the caspase 1 inhibitor YVAD-CHO failed to inhibit the development of DNA fragmentation in CaCo-2 cells treated with this lower CPE dose (Fig. 3B).
When confluent CaCo-2 cells were treated with 10 μg of CPE per ml, smeared DNA cleavage (Fig. 4) was observed instead of the distinct laddering pattern of DNA fragments caused by the low CPE dose (Fig. 3). The smeared DNA cleavage induced by this higher-dose CPE treatment could be inhibited for 30 min by the presence of 5 mM glycine but not by the presence of 50 μM Z-VAD-FMK (Fig. 4). However, the protective effect of 5 mM glycine on DNA cleavage induced by the higher CPE dose was transient, as CaCo-2 cultures treated with 10 μg of CPE per ml for 60 min in the presence of 5 mM glycine exhibited the same smeared DNA cleavage pattern observed in cells treated with that high CPE dose for 30 or 60 min in the absence of glycine (Fig. 4).
FIG. 4.

CPE-induced DNA cleavage in CaCo-2 cells treated with 10 μg of CPE per ml. Confluent CaCo-2 cells were pretreated for 2 h at 37°C with HBSS that contained or did not contain the broad-spectrum caspase inhibitor Z-VAD-FMK (50 μM final concentration) or oncosis inhibitor glycine (5 mM final concentration). Those cultures were then treated for 30 or 60 min (as indicated) at 37°C in HBSS with or without 10 μg of CPE per ml, along with the same inhibitor (if any) used during pretreatment. At the end of the treatment period, DNA was extracted from each sample and run on a 2% agarose gel, followed by staining with ethidium bromide. Control, CaCo-2 cells treated only with HBSS (no CPE). The arrows indicate the migration of the DNA size markers.
Preincubation of either the higher or lower CPE doses with CPE-neutralizing monoclonal antibody 3C9 prior to addition of the toxin to CaCo-2 cells blocked development of DNA cleavage and morphological damage (data not shown), confirming CPE as the active agent responsible for the effects shown in Fig. 1 to 4. Furthermore, while CaCo-2 cultures treated with 1 μg of rCPE per ml developed DNA cleavage and morphological damage (data not shown), both of those effects were absent in cultures treated with the same or higher doses of the nontoxic rCPE mutant G49D (data not shown); i.e., the cytotoxic activity of CPE is necessary for obtaining the DNA fragmentation and morphological damage shown in Fig. 1 to 4.
Effects of caspase and oncosis inhibitors on early steps in CPE action.
The ability of DEVD-CHO and Z-VAD-FMK to protect CaCo-2 cells from the morphological damage and DNA cleavage caused by 1 μg of CPE per ml dissolved in HBSS (as shown in Fig. 1 and 3) could indicate that this lower CPE dose triggers caspase 3/7-mediated apoptosis. Similarly, the ability of glycine to transiently protect CaCo-2 cells from the morphological damage and DNA cleavage caused by 10 μg of CPE per ml dissolved in HBSS (as shown in Fig. 2 and 4) could indicate that this higher CPE dose triggers oncosis.
To evaluate the alternative possibility that these protective caspase or oncosis inhibitors might alter CPE-induced morphological damage and DNA fragmentation in CaCo-2 cells simply by interfering with an early step specific to CPE action rather than by blocking a generalized cell death pathway, a series of studies were performed to evaluate the effects of the protective caspase and oncosis inhibitors on the earliest steps in CPE action. Those studies revealed that, within 15 min, neither caspase inhibitors nor glycine interferes with 125I-CPE specific binding to CaCo-2 cells (Fig. 5A) and that neither caspase inhibitors nor glycine inhibits the ability of either 1 or 10 μg of CPE per ml to induce membrane permeability alterations in CaCo-2 cells (Fig. 5B). Consistent with these results, formation of the ∼155-kDa CPE-containing large complex linked to membrane permeability changes was still detectable in CaCo-2 cultures pretreated with caspase or oncosis inhibitors prior to and during treatment with 1 μg (Fig. 5C) or 10 μg (not shown) of CPE per ml. DEVD-CHO and Z-VAD-FMK did inhibit large-complex formation by >6-fold after 1 h of low-dose CPE treatment (Fig. 5C) but had little effect (i.e., a <1.4-fold inhibition [data not shown]) on complex formation within the 15-min treatment period needed to induce membrane permeability changes in the experiments with the results shown in Fig. 5B.
FIG. 5.

Early CPE effects on CaCo-2 cells treated with toxin in the presence of caspase or oncosis inhibitors. (A) Specific binding of 125I-CPE to CaCo-2 cultures pretreated with a specified caspase or oncosisinhibitor and then treated, in the presence or absence of a 200-fold excess of native CPE, at RT for 15 min with 1 μg of 125I-CPE per ml dissolved in HBSS. The same concentration of inhibitor (if any) present during the preincubation step was also present during this binding reaction. Nanograms of 125I-CPE specifically bound per 106 CaCo-2 cells were then calculated as described in Materials and Methods. The data shown represent the means from three independent experiments. Error bars indicate the standard deviations. Data with no error bars had a standard deviation too small to depict. (B) Cytotoxic effect of CPE. 86Rb-labeled CaCo-2 cultures pretreated with a specified caspase or oncosis inhibitor were then treated for 15 min at 37°C with HBSS containing (as indicated) either 1 or 10 μg of CPE per ml, along with the same caspase or oncosis inhibitor (if any) used for preincubation. The bars represent means from three independent experiments. Error bars indicate the standard deviations; data with no error bars had a standard deviation too small to depict. (C) CPE Western immunoblot analysis of large-complex formation in CaCo-2 cells. Confluent CaCo-2 cells were pretreated with a specified caspase or oncosis inhibitor and then treated for 60 min at 37°C with HBSS that contained or did not contain 1 μg of CPE per ml along with the same caspase or oncosis inhibitor (if any) used for pretreatment. Cell lysates were prepared, electrophoresed, and CPE-Western immunoblotted as described in Materials and Methods. Control, CaCo-2 cells treated only with HBSS (no CPE). The arrows indicate the migrations of the ∼200- and ∼155-kDa CPE complexes.
Effects of CPE treatment on caspase activity in CaCo-2 cells.
Since the results shown in Fig. 5 indicate that caspase 3/7 inhibitors and glycine did not significantly block the early steps in CPE action, experiments were performed to evaluate directly the effects of lower and higher CPE doses on activation of caspase 1 or caspase 3/7 activity in CaCo-2 cells. In these studies, treatment with 1 μg of CPE per ml started increasing caspase 3/7 activity in CaCo-2 cells within 30 min (data not shown), and that caspase 3/7 activation reached ∼10-fold by 1 h (Fig. 6A). Furthermore, the caspase 3/7 activation induced by this lower CPE dose could be inhibited by incubating CaCo-2 cells with the caspase 3/7-specific inhibitor DEVD-CHO before and during this toxin treatment (Fig. 6A). The 1-μg/ml CPE treatment also caused a modest increase in CaCo-2 cell caspase 1 activity, with that effect becoming detectable at 30 min (data not shown) and reaching an ∼2- to 3-fold activation level by 1 h (Fig. 6B). That caspase 1 activation could be inhibited by incubating CaCo-2 cells with the caspase 1 inhibitor YVAD-CHO before and during this lower-dose CPE treatment (Fig. 6B).
FIG. 6.
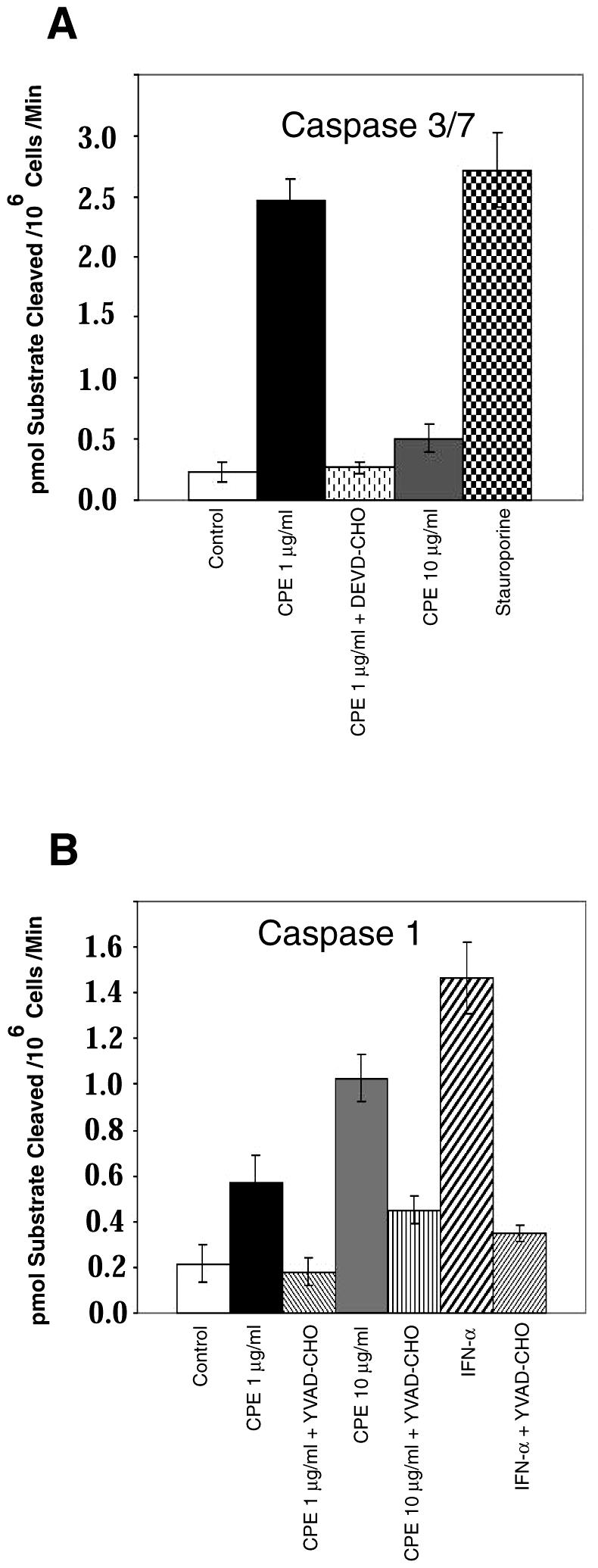
CPE-induced caspase activation in CaCo-2 cells. Confluent CaCo-2 cells were pretreated with HBSS that contained or did not contain a 50 μM final concentration of either the caspase 3/7 inhibitor DEVD-CHO (A) or the caspase 1 inhibitor YVAD-CHO (B) for 2 h at 37°C. Those cultures were then treated for 1 h at 37°C in HBSS with or without 1 or 10 μg of CPE per ml. After treatment, cells were processed for caspase activity by using the caspase 1-specific substrate Ac-YVAD-pNA, while caspase 3/7 activity was measured by using the caspase 3/7-specific substrate Ac-DEVD-pNA. CaCo-2 cells treated with 100 U of IFN-α per ml for 8 h served as a specific positive control for caspase 1 induction, while CaCo-2 cells treated with 2 μM staurosporine for 8 h served as a specific positive control for caspase 3/7 induction. Control, CaCo-2 cells treated only with HBSS (no CPE). The data represent means from four independent experiments. Error bars represent standard deviations.
In contrast, treatment with 10 μg of CPE per ml for 30 min (data not shown) or 60 min (Fig. 6A) caused only a minimal (<2-fold) increase in caspase 3/7 activity in CaCo-2 cultures. However, caspase 1 activity increased considerably in CaCo-2 cells treated with this higher CPE dose, with the activation increasing by fivefold within 1 h (Fig. 6B). The increase in caspase 1 activity caused by treatment with 10 μg of CPE per ml was substantially reduced by the presence of YVAD-CHO before and during CPE treatment (Fig. 6B). The presence of glycine during treatment of CaCo-2 cultures with either the higher or lower CPE dose had no effect on caspase 3/7 or caspase 1 activation at either 30 or 60 min (data not shown).
To confirm the validity of these CPE-induced caspase activity results, control experiments were performed in which CaCo-2 cultures were treated with HBSS containing either a 2 μM concentration of staurosporine (a known caspase 3/7 activator) or a 100-U/ml concentration of IFN-α (a known caspase 1 activator). As shown in Fig. 6, those two agents increased caspase 3/7 and caspase 1 activities in CaCo-2 cells by ∼11- and ∼7.5-fold, respectively. As expected, the IFN-α-induced activation of caspase 1 was inhibited by YVAD-CHO (Fig. 6B), while the staurosporine-induced activation of caspase 3/7 was inhibited by DEVD-CHO (data not shown).
Evaluation of mitochondrial involvement in CPE-induced apoptosis.
Apoptotic induction sometimes results from mitochondrial membrane depolarization (8). To investigate whether mitochondria are involved in CPE-induced cell death, the dye JC-1 (42, 53) was used to assess mitochondrial membrane depolarization. As shown in Fig. 7, CaCo-2 cells treated with HBSS containing 1 μg of CPE per ml exhibited a substantial increase in mitochondrial depolarization; i.e., the percentage of CaCo-2 cells testing positive for depolarized mitochondria increased from ∼20% of control cells to ∼82% of cells treated with the lower CPE dose. Consistent with our earlier figures indicating that 10 μg of CPE per ml induces oncosis rather than apoptosis, only 23% of CaCo-2 cells treated with HBSS containing that higher CPE dose tested positive for mitochondrial depolarization.
FIG. 7.

Effects of CPE on mitochondrial membrane depolarization. Confluent CaCo-2 cells that had been pretreated with a specified caspase or oncosis inhibitor were treated for 15 min at 37°C with HBSS containing 1 or 10 μg of CPE per ml. After treatment, the cells were incubated with JC-1 dye and mitochondrial depolarization was demonstrated by using a FACSCalibur flow cytometer (depolarization is indicated in the lower right quadrant). CaCo-2 cells treated with 2 μM staurosporine for 6 h at 37°C served as a positive control for mitochondrial depolarization. Control, CaCo-2 cells treated only with HBSS (no CPE). The data shown are a representative result from one of three experiments.
Previous studies (8) have shown that during apoptosis, mitochondrial depolarization can either result from caspase activation (i.e., be a caspase-dependent process) or can itself trigger caspase activation (i.e., be a caspase-independent process). Therefore, experiments were performed to investigate whether the lower-CPE-dose-induced caspase activation detected in Fig. 6 is a cause or a consequence of mitochondrial depolarization. Those studies demonstrated that the presence of caspase or oncosis inhibitors prior to and during treatment with 1 μg of CPE per ml had no effect on lower-dose-CPE-induced mitochondrial depolarization (Fig. 7), i.e., that the mitochondrial depolarization induced by this low CPE dose is caspase-independent.
Depolarization of mitochondria releases several apoptogenic proteins, most notably cytochrome c, from the mitochondria into the cytosol (8). Once in the cytosol, the released cytochrome c binds to other proteins, producing a complex that activates caspase cascades (8). To confirm that treatment with lower CPE doses releases cytochrome c from mitochondria into the cytosol of CaCo-2 cells, mitochondrial and cytosolic fractions of CPE-treated cells were prepared and subjected to Western immunoblot analysis with an anti-cytochrome c monoclonal antibody. The results from those Western immunoblotting analyses demonstrated that after treatment with 1 μg of CPE per ml, the presence of cytochrome c in the cytosol started within 15 min (data not shown) and became obvious by 30 min (Fig. 8). As expected from our earlier results indicating that higher CPE doses induce oncosis rather than apoptosis, the presence of cytochrome c was not detected in the cytosolic fraction of CaCo-2 cells treated for either 15 or 30 min with 10 μg of CPE per ml.
FIG. 8.

CPE-induced cytochrome c release from mitochondria into the cytosol in CaCo-2 cells. Confluent CaCo-2 cells were treated for 30 min at 37°C with HBSS that contained either 1 or 10 μg of CPE per ml. After that treatment, cells were processed for Western immunoblotting to detect cytochrome c release from mitochondria into the cytosol. Control, CaCo-2 cells treated only with HBSS (no CPE). The arrow indicates the location of cytochrome c in a lane, when present.
DISCUSSION
Prior to the present study, C. perfringens enterotoxin was known to kill the enterocyte-like CaCo-2 human cell line (46, 47), but the cell death pathway(s) involved had not yet been evaluated. The results presented in this study now demonstrate that a lower dose of CPE can induce apoptosis in CaCo-2 cells within 1 h, which is rapid, but not unprecedented, for apoptosis development (12). Furthermore, this CPE-induced apoptotic process was determined to involve a classical pathway mediated by mitochondrial perturbations and requiring caspase 3/7, but not caspase 1, activation.
In contrast, a higher CPE dose was found to induce cell death by a markedly different pathway that is independent of caspase 3/7 activation. Although the present study demonstrated a higher CPE dose causes substantial activation of caspase 1, the caspase 1 inhibitor YVAD-CHO was unable to block either the morphological damage or DNA cleavage induced by that higher CPE dose. Coupling that finding with our observation that glycine can inhibit both the morphological damage and DNA cleavage induced in CaCo-2 cells by 30 min if treatment with a higher CPE dose establishes the cell death pathway induced by that CPE dose as oncosis, rather than the recently recognized, caspase 1-dependent oncosis subtype referred to as pyrotosis (3, 5, 6). It is also notable that even when no longer protected by glycine following 60 min of treatment with 10 μg of CPE per ml, CaCo-2 cells still exhibited characteristics of CPE-induced oncosis, including smeared DNA cleavage patterns and little caspase 3/7 activation.
Determining that a lower CPE dose causes apoptosis while a higher enterotoxin dose causes oncosis has potential relevance for understanding the pathogenesis of CPE-associated GI disease. Previous studies of diarrheic feces from patients with CPE-associated GI diseases detected the presence of enterotoxin at concentrations ranging from >10 ng/ml to >100 μg/ml (1, 2). Therefore, the CPE concentrations used in the present study to demonstrate apoptosis (1 μg/ml) and oncosis (10 μg/ml) have pathophysiologic relevance, which suggests that both oncosis and apoptosis may occur in the intestines during CPE-associated GI disease. This finding could help clarify some apparent confusion in the literature regarding whether CPE has proinflammatory properties. Some previous in vivo studies using rabbit ileal loops reported that CPE induces inflammatory cell (mainly polymorphonuclear leukocyte) infiltration (34), while other previous studies using the same CPE-treated animal model failed to detect that effect (44, 45). Interestingly, an older study investigating the effects of various CPE doses on rabbit ileal loops noted that inflammatory cell infiltration is CPE dose dependent (36). That previous observation is fully consistent with our present findings indicating that a lower CPE dose causes apoptosis, a noninflammatory process, while a higher CPE dose induces the proinflammatory process of oncosis. Since inflammation contributes to the symptoms of other GI diseases (3, 5), it is possible that the proinflammatory consequences of oncosis caused by higher CPE doses contribute to intestinal pathology in some cases of CPE-induced GI disease.
Recent studies (43, 46, 47, 49) have revealed that CPE is a multifunctional toxin whose action consists of, at a minimum, (i) altering plasma membrane permeability and (ii) internalizing tight-junction proteins to disrupt tight junctions. Given that staphylococcal α toxin, a pore-forming toxin that alters plasma membrane permeability properties, resembles CPE in causing apoptosis at lower doses but necrosis (presumably oncosis) at higher doses (52), our present results are consistent with previous suggestions (13) that the primary cytotoxic action of CPE involves the induction of membrane permeability alterations, possibly via pore formation (10).
The results from our present study also indicate that early steps occurring in the first 15 min of CPE action (i.e., CPE binding, initial formation of the ∼155-kDa complex, and the onset of membrane permeability alterations) are not substantially blocked by caspase or oncosis inhibitors, which confirms that those inhibitors are acting primarily on downstream events in apoptosis or oncosis activation pathways in CPE-treated cells. Determining that glycine transiently blocks DNA degradation or morphological damage, but not CPE-induced membrane permeability alterations, in CaCo-2 cells treated with a higher CPE dose is interesting given that the mechanism by which glycine protects mammalian cells from oncosis is generally attributed to “plasma membrane stabilization” (7).
Similarly interesting are the results in Fig. 5C indicating that in CaCo-2 cells treated for longer, 1-h periods with 1 μg of CPE per ml, a caspase 3/7 inhibitor substantially decreases formation of the ∼155-kDa CPE complex and blocks formation of the ∼200 kDa complex. Those substantial inhibitory effects on ∼155- and ∼200-kDa complex formation, which appear after the occurrence of plasma membrane permeability changes (Fig. 5B), are likely attributable to the ability of the caspase 3/7 inhibitor to protect CaCo-2 cells from morphological damage caused by the low CPE dose (as shown in Fig. 1). Previous (46) studies have demonstrated that the development of morphological damage after 30 min of lower-dose CPE treatment exposes the basolateral membrane of CaCo-2 cells, which allows additional CPE binding to form more ∼155-kDa complex and to interact with occludin to form the ∼200-kDa complex.
The results shown in Fig. 5 also suggest that apoptosis not only promotes ∼155-kDa complex formation by increasing CPE binding levels in CaCo-2 cells but may also facilitate the localization of bound CPE into the ∼155-kDa complex (compare the levels of immunoreactive material present at the bottom of the lanes in Fig. 5C with versus without caspase 3/7 inhibitor treatment). Since ∼155-kDa CPE complex formation is apparently influenced by membrane fluidity (30), the ability of apoptosis to promote the localization of bound CPE into the ∼155-kDa complex in Fig. 5 might result from the effects of apoptosis on plasma membrane structure and function (5). Consistent with the possibility of CPE-induced apoptosis affecting plasma membranes, 1 μg of CPE per ml was found to increase the percentage of CaCo-2 cells staining annexin V positive (data not shown), a finding demonstrating that apoptosis-induced flipping of phosphatidylserine from the inner to the outer leaflet occurs in CPE-treated plasma membranes (5).
Further studies are under way to determine the mechanism by which CPE treatment activates biochemical pathways leading to apoptosis or oncosis and to clarify the relevance of these effects for CPE action in vivo.
Acknowledgments
This research was generously supported by Public Health Service grant AI19844 from the National Institute of Allergy and Infectious Diseases.
We thank William Goins for use of his fluorescence microscope, Surojit Sarkar for help with fluorescence-activated cell sorter analysis, and Usha Singh for help with electrophoresis studies.
Editor: J. T. Barbieri
REFERENCES
- 1.Batholomew, B. A., M. F. Stringer, G. N. Watson, and R. J. Gilbert. 1985. Development and application of an enzyme-linked immunosorbent assay for Clostridium perfringens type A enterotoxin. J. Clin. Pathol. 38:222-228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Birkhead, G., R. L. Vogt, E. M. Heun, J. T. Snyder, and B. A. McClane. 1988. Characterization of an outbreak of Clostridium perfringens food poisoning by quantitative fecal culture and fecal enterotoxin measurement. J. Clin. Microbiol. 26:471-474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boise, L. H., and C. M. Collins. 2001. Salmonella-induced cell death: apoptosis, necrosis or programmed cell death. Trends Microbiol. 9:64-67. [DOI] [PubMed] [Google Scholar]
- 4.Bossy-Wetzel, E., D. D. Newmeyer, and D. R. Green. 1998. Mitochondrial cytochrome C release in apoptosis occurs upstream of DEVD-specific caspase activation and independently of mitochondrial transmembrane depolarization. EMBO J. 17:37-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brennan, M., and B. T. Cookson. 2000. Salmonella induces macrophage death by caspase-1 dependent necrosis. Mol. Microbiol. 38:31-40. [DOI] [PubMed] [Google Scholar]
- 6.Cookson, B. T., and M. A. Brennan. 2001. Pro-inflammatory programmed cell death. Trends Microbiol. 9:113-114. [DOI] [PubMed] [Google Scholar]
- 7.Dong, Z., P. Saikumar, J. M. Weinberg, and M. A. Venkatachalam. 1997. Internucleosomal DNA cleavage triggered by plasma membrane damage during necrotic cell death. Am. J. Pathol. 151:1205-1213. [PMC free article] [PubMed] [Google Scholar]
- 8.Dorrie, J., H. Gerauer, Y. Wachter, and S. J. Zunino. 2001. Resveratrol induces extensive apoptosis by depolarizing mitochondrial membranes and activating caspase-9 in acute lymphoblastic leukemia cells. Cancer Res. 61:4731-4739. [PubMed] [Google Scholar]
- 9.Enright, H., R. P. Hebbel, and K. A. Nath. 1994. Internucleosomal cleavage of DNA as the sole criterion for apoptosis may be artifactual. J. Lab. Clin. Med. 124:63-68. [PubMed] [Google Scholar]
- 10.Hardy, S. P., M. Denmead, N. Parekh, and P. E. Granum. 1999. Cationic currents induced by Clostridium perfringens type A enterotoxin in human intestinal Caco-2 cells. J. Med. Microbiol. 48:235-243. [DOI] [PubMed] [Google Scholar]
- 11.Hockenbery, D. 1995. Defining apoptosis. Am. J. Pathol. 146:16-19. [PMC free article] [PubMed] [Google Scholar]
- 12.Hogquist, K. A., M. A. Nett, E. R. Unanue, and D. D. Chaplin. 1991. Interleukin 1 is processed and released during apoptosis. Proc. Natl. Acad. Sci. USA 88:8485-8489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hulkower, K. I., A. P. Wnek, and B. A. McClane. 1989. Evidence that alterations in small molecule permeability are involved in the Clostridium perfringens type-A enterotoxin-induced inhibition of macromolecular synthesis in Vero cells. J. Cell. Physiol. 140:498-504. [DOI] [PubMed] [Google Scholar]
- 14.Katahira, J., N. Inoue, Y. Horiguchi, M. Matsuda, and N. Sugimoto. 1997. Molecular cloning and functional characterization of the receptor for Clostridium perfringens enterotoxin. J. Cell Biol. 136:1239-1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Katahira, J., H. Sugiyama, N. Inoue, Y. Horiguchi, M. Matsuda, and N. Sugimoto. 1997. Clostridium perfringens enterotoxin utilizes two structurally related membrane proteins as functional receptors in vivo. J. Biol. Chem. 272:26652-26658. [DOI] [PubMed] [Google Scholar]
- 16.Kokai-Kun, J. F., K. Benton, E. U. Wieckowski, and B. A. McClane. 1999. Identification of a Clostridium perfringens enterotoxin region required for large complex formation and cytotoxicity by random mutagenesis. Infect. Immun. 67:6534-6541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li, P., D. Nijhawan, I. Budihardjo, S. M. Srinivasula, M. Ahmad, E. S. Alnemri, and X. Wang. 1997. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 91:479-489. [DOI] [PubMed] [Google Scholar]
- 18.Ma, F., C. Zhang, K. V. S. Prasad, G. J. Freeman, and S. F. Schlossman. 2001. Molecular cloning of porimin, a novel cell surface receptor mediating oncotic cell death. Proc. Natl. Acad. Sci. USA 98:9778-9783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Majno, G., and I. Joris. 1995. Apoptosis, oncosis, and necrosis. An overview of cell death. Am. J. Pathol. 146:3-15. [PMC free article] [PubMed] [Google Scholar]
- 20.Martin, S. J., and T. G. Cotter. 1990. Disruption of microtubules induces an endogenous suicide pathway in human leukaemia HL-60 cells. Cell Tissue Kinet. 23:545-559. [DOI] [PubMed] [Google Scholar]
- 21.Matsubara, K., M. Kubota, S. Adachi, K. Kuwakado, H. Hirota, Y. Wakazono, Y. Akiyama, and H. Mikawa. 1994. Different mode of cell death induced by calcium ionophore in human leukemia cell lines: possible role of constitutive endonuclease. Exp. Cell Res. 210:19-25. [DOI] [PubMed] [Google Scholar]
- 22.Matsuda, M., and N. Sugimoto. 1979. Calcium-independent and calcium-dependent steps in action of Clostridium perfringens enterotoxin. Biochem. Biophys. Res. Commun. 91:629-636. [DOI] [PubMed] [Google Scholar]
- 23.Matsumura, H., Y. Shimizu, Y. Ohsawa, A. Kawahara, Y. Uchiyama, and S. Nagata. 2000. Necrotic death pathway in Fas receptor signaling. J. Cell Biol. 151:1247-1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McClane, B. A. 2001. Clostridium perfringens, p. 351-372. In M. P. Doyle, L. R. Beuchat, and T. J. Montville (ed.), Food microbiology: fundamentals and frontiers, 2nd ed. ASM Press, Washington, D.C.
- 25.McClane, B. A. 1984. Osmotic stabilizers differentially inhibit permeability alterations induced in Vero cells by Clostridium perfringens enterotoxin. Biochim. Biophys. Acta 777:99-106. [DOI] [PubMed] [Google Scholar]
- 26.McClane, B. A., D. M. Lyerly, J. S. Moncrief, and T. D. Wilkins. 2000. Enterotoxic clostridia: Clostridium perfringens type A and Clostridium difficile, p. 551-562. In V. A. Fischetti, R. P. Novick, J. J. Ferretti, D. A. Portnoy, and J. Rood (ed.), Gram-positive pathogens. ASM Press, Washington, D.C.
- 27.McClane, B. A., and J. L. McDonel. 1980. Characterization of membrane permeability alterations induced in Vero cells by Clostridium perfringens enterotoxin. Biochim. Biophys. Acta 600:974-985. [DOI] [PubMed] [Google Scholar]
- 28.McClane, B. A., and J. L. McDonel. 1979. The effects of Clostridium perfringens enterotoxin on morphology, viability and macromolecular synthesis. J. Cell Physiol. 99:191-200. [DOI] [PubMed] [Google Scholar]
- 29.McClane, B. A., and J. L. McDonel. 1981. Protective effects of osmotic stabilizers on morphological and permeability alterations induced in Vero cells by Clostridium perfringens enterotoxin. Biochim. Biophys. Acta 641:401-409. [DOI] [PubMed] [Google Scholar]
- 30.McClane, B. A., and A. P. Wnek. 1990. Studies of Clostridium perfringens enterotoxin action at different temperatures demonstrate a correlation between complex formation and cytotoxicity. Infect. Immun. 58:3109-3115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McClane, B. A., A. P. Wnek, K. I. Hulkower, and P. C. Hanna. 1988. Divalent cation involvement in the action of Clostridium perfringens type A enterotoxin. J. Biol. Chem. 263:2423-2435. [PubMed] [Google Scholar]
- 32.McDonel, J. L. 1980. Binding of Clostridium perfringens 125I-enterotoxin to rabbit intestinal cells. Biochemistry 21:4801-4807. [DOI] [PubMed] [Google Scholar]
- 33.McDonel, J. L. 1974. In vivo effects of Clostridium perfringens enterotoxin on the rat ileum. Infect. Immun. 10:1156-1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McDonel, J. L., L. W. Chang, J. L. Pounds, and C. L. Duncan. 1978. The effects of Clostridium perfringens enterotoxin on rat and rabbit ileum: an electron microscopy study. Lab. Investig. 39:210-218. [PubMed] [Google Scholar]
- 35.McDonel, J. L., and G. W. Demers. 1982. In vivo effects of enterotoxin from Clostridium perfringens type A in rabbit colon: binding vs. biologic activity. J. Infect. Dis. 145:490-494. [DOI] [PubMed] [Google Scholar]
- 36.McDonel, J. L., and C. L. Duncan. 1975. Histopathological effect of Clostridium perfringens enterotoxin in the rabbit ileum. Infect. Immun. 12:1214-1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McDonel, J. L., and C. L. Duncan. 1977. Regional localization of activity of Clostridium perfringens type A enterotoxin in the rabbit ileum, jejunum and duodenum. J. Infect. Dis. 136:661-666. [DOI] [PubMed] [Google Scholar]
- 38.McDonel, J. L., and B. A. McClane. 1988. Production, purification and assay of Clostridium perfringens enterotoxin. Methods Enzymol. 165:94-103. [DOI] [PubMed] [Google Scholar]
- 39.Mills, E. M., D. Xu, M. M. Fergusson, C. A. Combs, Y. Xu, and T. Finkel. 2002. Regulation of cellular oncosis by uncoupling protein 2. J. Biol. Chem. 277:27385-27392. [DOI] [PubMed] [Google Scholar]
- 40.Nakagawa, I., M. Nakata, S. Kawabata, and S. Hamada. 2001. Cytochrome c-mediated caspase-9 activation triggers apoptosis in Streptococcus pyogenes-infected epithelial cells. Cell. Microbiol. 3:395-405. [DOI] [PubMed] [Google Scholar]
- 41.Pirhonen, J., T. Sareneva, I. Julkunen, and S. Matikainen. 2001. Virus infection induces proteolytic processing of IL-18 in human macrophages via caspase-1 and caspase-3 activation. Eur. J. Immunol. 31:726-733. [DOI] [PubMed] [Google Scholar]
- 42.Polla, B. S., S. Kantengwa, D. Francois, S. Salvioli, C. Franceschi, C. Marsac, and A. Cossarizza. 1996. Mitochondria are selective targets for the protective effects of heat shock against oxidative injury. Proc. Natl. Acad. Sci. USA 93:6458-6463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rahner, C., L. L. Mitic, B. A. McClane, and J. M. Anderson. 1999. Clostridium perfringens enterotoxin impairs bile flow in the isolated perfused rat liver and induces fragmentation of tight junction fibrils. Hepatology 30:326A.
- 44.Sarker, M. R., R. J. Carman, and B. A. McClane. 1999. Inactivation of the gene (cpe) encoding Clostridium perfringens enterotoxin eliminates the ability of two cpe-positive C. perfringens type A human gastrointestinal disease isolates to affect rabbit ileal loops. Mol. Microbiol. 33:946-958. [DOI] [PubMed] [Google Scholar]
- 45.Sherman, S., E. Klein, and B. A. McClane. 1994. Clostridium perfringens type A enterotoxin induces concurrent development of tissue damage and fluid accumulation in the rabbit ileum. J. Diarrheal Dis. Res. 12:200-207. [PubMed] [Google Scholar]
- 46.Singh, U., L. L. Mitic, E. Wieckowski, J. M. Anderson, and B. A. McClane. 2001. Comparative biochemical and immunochemical studies reveal differences in the effects of Clostridium perfringens enterotoxin on polarized CaCo-2 cells versus Vero cells. J. Biol. Chem. 276:33402-33412. [DOI] [PubMed] [Google Scholar]
- 47.Singh, U., C. M. Van Itallie, L. L. Mitic, J. M. Anderson, and B. A. McClane. 2000. CaCo-2 cells treated with Clostridium perfringens enterotoxin form multiple large complex species, one of which contains the tight junction protein occludin. J. Biol. Chem. 275:18407-18417. [DOI] [PubMed] [Google Scholar]
- 48.Songer, J. G. 1996. Clostridial enteric diseases of domestic animals. Clin. Microbiol. Rev. 9:216-234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sonoda, N., M. Furuse, H. Sasaki, S. Yonemura, J. Katahira, Y. Horiguchi, and S. Tsukita. 1999. Clostridium perfringens enterotoxin fragments removes specific claudins from tight junction strands: evidence for direct involvement of claudins in tight junction barrier. J. Cell Biol. 147:195-204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Steller, H. 1995. Mechanisms and genes of cellular suicide. Science 267:1445-1449. [DOI] [PubMed] [Google Scholar]
- 51.Wang, G. Q., B. R. Gastman, E. Wieckowski, L. A. Goldstein, A. Gambotto, T. H. Kim, B. Fang, A. Rabinovitz, X. M. Yin, and H. Rabinowich. 2001. A role for mitochondrial Bak in apoptotic response to anticancer drugs. J. Biol. Chem. 276:34307-34317. [DOI] [PubMed] [Google Scholar]
- 52.Weinrauch, Y., and A. Zychlinsky. 1999. The induction of apoptosis by bacterial pathogens. Annu. Rev. Microbiol. 53:155-187. [DOI] [PubMed] [Google Scholar]
- 53.Weiss, D. S., and A. Zychlinsky. 2002. Methods for studying bacteria-induced host cell death, p. 439-460. In P. Sansonetti and A. Zyclinsky (ed.), Molecular cellular microbiology, vol. 31. Academic Press, San Diego, Calif.
- 54.Wieckowski, E. U., A. P. Wnek, and B. A. McClane. 1994. Evidence that an ∼50kDa mammalian plasma membrane protein with receptor-like properties mediates the amphiphilicity of specifically bound Clostridium perfringens enterotoxin. J. Biol. Chem. 269:10838-10848. [PubMed] [Google Scholar]
- 55.Wnek, A. P., and B. A. McClane. 1986. Comparison of receptors for Clostridium perfringens type A and cholera enterotoxins in isolated rabbit intestinal brush border membranes. Microb. Pathog. 1:89-100. [DOI] [PubMed] [Google Scholar]