

Abstract
Anaplasma phagocytophilum propagates within neutrophils and causes a disease marked by inflammatory tissue injury or complicated by opportunistic infections. We hypothesized that infection with A. phagocytophilum modifies the binding of neutrophils to endothelial cells and the expression of neutrophil adhesion molecules and studied these changes in vitro. Infected dimethyl sulfoxide-differentiated HL-60 cells and neutrophils showed reduced binding to cultured brain and systemic endothelial cells and lost expression of P-selectin glycoprotein ligand 1 (PSGL-1, CD162) and L-selectin (CD62L) (to 33 and 5% of control values, respectively), at a time when the levels of β2 integrin and immunoglobulin superfamily adhesion molecules and activation markers Mac-1 and intercellular adhesion molecule 1 increased (5 to 10 times that of the control). The loss of CD162 and CD62L expression was inhibited by EDTA, which suggests that neutrophil activation and sheddase cleavage occurred. The loss of selectin expression and the retained viability of the neutrophils persisted for at least 18 h with A. phagocytophilum infection, whereas Escherichia coli and Staphylococcus aureus rapidly killed neutrophils. The adhesion defect might increase the numbers of infected cells and their persistence in the blood prior to tick bites. However, decreased CD162 expression and poor endothelial cell binding may partly explain impaired host defenses, while simultaneous neutrophil activation may aggravate inflammation. These observations may help us to understand the modified biological responses, host inflammation, and immune response that occur with A. phagocytophilum infections.
Human anaplasmosis or granulocytic ehrlichiosis is an emerging, tick-borne disease that ranges from mild to fatal. It is caused by Anaplasma phagocytophilum, an obligate intracellular bacterium that is unique because it propagates within neutrophil vacuoles (12, 16, 39). Human anaplasmosis and related veterinary diseases can result in opportunistic infections that usually occur after T- or B-cell immune suppression or neutrophil dysfunction (16, 39). Clinical findings often mimic toxin- or cytokine-mediated disease. However, A. phagocytophilum is infrequently demonstrated in tissues to a degree that explains the histopathology or illness accompanying infection (16, 39), which suggests that A. phagocytophilum disease results from inflammation or immunity in the host rather than from direct bacterial injury (28, 29).
Neutrophil function requires the orchestration of consecutive events, including the arrest and transmigration of neutrophils through endothelial cells and activation for phagocytosis and microbial killing. The initial binding of neutrophils to the endothelium is critical for the progression of this response in vivo (26). Although the precise mechanism of neutrophil arrest on the endothelium is not well understood, the principles of selectin-mediated rolling, cellular activation, and integrin-mediated, tight binding are now well accepted (26). Without selectin-mediated rolling, neutrophil arrest on endothelial cell surface integrins is poor and inflammation is modified (4, 10, 24, 32). Paradoxically, the loss of normal neutrophil function can even exacerbate inflammation because of the regulatory effects of neutrophil phagocyte oxidase (33).
Along with the A. phagocytophilum-induced defects noted previously in the phagocyte oxidase and Rac-associated functions of neutrophils (2, 11, 30) and the evidence of partial neutrophil activation with chemokine production (25), we hypothesized that other neutrophil functions would be altered by A. phagocytophilum infection. We examined neutrophil binding to endothelial cells and the changes in adhesion molecules associated with this process. The results of this study indicate that A. phagocytophilum-infected neutrophils have a reduced capacity for binding to endothelial cells, a finding closely linked to the neutrophil-derived metalloprotease shedding of CD62L (L-selectin) and extracellular P-selectin glycoprotein ligand 1 (PSGL-1, CD162) domains. Although selectin shedding is a well-known response of neutrophils to inflammatory stimuli, in the context of a bacterium that has adapted for survival within neutrophils, this shedding would lead to the prolonged functional incapacity of the infected cells at a time when the neutrophils were partly activated for promoting inflammation.
MATERIALS AND METHODS
In vitro growth and preparation of cell-free A. phagocytophilum.
A. phagocytophilum strain Webster was cultivated on HL-60 cells in RPMI 1640 medium (GIBCO, Grand Island, N.Y.) with 1% fetal bovine serum (FBS; GIBCO) and 2 mM l-glutamine (GIBCO) (21). Cell-free A. phagocytophilum was prepared from approximately 107 HL-60 cells when >90% were infected, as determined by Romanowsky staining (HEMA 3; Biochemical Science Inc., Swedesboro, N.J.). Infected HL-60 cells were lysed by five passages through a 25-gauge needle, and cellular debris was removed by centrifugation at 750 × g for 10 min. The supernatant was centrifuged (2,500 × g for 15 min) to remove extraneous soluble contaminants. Pellets containing the cell-free A. phagocytophilum were immediately used to infect 107 human peripheral blood neutrophils. Granulocytic differentiation was induced in HL-60 cells by cultivation for 3 days with 1.25% dimethyl sulfoxide (DMSO) in RPMI 1640 medium.
Neutrophil isolation.
Human peripheral blood neutrophils were isolated from the EDTA-anticoagulated blood of healthy donors by dextran sedimentation and density gradient centrifugation (Histopaque 1077; Sigma). Contaminating erythrocytes were lysed in hypotonic (0.2%) NaCl for 30 s and then neutralized with hypertonic (1.6%) NaCl. Neutrophil purity and viability were always >95% as assessed by Romanowsky staining and trypan blue staining, respectively.
Endothelial cell cultures.
Two different endothelial cell lines were cultured to confluence in 24-well plates. Human brain microvascular endothelial cells (BMEC) transformed by simian virus 40 (36) were propagated in RPMI 1640 medium with 20% heat-inactivated FBS (Omega Scientific Inc., Tarzana, Calif.), 2 mM l-glutamine, 1 mM minimal essential medium (MEM)-sodium pyruvate (GIBCO), 1× MEM-nonessential amino acid solution (Sigma), and 1× MEM-vitamin solution (Sigma). EA.hy926 cells, a fusion of A549 cells with human umbilical vein endothelial cells (18), were cultivated in high-glucose (4.5 g/liter) Dulbecco's modified Eagle medium (GIBCO) with 10% heat-inactivated FBS and 1× hypoxanthine-thymidine supplement (GIBCO). After 4 h of stimulation with recombinant human tumor necrosis factor-α (rTNF-α; R&D Systems, Inc., Minneapolis, Minn.), expression of P-selectin (CD62P) and E-selectin (CD62E) was demonstrated on BMEC and EA.hy926 cells, respectively, by flow cytometry.
Adhesion assay.
To ascertain changes in neutrophil adhesion to endothelial cells or changes in the expression of surface adhesion molecules, we used neutrophils that were uninfected, lipopolysaccharide (LPS) stimulated, or A. phagocytophilum infected and HL-60 cells that were DMSO differentiated and uninfected or A. phagocytophilum infected. Some endothelial cell cultures were stimulated with 80 ng of rTNF-α/ml for 4 h at 37°C. Prior to incubation with endothelial cells, neutrophils and HL-60 cells were labeled with PKH67 green fluorescent dye (Sigma), washed three times per the manufacturer's instructions, and then resuspended at 5 × 105 cells/ml; no change in cell viability was observed after fluorescence labeling. The fluorescently labeled cells (5 × 105) were incubated on confluent endothelial cell monolayers for 1 h at 37°C in 5% CO2 under static conditions (45 min) or under conditions of low-shear force (15 min at 4°C on a constantly rotating platform), as previously described (35). The nonadherent cells were removed, and the monolayers were washed twice with phosphate-buffered saline (PBS). Adherent cells were fixed with 1% paraformaldehyde (PFA) in PBS; fluorescence was immediately determined with a Fluorimager IS (Vistra Systems, Sunnyvale, Calif.), and data were analyzed with Molecular Dynamics Image QuaNT software (version 4.2a). Relative fluorescence intensities among replicate assays, groups, and controls were compared to determine differences in adhesion. The validity of the static and low-shear-force adhesion assays for CD162 (PSGL-1)-dependent leukocyte adhesion was tested by the method of Snapp et al. (35) with the fluorescence measurement of adhesion confirmed by microscopic inspection. At this point, DMSO-differentiated HL-60 cell cultures were incubated with saturating concentrations (10 μg/ml) of the CD162 adhesion-blocking monoclonal antibodies (MAbs) PL-1 (27) and KPL-1 (35). The adhesion of uninfected cells incubated with blocking antibodies was compared with that obtained for cells in the presence of an isotype-matched control antibody by static and low-shear-force assays.
Flow cytometric analysis.
Flow cytometry was used to quantitate the expression of surface molecules on uninfected or A. phagocytophilum-infected cells. Cells were cultivated for 3 h at 37°C without any additional stimulation or with 10 μg of LPS (from Escherichia coli O111:B4; Sigma) per ml, with 108 Staphylococcus aureus (ATCC 25290) or E. coli (ATCC 25922) cells, or with cell-free A. phagocytophilum derived from 107 infected HL-60 cells (approximately 108 bacteria) that were freshly prepared or fixed in 4% PFA. The cells were then washed twice with cold PBS containing 0.5% bovine serum albumin and 0.02% sodium azide. Fluorescence-labeled anti-CD11b, anti-CD18, anti-CD54 (intercellular adhesion molecule 1 [ICAM-1]), anti-CD62L, and anti-CD162 (KPL-1 or PL-1) antibodies or immunoglobulin G1(κ) [IgG1(κ)] isotype-matched controls were incubated with the cells for 30 min on ice. After incubation, the cells were washed, fixed with 1% PFA in PBS, and analyzed by flow cytometry (BD, San Diego, Calif.). Neutrophils were manually gated based on characteristic forward and side light scatter properties. The data were analyzed with CellQuest software (BD).
Cell sorting of A. phagocytophilum-infected HL-60 cells.
To separate CD162-expressing cells and non-CD162-expressing cells, DMSO-differentiated A. phagocytophilum-infected cells and uninfected HL-60 cells were incubated with phycoerythrin-conjugated anti-CD162 (BD Pharmingen, San Diego, Calif.) for 30 min on ice and resuspended in PBS. The cells were sorted with a FACS Vantage SE cell sorter system (Becton Dickinson) on the basis of CD162 expression. After centrifugation (300 × g for 5 min), the cells were examined by Romanowsky staining.
Analysis of CD162 shedding.
Neutrophils (107) were incubated for 3 h at 37°C with 10 μg of LPS/ml (Sigma), cell-free A. phagocytophilum, or medium only. Uninfected and A. phagocytophilum-infected DMSO-differentiated HL-60 cells (108) were incubated for 3 days in RPMI 1640 medium supplemented with 1.25% DMSO. To determine if CD162 was released into the external milieu, the supernatants were harvested and a protease inhibitor cocktail containing aprotinin, bestatin, leupeptin, E-64, and pepstatin A (Sigma) designed for tissue culture use was added. To identify reductions in cell-associated CD162 with A. phagocytophilum infection, the cell pellets were washed three times in PBS and lysed in lysis buffer (50 mM Tris-HCl [pH 8.0], 150 mM NaCl, 1% Nonidet P-40, protease inhibitor cocktail) for 30 min on ice. Lysates were clarified by centrifugation at 10,000 × g for 15 min. For immunoprecipitation of cell-associated CD162 from cell lysates or CD162 released into the supernatants, samples were precleared with protein G-Sepharose beads (Pharmacia Biotech, Uppsala, Sweden) for 1 h at 4°C. Thereafter, samples were incubated with 5 μg of CD162 MAb PL-1 (Ancell Corp., Bayport, Minn.) for 3 h at 4°C, which was followed by the addition of protein G-Sepharose beads and centrifugation. The CD162 MAb-loaded protein G-Sepharose beads were washed five times with cold lysis buffer, and proteins were separated on a sodium dodecyl sulfate-8% polyacrylamide gel electrophoresis gel under reducing conditions, transferred to nitrocellulose membranes, and reacted with CD162 MAb PL-1. Reactions were detected by using alkaline phosphatase-conjugated goat anti-mouse IgG secondary antibody and enhanced chemiluminescence (Bio-Rad Laboratories, Hercules, Calif.).
Effects of EDTA and sheddase inhibitors on CD162 (PSGL-1) and CD62L (L-selectin) expression.
To examine whether CD162 and CD62L loss occurred via cleavage by a previously described metalloproteinase sheddase (15), neutrophils were preincubated for 3 h at 37°C with cell-free A. phagocytophilum and then treated for 1 h with (i) a tissue culture protease inhibitor cocktail that included aprotinin, bestatin, leupeptin, E-64, and pepstatin A, as per the manufacturer's recommendations (Sigma); (ii) the metalloproteinase inhibitor batimastat (10 ng/ml; R&D Systems, Inc.); or (iii) the divalent cation chelator EDTA (1.43 and 14.3 mM). Changes in the surface expression of CD162 and CD62L were examined by flow cytometry, and results for infected cells and uninfected cells in the presence or absence of inhibitors were compared.
RESULTS
Adhesion of neutrophils and DMSO-differentiated HL-60 cells to endothelial cells.
For human BMEC, moderate but reproducible reductions in adherence of A. phagocytophilum-infected DMSO-differentiated HL-60 cells (P < 0.002) (Fig. 1A) and neutrophils (P < 0.001) were observed, especially after rTNF-α stimulation of endothelial cells. For the HUVEC-derived EA.hy926 cells, the adhesion levels of A. phagocytophilum-infected DMSO-differentiated HL-60 cells and neutrophils were lower than those of controls (P < 0.001) (Fig. 1B), whether endothelial cells were stimulated by rTNF-α (P < 0.019) or not (data not shown). The results were confirmed in at least seven independent experiments and were similar regardless of whether static or low-shear-force adhesion assays were used.
FIG. 1.

A. phagocytophilum infection of neutrophils and DMSO-differentiated HL-60 cells inhibits adhesion to rTNF-α-activated endothelial cell monolayers under static and low-shear-force conditions. Uninfected and infected cells were labeled with PKH67 green fluorescent dye and incubated on confluent endothelial cell monolayers for 1 h at 37°C (static assay) or for 45 min at 4°C under static conditions followed by 15 min on a rotating platform at 4°C (low-shear-force assay), after which nonadherent cells were removed. The fluorescence retained was measured and expressed as a percentage of that of the cells adherent to monolayers treated with uninfected neutrophils or DMSO-differentiated HL-60 cells. Cells were described as having a low infection rate when 10 to 20% of the HL-60 cells contained morulae and a high infection rate when >70 to 80% were infected. Error bars indicate standard errors of the means; static, static assay; shear force, low-shear-force assay. (A) Adhesion of A. phagocytophilum-infected DMSO-differentiated HL-60 cells and infected neutrophils to rTNF-α-activated human BMEC (Neutrophils-1 and -2 indicate the results of two representative experiments). (B) Adhesion of A. phagocytophilum-infected neutrophils and DMSO-differentiated HL-60 cells to rTNF-α-activated EA.hy926 cells (HL-60 static-1 and static-2 indicate the results of two representative experiments). (C) Adhesion of uninfected DMSO-differentiated HL-60 cells to rTNF-α-activated human BMEC and EA.hy926 cells under low shear force is inhibited (P < 0.03) by the function-blocking CD162 MAbs KPL-1 and PL-1. Separate experiments are designated by the endothelial cell line used and the suffix -1 or -2. KPL-1+PL-1, mixture of both blocking antibodies.
The static and low-shear-force adhesion assays were at least in part dependent upon CD162 interactions, as the binding of DMSO-differentiated HL-60 cells was reduced (P < 0.04) in the presence of the function-blocking MAbs KPL-1 and PL-1 (Fig. 1C).
Surface expression of selectin ligands is decreased on A. phagocytophilum-infected cells.
Neutrophils infected with A. phagocytophilum showed reduced expression of CD162 (32% of cells at 3 h) (Fig. 2A) and CD62L (4% of cells at 3 h) (Fig. 2B), results similar to those observed with other inflammatory stimuli such as LPS, E. coli, or S. aureus. Incubation of neutrophils with PFA-fixed A. phagocytophilum diminished CD162 expression, to only 84% of the cells (Fig. 2A), but did not inhibit CD62L loss (Fig. 2B). This observation suggested that viable bacteria are required for CD162 loss and that selectins may be lost by different mechanisms. Likewise, PFA fixation of both E. coli and S. aureus inhibited the reduction in CD162 expression (data not shown). The HL-60 cells used here did not express CD62L even when they were uninfected (data not shown). However, like neutrophils, A. phagocytophilum-infected DMSO-differentiated HL-60 cells had decreased CD162 expression, albeit not as pronounced, relative to that of uninfected cells (Fig. 2C). The loss of selectin expression lasted for >18 h in neutrophils infected with A. phagocytophilum or stimulated by LPS with only moderate cell death; in contrast, live E. coli and S. aureus rapidly killed neutrophils (Fig. 3).
FIG. 2.

Changes in expression of CD162 and CD62L on A. phagocytophilum-infected cells. Surface expression of CD162 and CD62L was analyzed by flow cytometry. (A and B) Neutrophils were either not stimulated or stimulated with LPS (10 μg/ml) or live bacteria (E. coli, S. aureus) and infected with cell-free A. phagocytophilum or stimulated with PFA-fixed A. phagocytophilum for 3 h at 37°C. Surface expression of both CD162 (A) and CD62L (B) was decreased on A. phagocytophilum-infected neutrophils. (C) HL-60 cells were cultivated with 1.25% DMSO for 3 days to induce granulocytic differentiation and to achieve various degrees of infection. Expression of CD162 was reduced in A. phagocytophilum-infected DMSO-differentiated HL-60 cells after 3 days. SSC-H, side scatter.
FIG. 3.

A. phagocytophilum-infected and LPS-stimulated neutrophil cultures that lose selectin expression remain viable for at least 18 h after infection, whereas infection by E. coli and S. aureus rapidly kills neutrophils. Viability is expressed as the ratio of viable cells (as determined by trypan blue staining) at each interval to the total quantity of viable cells added at the beginning of the experiments. The data represent the means ± standard errors of the means (error bars) of determinations performed in at least two and up to five separate experiments.
By sorting cells based on expression levels, we found that loss of CD162 was predominantly associated with heavy A. phagocytophilum infection (multiple morulae) of DMSO-differentiated HL-60 cells (Fig. 4), yet approximately 20% of non-CD162-expressing cells were slightly infected or uninfected. Most CD162-expressing cells (>75%) were uninfected or had only small numbers of bacteria and individual morulae. Viability was usually >95% for both populations.
FIG. 4.

CD162-expressing and non-CD162-expressing A. phagocytophilum-infected differentiated HL-60 cells have different levels of infection. More non-CD162-expressing cells (A) were infected and contained larger numbers of morulae than did CD162-expressing cells (B), as determined by flow cytometry and cell sorting (Romanowsky stain; magnification, ×25). However, approximately 20 to 25% of uninfected cells also lacked CD162 expression (A, arrows).
Increased surface β2 integrin and ICAM-1 expression on A. phagocytophilum-infected cells.
The expression of Mac-1 and ICAM-1 increased in A. phagocytophilum-infected cells after 3 days of culture, up to 4.25 times that of DMSO-differentiated HL-60 cells (Fig. 5A and B). Most neutrophils constitutively expressed Mac-1 and thus showed only a moderate proportional increase in CD18- expressing cells with A. phagocytophilum infection or LPS stimulation. However, the intensity of expression increased approximately 5- to 10-fold for these cells compared with that of uninfected neutrophils (Fig. 5C). Likewise, the proportion of A. phagocytophilum-infected neutrophils that expressed ICAM-1 after 3 h increased 10-fold compared to that of uninfected neutrophils, a result similar to that for cells stimulated by LPS, E. coli, or S. aureus (Fig. 5D). No change in the expression of Mac-1 (data not shown) or ICAM-1 was observed in PFA-fixed A. phagocytophilum-stimulated neutrophils (Fig. 5D).
FIG. 5.

Surface expression of Mac-1 (CD-18) and ICAM-1 (CD54) in A. phagocytophilum-infected cells detected by flow cytometry; the results of CD18 staining are shown, but similar results were obtained for CD11b. The panels show the results of representative flow cytometric analyses of experiments conducted at least three times. Expression levels of Mac-1 (A) and ICAM-1 (B) were significantly increased on A. phagocytophilum-infected DMSO-differentiated HL-60 cells. The intensity of constitutively expressed Mac-1 (C) on A. phagocytophilum-infected neutrophils was increased, and new ICAM-1 expression (D) was markedly increased. Similarly, stimulation with live E. coli or S. aureus resulted in moderate increases in ICAM-1 expression, but stimulation with PFA-inactivated A. phagocytophilum did not. FITC, fluorescein isothiocynate; PE, phycoerythrin; SSC-H, side scatter.
CD162 shedding from A. phagocytophilum-infected cells.
Inflammatory stimuli such as LPS or phorbol myristate acetate induce shedding of CD62L (L-selectin) and extracellular CD162 (PSGL-1) domains on neutrophils (14). To determine whether infection with A. phagocytophilum induced shedding, CD162 was immunoprecipitated from culture supernatants and detergent-solubilized cell lysates of uninfected neutrophils, of LPS-stimulated and A. phagocytophilum-infected neutrophils, and of uninfected and A. phagocytophilum-infected DMSO-differentiated HL-60 cells. Under reducing conditions, a 110-kDa band corresponding to the extracellular domain of CD162 was observed in cell lysates and it but appeared only weakly or not at all in supernatants of uninfected cells (Fig. 6). However, after infection with A. phagocytophilum, the level of CD162 increased in infected cell supernatants and was accompanied by a simultaneous decrease in the level of CD162 in whole-cell lysates, thus confirming the shedding of CD162 in both but to a greater degree in differentiated HL-60 cells than in neutrophils.
FIG. 6.
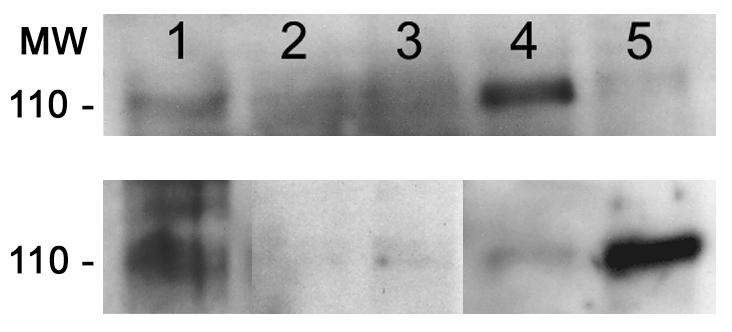
CD162 (PSGL-1) is shed from surfaces of A. phagocytophilum-infected neutrophils and DMSO-differentiated HL-60 cells in the culture supernatant. (Top) CD162 is lost from cells with A. phagocytophilum infection. Lane 1, uninfected neutrophil lysates; lane 2, LPS-stimulated neutrophil lysates; lane 3, A. phagocytophilum-infected neutrophil lysates; lane 4, uninfected DMSO-differentiated HL-60 cell lysates; lane 5, A. phagocytophilum-infected DMSO-differentiated HL-60 cell lysates. (Bottom) CD162 appears in cell culture supernatants after A. phagocytophilum infection. Lane 1, recombinant CD162; lane 2, uninfected neutrophils; lane 3, A. phagocytophilum-infected neutrophils; lane 4, uninfected DMSO-differentiated HL-60 cells; lane 5, A. phagocytophilum-infected DMSO-differentiated HL-60 cells. MW, molecular weight (in thousands).
EDTA inhibits CD162 loss in A. phagocytophilum-infected neutrophils.
The shedding of CD162 by neutrophils stimulated with LPS occurs via an unidentified EDTA-sensitive metalloprotease (15). With infected neutrophils, incubation in the presence of 14.3 mM EDTA inhibited CD162 down-expression by at least 5 to 28%, an effect not observed with 1.43 mM EDTA (Fig. 7). As reported previously for LPS stimulation (15), loss of CD162 expression in A. phagocytophilum-infected neutrophils was not inhibited in the presence of a serine-cysteine-aminopeptidase inhibitor cocktail or with the metalloprotease inhibitor batimastat (data not shown). As expected, a slight inhibition of CD62L loss was shown for the batimastat treatment only. EDTA treatment also partly inhibited the loss of CD62L, a result consistent with the effect on sheddase activity (data not shown). These data suggest that the release of CD162 from A. phagocytophilum-infected cell surfaces probably occurs via an unidentified sheddase (15) and that selectin loss comes from host cell sheddase (metalloprotease) release.
FIG. 7.

The divalent cation chelator EDTA inhibits the reduction in surface expression of CD162 in A. phagocytophilum-infected neutrophils. Neutrophils were preincubated with cell-free A. phagocytophilum for 3 h and then treated with either 14.3 or 1.43 mM EDTA. Incubation in the presence of 14.3 mM EDTA inhibits the loss of CD162 expression after A. phagocytophilum infection. Error bars, standard errors of the means.
DISCUSSION
Neutrophil adhesion to endothelial cells is a pivotal event in neutrophil migration and subsequent tissue inflammation and is initiated by endothelial cell P- and E-selectins and by leukocyte CD62L (L-selectin), all of which bind loosely to neutrophils via CD162 (PSGL-1) (26). Without prior selectin binding, the β2 integrin-mediated arrest of neutrophils on endothelial cells and Ig superfamily molecules (e.g., ICAM-1) may be completely or significantly reduced and inflammatory responses may be altered (4, 10, 37, 38, 40). Here, it is demonstrated that A. phagocytophilum-infected cells have reduced adherence to rTNF-α-activated endothelial cell lines and that this reduction is associated with the loss of neutrophil selectins. Reduced adhesion during intervals when CD11b/CD18 and ICAM-1 expression increased suggests that β2 integrins and Ig superfamily molecules are unlikely to be involved in the adhesion defect. Although defined shear stress assays should be conducted to confirm the adhesion defect, the inhibition of binding by CD162-blocking antibodies (27, 35) supports a linkage between selectin loss and reduced adhesion. Kinetic analysis demonstrates a prolonged reduction in CD162 surface expression despite persistent neutrophil activation and degranulation. Alone, each observation is not entirely unique; taken together, these findings suggest that infection with A. phagocytophilum promotes a sustained neutrophil presence in the blood and that neutrophil viability interferes with critical microbicidal neutrophil activity, yet potentially augments neutrophil responses that cause inflammatory tissue injury.
A significant mechanism for the regulation of inflammatory cell infiltrates and the resulting tissue injury involves the loss of surface adhesion molecules by sheddases (8, 23). Thus, removal of CD162 and CD62L from the surface of neutrophils infected with A. phagocytophilum is an example of such a regulatory deadhesive event. When stimulated by inflammatory molecules, neutrophils degranulate and secrete many enzymes, including an unidentified EDTA-sensitive metalloprotease that cleaves an extracellular domain of CD162 (15). A. phagocytophilum-infected cells also shed the 110-kDa extracellular domain of CD162 into culture supernatants, a process inhibited by EDTA that is consistent with the action of a metalloprotease sheddase. Loss of CD162 even from uninfected neutrophils provides evidence that the metalloprotease may affect other cells and suggests the potential for additional local injury.
In contrast, the expression of β2 integrin (Mac-1, CD11b/CD18) and ICAM-1 (CD54) adhesion molecules increased on A. phagocytophilum-infected cells. Mac-1 is found both on the surface of neutrophils and within specific cytoplasmic granules. Numerous signals, including cytokines such as gamma interferon, chemotactic factors, and ligation and cross-linking of CD62L or CD162 result in increased surface β2 integrin expression (a marker of neutrophil activation) and thereby increase neutrophil adhesion to ICAM-1 on endothelial cells and perhaps on other cells after rolling on selectins (5, 9, 34, 43). Although expression of β2 integrins is also increased in vivo, these molecules are not necessary for the adhesion of infected cells in dermal wounds, which in turn allows A. phagocytophilum-infected cells to be ingested by ticks (6, 7). However, integrins are important signal transducers for vital responses such as motility, growth and differentiation, and cell survival and for specialized functions such as degranulation and oxidant production in immune cells (3, 17, 20, 37, 38), and they probably contribute to inflammation and immunity in cases of A. phagocytophilum infection (6). In contrast, the role of ICAM-1 in neutrophils is less clear, but the binding of neutrophils to both CD62L and β2 integrins suggests participation of ICAM-1 in leukocyte-leukocyte or leukocyte-platelet interactions. Regardless of the role of ICAM-1, A. phagocytophilum-infected neutrophils and DMSO-differentiated HL-60 cells enter a prolonged proinflammatory state of activation and degranulation.
Several other defects in A. phagocytophilum-infected neutrophils are known. A. phagocytophilum infection disrupts phagocyte oxidase function by repression of rac2 mRNA and down-regulation of gp91phox mRNA or by degradation of p22phox, leading to the inhibition of superoxide anion generation (2, 11, 30). The net effect may be a transient condition that mimics chronic granulomatous disease with the inability to control some infectious agents. Similarly, defects in leukocyte adhesion, as demonstrated here, also contribute to an immunosuppressive state (32). Defects in phagocytosis, microbicidal activity, and motility have also been described for A. phagocytophilum-infected cells (19, 41). The possibility that these abnormalities support clinical manifestations is suggested by opportunistic infections in humans and horses and more dramatically by the occurrence of frequently fatal infections such as pyemia (disseminated staphylococcal infection) in pasture-raised sheep and goats after tick-borne fever (16, 39, 41).
A potentially more profound effect of neutrophil infection with A. phagocytophilum may occur as a result of the dichotomy of neutrophil activation (with protease release, degranulation, and secretion of chemokines) (1, 6, 25) and neutrophil dysfunction (with defects in adhesion, phagocytosis, and generation of respiratory burst) (2, 41) that leads to an activated-deactivated state. Paradoxically, defective phagocyte oxidase is associated with increased inflammation because it leads to an inability to metabolize proinflammatory and chemoattractant molecules such as leukotrienes, complement components, and N-formyl peptides (13, 31, 33). Thus, A. phagocytophilum-infected neutrophils may promote inflammation via chemokines that recruit neutrophils and other mononuclear cells and may also degranulate to release proinflammatory substances (1, 25). Yet, these infected cells are unable to emigrate or generate components for intracellular killing and contribute little toward infection control and regulation of inflammation.
How does A. phagocytophilum benefit from neutrophil disturbances? Most evidence suggests that humans are unnatural dead-end hosts that develop low-level bacteremia of limited duration (12, 16). However, it is likely that some animals and reservoir hosts develop low-level persistent or remittent bacteremia. Disturbances in endothelial cell adhesion coupled with the prolonged viability and perhaps delayed sequestration and apoptosis of A. phagocytophilum-infected neutrophils (22, 42) may potentially increase the pool of infected cells available to ticks in a blood meal.
In summary, we found that (i) A. phagocytophilum-infected neutrophils lose surface expression of CD162 and CD62L, which is accompanied by reduced neutrophil adhesion to endothelial cells; (ii) the decrease of CD162 surface expression in A. phagocytophilum-infected neutrophils may be due to cleavage by a sheddase; and (iii) the infected cells display up-regulated expression of Mac-1, an indication of neutrophil activation and active degranulation. If these findings are confirmed in vivo, they suggest that early and prolonged reduction of the expression of CD162 may reduce the interaction of neutrophils with vasculature and diminish the capacity for a neutrophil-mediated defense against opportunistic pathogens while neutrophil activation may promote increased inflammation and disease. A more complete understanding of A. phagocytophilum-modified biological processes and the manner by which these influence inflammation and immune responses in the host should be sought.
Acknowledgments
This work was supported by grant R01-AI44102 from the National Institutes of Allergy and Infectious Diseases. K.-S. Choi and J. Park were supported in part by grants from the Korea Science and Engineering Foundation (KOSEF).
We acknowledge Dennis Grab for helpful suggestions and careful reading of the manuscript, Lee Blossner for help with flow cytometry and cell sorting, and Ki Jun Kim for assistance with digital photography. Recombinant CD162-Fc fusion protein was provided courtesy of Kristin Murray, Genetics Institute, Andover, Mass. The BMEC and EA.hy926 endothelial cell lines were provided courtesy of Dennis Grab and Kwang Sik Kim, The Johns Hopkins University School of Medicine, Baltimore, Md., and Cora Jean Edgell, University of North Carolina, Chapel Hill, respectively. We also are grateful for the contributions of Max Maurin.
Editor: W. A. Petri, Jr.
REFERENCES
- 1.Akkoyunlu, M., S. E. Malawista, J. Anguita, and E. Fikrig. 2001. Exploitation of interleukin-8-induced neutrophil chemotaxis by the agent of human granulocytic ehrlichiosis. Infect. Immun. 69:5577-5588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Banerjee, R., J. Anguita, D. Roos, and E. Fikrig. 2000. Cutting edge: infection by the agent of human granulocytic ehrlichiosis prevents the respiratory burst by down-regulating gp91phox. J. Immunol. 164:3946-3949. [DOI] [PubMed] [Google Scholar]
- 3.Berton, G., S. R. Yan, L. Fumagalli, and C. A. Lowell. 1996. Neutrophil activation by adhesion: mechanisms and pathophysiological implications. Int. J. Clin. Lab. Res. 26:160-177. [DOI] [PubMed] [Google Scholar]
- 4.Bevilacqua, M. P. 1993. Endothelial-leukocyte adhesion molecules. Annu. Rev. Immuol. 11:767-804. [DOI] [PubMed] [Google Scholar]
- 5.Blanks, J. E., T. Moll, R. Eytner, and D. Vestweber. 1998. Stimulation of P-selectin glycoprotein ligand-1 on mouse neutrophils activates beta 2-integrin mediated cell attachment to ICAM-1. Eur. J. Immunol. 28:433-443. [DOI] [PubMed] [Google Scholar]
- 6.Borjesson, D. L., S. I. Simon, E. Hodzic, C. M. Ballantyne, and S. W. Barthold. 2002. Kinetics of CD11b/CD18 up-regulation during infection with the agent of human granulocytic ehrlichiosis in mice. Lab. Investig. 82:303-311. [DOI] [PubMed] [Google Scholar]
- 7.Borjesson, D. L., S. I. Simon, E. Hodzic, H. E. DeCock, C. M. Ballantyne, and S. W. Barthold. 2003. Roles of neutrophil β2 integrins in kinetics of bacteremia, extravasation, and tick acquisition of Anaplasma phagocytophila in Mice. Blood 101:3257-3264. [DOI] [PubMed]
- 8.Borland, G., G. Murphy, and A. Ager. 1999. Tissue inhibitor of metalloproteinases-3 inhibits shedding of L-selectin from leukocytes. J. Biol. Chem. 274:2810-2815. [DOI] [PubMed] [Google Scholar]
- 9.Borregaard, N., and J. B. Cowland. 1997. Granules of the human neutrophilic polymorphonuclear leukocyte. Blood 89:3503-3521. [PubMed] [Google Scholar]
- 10.Carlos, T. M., and J. M. Harlan. 1994. Leukocyte-endothelial adhesion molecules. Blood 84:2068-2101. [PubMed] [Google Scholar]
- 11.Carlyon, J. A., W. T. Chan, J. Galan, D. Roos, and E. Fikrig. 2002. Repression of rac2 mRNA expression by Anaplasma phagocytophila is essential to the inhibition of superoxide production and bacterial proliferation. J. Immunol. 169:7009-7018. [DOI] [PubMed] [Google Scholar]
- 12.Chen, S.-M., J. S. Dumler, J. S. Bakken, and D. H. Walker. 1994. Identification of a granulocytotropic Ehrlichia species as the etiologic agent of human disease. J. Clin. Microbiol. 32:589-595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Conti, P., M. Reale, R. C. Barbacane, M. R. Panara, M. Bongrazio, S. Fiore, J. W. Mier, and R. A. Dempsey. 1992. Granulocyte-macrophage colony stimulating factor potentiates human polymorphonuclear leukocyte aggregation responses to formyl-methionyl-leucyl-phenylalanine. Immunol. Lett. 32:71-79. [DOI] [PubMed] [Google Scholar]
- 14.Coughlan, A. F., H. Hau, L. C. Dunlop, M. C. Berndt, and W. W. Hancock. 1994. P-selectin and platelet-activating factor mediate initial endotoxin-induced neutropenia. J. Exp. Med. 179:329-334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Davenpeck, K. L., M. E. Brummet, S. A. Hudson, R. J. Mayer, and B. S. Bochner. 2000. Activation of human leukocytes reduces surface P-selectin glycoprotein ligand-1 (PSGL-1, CD162) and adhesion to P-selectin in vitro. J. Immunol. 165:2764-2772. [DOI] [PubMed] [Google Scholar]
- 16.Dumler, J. S., and J. S. Bakken. 1998. Human ehrlichioses: newly recognized infections transmitted by ticks. Annu. Rev. Med. 49:201-213. [DOI] [PubMed] [Google Scholar]
- 17.Dusi, S., V. Della Bianca, M. Donini, K. A. Nadalini, and F. Rossi. 1996. Mechanisms of stimulation of the respiratory burst by TNF in nonadherent neutrophils: its independence of lipidic transmembrane signaling and dependence on protein tyrosine phosphorylation and cytoskeleton. J. Immunol. 157:4615-4623. [PubMed] [Google Scholar]
- 18.Edgell, C. J., C. C. McDonald, and J. B. Graham. 1983. Permanent cell line expressing human factor VIII-related antigen established by hybridization. Proc. Natl. Acad. Sci. USA 80:3734-3737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Foster, W. N. M., and A. E. Cameron. 1970. Observations on the functional integrity of neutrophil leucocytes infected with tick-borne fever. J. Comp. Pathol. 80:487-491. [DOI] [PubMed] [Google Scholar]
- 20.Gao, J. X., and A. C. Issekutz. 1995. Polymorphonuclear leucocyte migration through human dermal fibroblast monolayers is dependent on both beta 2-integrin (CD11/CD18) and beta 1-integrin (CD29) mechanisms. Immunology 85:485-494. [PMC free article] [PubMed] [Google Scholar]
- 21.Goodman, J. L., C. Nelson, B. Vitale, J. E. Madigan, J. S. Dumler, T. J. Kurtti, and U. G. Munderloh. 1996. Direct cultivation of the causative agent of human granulocytic ehrlichiosis. N. Engl. J. Med. 334:209-215. [DOI] [PubMed] [Google Scholar]
- 22.Hsieh, T. C., M. E. Aguero-Rosenfeld, J. M. Wu, C. Ng, N. A. Papanikolaou, S. A. Varde, I. Schwartz, J. G. Pizzolo, M. Melamed, H. W. Horowitz, R. B. Nadelman, and G. P. Wormser. 1997. Cellular changes and induction of apoptosis in human promyelocytic HL-60 cells infected with the agent of human granulocytic ehrlichiosis (HGE). Biochem. Biophys. Res. Commun. 232:298-303. [DOI] [PubMed] [Google Scholar]
- 23.Kahn, J., R. H. Ingraham, F. Shirley, G. I. Migaki, and T. K. Kishimoto. 1994. Membrane proximal cleavage of L-selectin: identification of the cleavage site and a 6-kD transmembrane peptide fragment of L-selectin. J. Cell Biol. 125:461-470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kansas, G. S. 1996. Selectins and their ligands: current concepts and controversies. Blood 88:3259-3287. [PubMed] [Google Scholar]
- 25.Klein, M. B., S. Hu, C. C. Chao, and J. L. Goodman. 2000. The agent of human granulocytic ehrlichiosis induces the production of myelosuppressing chemokines without induction of proinflammatory cytokines. J. Infect. Dis. 182:200-205. [DOI] [PubMed] [Google Scholar]
- 26.Ley, K. 2002. Integration of inflammatory signals by rolling neutrophils. Immunol. Rev. 186:8-18. [DOI] [PubMed] [Google Scholar]
- 27.Li, F., H. P. Erickson, J. A. James, K. L. Moore, R. D. Cummings, and R. P. McEver. 1996. Visualization of P-selectin glycoprotein ligand-1 as a highly extended molecule and mapping of protein epitopes for monoclonal antibodies J. Biol. Chem. 271:6342-6348. [DOI] [PubMed] [Google Scholar]
- 28.Martin, M. E., J. E. Bunnell, and J. S. Dumler. 2000. Pathology, immunohistology, and cytokine responses in early phases of human granulocytic ehrlichiosis in a murine model. J. Infect. Dis. 181:374-378. [DOI] [PubMed] [Google Scholar]
- 29.Martin, M. E., K. Caspersen, and J. S. Dumler. 2001. Immunopathology and ehrlichial propagation are regulated by interferon gamma (IFNγ) and interleukin-10 (IL-10) in a murine model of human granulocytic ehrlichiosis (HGE). Am. J. Pathol. 158:1881-1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mott, J., Y. Rikihisa, and S. Tsunawaki. 2002. Effects of Anaplasma phagocytophilum on NADPH oxidase components in human neutrophils and HL-60 cells. Infect. Immun. 70:1359-1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rochon, Y. P., and M. M. Frojmovic. 1993. Regulation of human neutrophil aggregation: comparable latent times, activator sensitivities, and exponential decay in aggregability for FMLP, platelet-activating factor, and leukotriene B4. Blood 82:3460-3468. [PubMed] [Google Scholar]
- 32.Roos, D., and S. K. Law. 2001. Hematologically important mutations: leukocyte adhesion deficiency. Blood Cells Mol. Dis. 27:1000-1004. [DOI] [PubMed] [Google Scholar]
- 33.Segal, B. H., T. L. Leto, J. I. Gallin, H. L. Malech, and S. M. Holland. 2000. Genetic, biochemical, and clinical features of chronic granulomatous disease. Medicine 79:170-200. [DOI] [PubMed] [Google Scholar]
- 34.Simon, S. I., A. R. Burns, A. D. Taylor, P. K. Gopalan, E. B. Lynam, L. A. Sklar, and C. W. Smith. 1995. L-selectin (CD62L) cross-linking signals neutrophil adhesive functions via the Mac-1 (CD11b/CD18) beta 2-integrin. J. Immunol. 155:1502-1514. [PubMed] [Google Scholar]
- 35.Snapp, K. R., H. Ding, K. Atkins, R. Warnke, F. W. Luscinkas, and G. S. Kansas. 1998. A novel P-selectin glycoprotein ligand-1 (PSGL-1) monoclonal antibody recognizes an epitope within the tyrosine sulfate motif of human PSGL-1 and blocks recognition of both P- and L-selectin. Blood 91:154-164. [PubMed] [Google Scholar]
- 36.Stins, M. F., J. Badger, and S. K. Kim. 2001. Bacterial invasion and transcytosis in transfected human brain microvascular endothelial cells. Microb. Pathog. 30:19-28. [DOI] [PubMed] [Google Scholar]
- 37.Todd, R. F., III, and H. R. Petty. 1997. Beta 2 (CD11/CD18) integrins can serve as signaling partners for other leukocyte receptors. Lab. Clin. Med. 129:492-498. [DOI] [PubMed] [Google Scholar]
- 38.van Eeden, S. F., M. E. Klut, B. A. Walker, and J. C. Hogg. 1999. The use of flow cytometry to measure neutrophil function. J. Immunol. Methods. 232:23-43. [DOI] [PubMed] [Google Scholar]
- 39.Walker, D. J., and J. S. Dumler. 1997. Human monocytic and granulocytic ehrlichiosis. Arch. Pathol. Lab. Med. 121:785-791. [PubMed] [Google Scholar]
- 40.Williams, M. A., and J. S. Solomkin. 1999. Integrin-mediated signaling in human neutrophil functioning. J. Leukoc. Biol. 65:725-736. [DOI] [PubMed] [Google Scholar]
- 41.Woldehiwet, Z. 1987. The effects of tick-borne fever on some functions of polymorphonuclear cells of sheep. J. Comp. Pathol. 97:481-485. [DOI] [PubMed] [Google Scholar]
- 42.Yoshiie, K., H.-Y. Kim, J. Mott, and Y. Rikihisa. 2000. Intracellular infection by the human granulocytic ehrlichiosis agent inhibits human neutrophil apoptosis. Infect. Immun. 68:1125-1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yoshitake, H., Y. Takeda, T. Nitto, and F. Sendo. 2002. Cross-linking of GPI-80, a possible regulatory molecule of cell adhesion, induces up-regulation of CD11b/CD18 expression on neutrophil surfaces and shedding of L-selectin. J. Leukoc. Biol. 71:205-211. [PubMed] [Google Scholar]