

Abstract
Ramón y Cajal proposed 100 years ago that memory formation requires the growth of nerve cell processes. One-half century later, Hebb suggested that growth of presynaptic axons and postsynaptic dendrites consequent to coactivity in these synaptic elements was essential for such information storage. In the past 25 years, candidate growth genes have been implicated in learning processes, but it has not been demonstrated that they in fact enhance them. Here, we show that genetic overexpression of the growth-associated protein GAP-43, the axonal protein kinase C substrate, dramatically enhanced learning and long-term potentiation in transgenic mice. If the overexpressed GAP-43 was mutated by a Ser → Ala substitution to preclude its phosphorylation by protein kinase C, then no learning enhancement was found. These findings provide evidence that a growth-related gene regulates learning and memory and suggest an unheralded target, the GAP-43 phosphorylation site, for enhancing cognitive ability.
High-resolution imaging studies of altered nerve cell structure under the influence of synaptic input (1–3) provide a cellular basis for the view that learning involves structural modification of synapses (4, 5). One molecule that has been implicated in input-dependent alterations of synaptic morphology is the growth-associated GAP-43 protein (6), a protein kinase C (PKC) (7, 8) substrate and an intrinsic determinant of structural change at the synapse. GAP-43, previously implicated in memory storage processes (9–16), binds to actin (17) and fodrin (18), and by such protein–protein interactions may affect morphological change.
To determine whether the neuron-specific GAP-43 growth protein in fact regulates memory formation, we studied the effect on learning and synaptic potentiation of its overexpression in transgenic mice. The GAP-43-null mutation is lethal (19). Because evidence from this and other laboratories indicated that learning increases GAP-43 phosphorylation (9–16), one might expect that a transgenic mouse that overexpresses phosphorylatable GAP-43 would demonstrate enhanced learning. A critical corollary of this prediction is that such genetically enhanced learning would not occur if the PKC site of the overexpressed GAP-43 were mutated to prevent its phosphorylation.
Materials and Methods
Animals.
Transgenic mice production has been described in detail elsewhere (20). Briefly, to construct the expression cassette, an 8.2-kb EcoRI GAP-43 genomic fragment including the Thy-1.2 promoter was used. Germline-transmitting chimeras were obtained by standard injection into C57BL/6 blastocysts, and the mutation was crossed into either C57BL/6 or C2D2/DBA genetic backgrounds. G-Phos is the S42wt line, G-NonP the S42A line, and G-Perm the S42D line. Nontransgenic, wild-type (WT) mice from the breeding program were used as controls. Transgenic animals were screened by slot blot hybridization.
Slot Blot and in Situ Hybridization.
Genomic DNA purified from mouse tail was used for slot blot hybridization by using a 32P-labeled chick cDNA probe (21). Chick GAP-43 cDNA (1.0 kb), obtained from Lawrence Baizer (ref. 22; R.S. Dow Neurological Science Institute; Portland, Oregon), was subcloned into the EcoRI site of pGEM3Z plasmid (Promega). The 0.5-kb HindIII fragment of chick GAP-43 cDNA, which contains the C-terminal coding region of GAP-43, was isolated (22) and used for preparing a 32P-labeled chick cDNA probe by nick translation (21). This DNA fragment does not include the DNA site of point mutation (amino acid 42; Ser → Ala or Ser → Asp) near the GAP-43 N terminus (6), and therefore detects the transgene sequence that is identical in the three transgenic mouse strains. The cloned whole sizes of chick and mouse GAP-43 cDNA are 1028 bp and 1227 bp, respectively (22, 23). Comparison of mouse and chick sequences shows 58% identity. Identity in the coding region is 71.7%. There is a 30-nt deletion in the middle of the coding region in chick cDNA so sequence identity is higher when the deletion region is excluded for comparison. Because even this sequence identity was randomly distributed over the 0.5-kb region, the possibility of cross-hybridization of chick GAP-43 cDNA with endogenous mouse GAP-43 genomic DNA would be low. Indeed, no hybridization signal was detected in nontransgenic control mice (see Fig. 3). Genomic DNA hybridization and in situ hybridization were carried out essentially as described previously (refs. 21 and 24, respectively).
Figure 3.

(A and B) Immunohistochemical staining with 7B10 Ab, which recognizes both the endogenous and the transgenic GAP-43 protein in a WT (A) and G-Perm (B) mouse. Note that, in dentate gyrus, increased staining is observed in the perforant path target zone (oml and mml, outer and middle molecular layers, respectively), the mossy cell target zone (iml, inner molecular layer), and the mossy fibers (stratum lucidum). (C–F) Similar GAP-43 overexpression among the three transgenic mouse lines in the major cell fields in hippocampus. Darkfield photomicrographs of in situ hybridization with riboprobe that recognizes transgenic chick GAP-43 mRNA but not endogenous mouse GAP-43 mRNA. Note similar levels of expression in the three transgenic lines (D–F) in the major subfields of the hippocampus and absence of transgene in WT mice (C). Abbreviations: h, hilus; so, stratum oriens; slm, stratum lacunosum moleculare; sr, stratum radiatum; gcl, granule cell layer. Bar = 150 μm.
Immunohistochemistry.
Detection of transgenic GAP-43 protein by immunohistochemistry was carried out as previously described (25) by using Abs described by Meiri et al. (26). Ab 7B10 crossreacted with both chick and mouse GAP-43, whereas 10E81E7 recognized only mouse GAP-43.
Behavior.
Behavioral testing was performed as previously described (27). Briefly, the mice were maintained on a 12-h light/dark cycle and were given unlimited mouse chow and water. Twenty inbred mice (n = 5 per group) ranging from 17–25 g were separated into individual cages before testing. Ages ranged from 50–100 days. A small cup (d = 2 cm, h = .5 cm) into which food reward could be placed was fixed at the end of each of the 8 arms (l = 39 cm, w = 11.5 cm) of the Olton maze. At the start of each trial the animals were placed at the center of the hub (d = 39 cm) in an opaque cylindrical container (d = 12 cm, h = 11 cm), which was removed after 5 s. The animals were then free to explore the maze. Activity was measured by the number of line crossings drawn as quadrants of the 39-cm hub. The animals were food-deprived, and their weights were reduced to 85%–90% original weight and maintained at that weight for all subsequent tasks. Familiarization occurred over the next 7 days, animals exploring the maze for 3 min/day. In the win-shift task with a 1-min delay carried out over 5 days, animals were removed from the maze after retrieving four food rewards on trial 1 and replaced after a 1-min delay. The maze was randomly rotated during the delay, and unretrieved food was moved to the spatial location before rotation. Animals were expected to retrieve the four remaining food rewards on trial 2. An error was scored when the animal entered an arm already visited on trial 1 or 2. This procedure was repeated in the subsequent win-shift task with a 20-min delay, which lasted 4 days. In the win-stay task, carried out for 23 days, only one arm was baited and was randomly selected each day. The animal explored the maze on trial 1 until it retrieved the food reward, after which it was removed for a period of 1 min. The maze was rotated during the delay, and food was replaced at the original spatial location. The animal again explored the maze on trial 2 until the single food reward was retrieved or after a maximum of 12 arm entries. Errors were scored when an entry was made into an arm other than the baited arm. A criterion was set for two or fewer errors on 4 consecutive days.
Electrophysiology.
In vivo long-term potentiation (LTP) in the intact mouse hippocampus was carried out as described in detail elsewhere (28, 29). Briefly, field potentials from the dentate gyrus in urethane-anesthetized (1 g/kg) mice maintained on a 37°C heating jacket, and conforming to approved American Association of Laboratory Animal Care standards, were evoked by perforant path stimulation. Stimulation and recording electrodes were positioned after observation of the characteristic molecular layer laminar profile. The recording electrode was placed in the dentate hilus where the maximum population spike potential was obtained. Baseline stimulation voltage of 0.1-ms pulses delivered at 0.1 Hz was adjusted so that the population spike amplitude was one-third the population excitatory postsynaptic potential (EPSP) amplitude. Stimulation intensity selected for tetanus (three 400-Hz trains at 0.1 Hz, with each train consisting of eight 0.4-ms pulses per train) was the minimum voltage required to elicit the maximum population spike. Treatment effects were evaluated by using a repeated-measures ANOVA, and individual comparisons were made with t test.
Results
We studied three different transgenic mouse lines that were under the control of the Thy-1 promoter as initially described (20). One transgenic line, here designated G-Phos, overexpressed the phosphorylatable form of chick GAP-43. A second line, designated G-NonP, overexpressed an unphosphorylatable form, in which alanine was substituted for serine-42, preventing PKC from phosphorylating GAP-43. The third line, designated G-Perm, overexpressed a permanently pseudophosphorylated form of chick GAP-43 in which aspartate was substituted for serine-42. WT mice that did not harbor the transgene were from the same breeding program as the transgenic mice and served as controls. It may be noted that all transgenic mice had levels of endogenous GAP-43 that were no different from WT controls.
The ability of each of these four groups to remember location of food was evaluated in the 8-arm Olton radial maze, first in a delayed nonmatching to sample (win-shift) task and then in a delayed matching to sample (win-stay) paradigm. Both paradigms test for spatial and working memory as described previously (27).
G-Phos animals performed the win-shift delay task with fewer errors than G-NonP mice, who performed at chance levels during training (Fig. 1A). This confirmed the difference predicted between G-Phos and G-NonP mouse strains. Both WT controls and G-Perm animals were superior to G-NonP mice. The three strains of transgenic mice thus demonstrated discernibly different behavioral phenotypes in this memory task.
Figure 1.
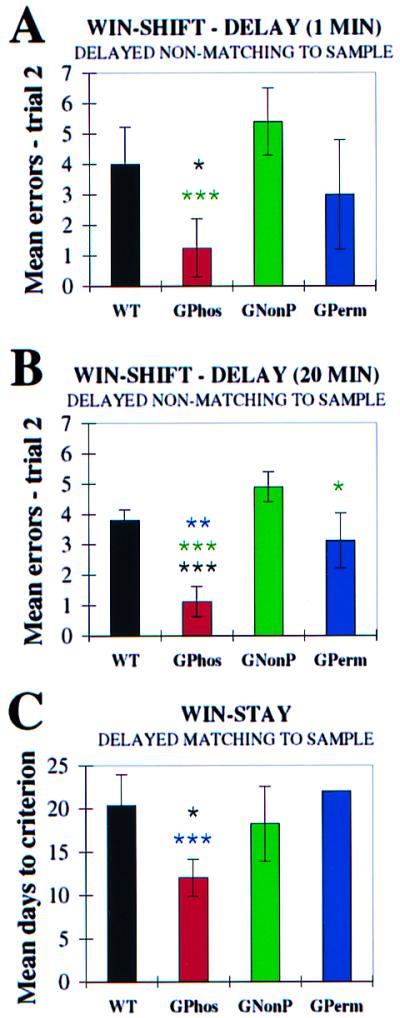
(A) Effect of GAP-43 overexpression on acquisition of 1-min delayed nonmatching to sample (win-shift) task. Chance performance is slightly greater than four arm entries. The mean errors on trial 2 are compared among the groups. G-Phos animals, with phosphorylatable GAP-43, committed significantly fewer errors (ANOVA followed by individual t tests) than WT controls and G-NonP, overexpressing nonphosphorylatable GAP-43. (B) In the 20-min delay win-shift task, G-Phos mice performance was significantly superior to the other 3 mouse lines. (C) Effect of GAP-43 overexpression on delayed-matching to sample (win-stay) task. Mean days to criterion, two errors or fewer on 4 consecutive days, is compared among the groups. G-Phos animals required significantly fewer days to reach criterion than either G-Perm, overexpressing permanently pseudophosphorylated GAP-43, or WT controls. Errors were scored when the animal entered an arm other than the one baited arm. All groups, except G-NonP, began the task committing more errors than at chance level (for A-C, *, P < 0.05; **, P < 0.01; and ***, P < 0.001).
When the delay interval in the win-shift task was then increased from 1 min to 20 min, increasing the temporal demands on spatial memory systems, the superiority of G-Phos mice relative to the other mouse strains was more clearly evident. Indeed G-Phos animals made significantly fewer errors relative to each of the other mouse strains (Fig. 1B, P < 0.02). G-NonP animals were, as in the 1-min task, unable to learn this 20-min delay task. G-Perm mice were significantly inferior to G-Phos animals (see Fig. 1B).
We then instituted a paradigm shift. Mice were now required to return to the same spatial location where the single arm was baited, i.e., a delayed matching-to-sample (or win-stay) task as opposed to the prior win-shift task. Because there were no intramaze cues that distinguished one arm from another, this was a spatial memory task, not one based on cued recall.
Striking behavioral phenotypic differences were again observed among the mice from the different lines. When the number of days required to unlearn the win-shift strategy was compared, WT controls achieved chance performance on day 7 whereas G-Perm animals, in contrast, required 22 days. G-NonP animals, in contrast to the other mouse lines, actually showed chance levels of performance on day 1 of win-stay training. This chance performance occurred because G-NonP mice learned win-shift poorly, if at all; hence, there was little requirement to unlearn the win-shift rule. G-Phos transgenics required 10 days to achieve chance performance.¶
Only the G-Phos animals, as a group, readily learned the spatial win-stay task and, as predicted, were superior to G-NonP mice. Because we had previously reported that, in contrast to rats, three different inbred strains of mice (C57/B6, B62DF1/J, and ICR/J) were unable to learn this win-stay spatial task (30), it was surprising that all of the G-Phos mice achieved criterion performance, and did so in a mean of 12 days (Fig. 1C). Surprising, too, was the fact that these mice took fewer days to achieve criterion than did rats (27, 30). In contrast, 13 of 15 of the G-NonP, G-Perm, and WT mice failed to reach criterion performance.
We sought to determine whether these striking phenotypic learning differences would demonstrate parallel differences in LTP, a physiological mechanism for learning (31). Drawing such parallels remains controversial (32), given the gene-targeting studies that show either positive (33) or negative (34) correlations between learning and LTP. Nonetheless, different LTP phenotypes can be predicted here based on earlier reports demonstrating both a selective LTP-induced increase in GAP-43 phosphorylation and its prevention by LTP-blocking N-methyl-d-aspartate (NMDA) receptor antagonists (35–37).
The G-Phos and G-Perm mice indeed demonstrated enhanced LTP compared with WT controls and G-NonP mice. We used the LTP paradigm in the intact mouse (28, 29) to study synaptic plasticity of the perforant path–granule cell synapse, part of the hippocampal circuitry involved in both win-shift and win-stay learning. G-Phos showed enhanced LTP to 700% of baseline as compared with the 400% of WT controls (Fig. 2A). G-Perm mice showed LTP enhancement exceeding 1000% of baseline. G-NonP mice showed, in contrast, no enhancement of the LTP response beyond that seen in WT controls (P > 0.50). Given that G-NonP mice overexpressed the same levels of GAP-43 transgenic protein as the G-Phos and G-Perm mice (see what follows), one may conclude that the state of the GAP-43 phosphorylation site regulated the level of LTP enhancement.
Figure 2.
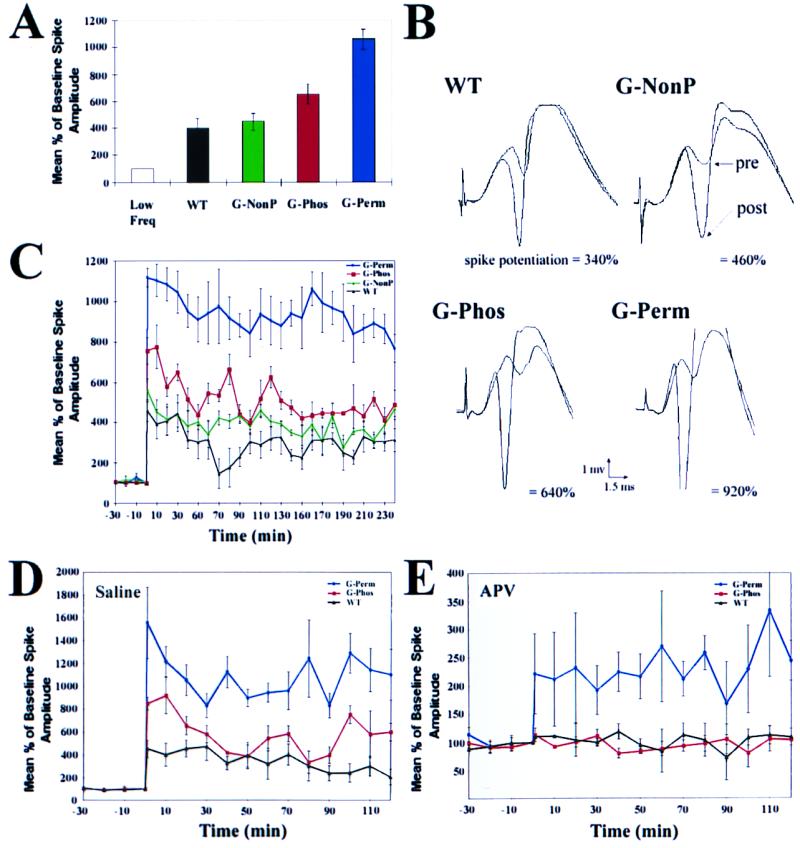
As compared with WT controls, overexpression of GAP-43 of either the G-Phos or G-Perm type led to an immediate and persistent enhancement of conventional LTP. In contrast, overexpression of G-NonP, the nonphosphorylatable GAP-43, did not enhance LTP beyond that seen in WT mice. (A) Comparison of low frequency (low freq) controls receiving an 0.1 Hz stimulus, with transgenic and WT mice receiving a high frequency tetanus. There was no difference in response to low frequency stimulation over the 4-h test period among the four mouse groups. Mean percentage increase relative to baseline before tetanus in population spike (±SEM) recorded from the granule cell layer of the dentate gyrus at 30 min was similar for WT and G-NonP animals but was enhanced by the G-Phos and G-Perm transgene. The latter two groups demonstrated significantly greater LTP than WT and G-NonP (P < 0.05 for G-Phos vs. WT and vs. G-NonP; P < 0.001 for G-Perm vs. WT and G-NonP; P < 0.02 for G-perm vs. G-Phos (ANOVA followed by t tests of individual comparisons). n = 4 per group. (B) Average of five consecutive waveforms taken from each of the four mouse strains before, and 1 h after, perforant path tetanus. Note that, before tetanus, waveform shape and amplitude are similar for the four lines. After tetanus, a dramatic enhancement was observed in the population spike (below each waveform is the percentage enhancement for that particular set of waveforms). (C) Kinetics of enhanced LTP over 4-h period. Note the persistence of enhanced LTP in G-Perm animals, and its decline in G-Phos mice, approaching the level of enhancement seen in WT and G-NonP animals. (D and E) Effect of NMDA receptor antagonist 2-amino-5-phosphonovaleric acid (APV) on G-Perm, G-Phos, and WT animals. Whereas LTP is blocked by APV in G-Phos (red line) and WT (black line) controls as expected, LTP is still present in G-Perm animals (blue line) after APV treatment. In D, note that the amplitude and kinetics of enhanced LTP essentially replicate the results in uninjected animals (C), even with injection of 21 nl injection of vehicle into the molecular layer 15 min before tetanus (n = 3 per group). Moreover, as in C, the decay kinetics of WT controls relative to G-Perm and G-Phos animals are similar. Data are expressed as a percentage of the mean baseline response over the 30 min before tetanus. Each response was the average of five individual waveforms.
The enhanced LTP response in G-Phos and G-Perm mice was unlikely to be due to a difference in sensitivity or reactivity of the perforant pathway to low frequency or tetanic stimulation because the voltages selected, based on specified response criteria, were comparable in all four mouse strains (see Table 1). Nor was the enhanced LTP response or the phenotypic differences in learning due to differential levels of GAP-43 overexpression in the lines studied, because prior whole brain analysis (20) and the present studies of hippocampal transgene protein and mRNA (Fig. 3) indicated that the three mouse strains did not differ. Indeed, we found that all three lines showed qualitatively similar levels of chick GAP-43 immunohistochemical staining comparable to that shown for the G-Perm mice in Fig. 3B. Moreover, transgene expression level was similar among the different mouse strains (Fig. 3 D–F), when measured by using a riboprobe for chick GAP-43 mRNA. This probe did not recognize endogenous mouse GAP-43 mRNA, as demonstrated by the lack of hybridization in WT controls (Fig. 3C).
Table 1.
Electrophysiological responses
| Line | Baseline voltage, V | Baseline spike response, mV | Tetanus voltage, V |
|---|---|---|---|
| WT | 4.0 ± 0.23 | 1.05 ± 0.2 | 6.0 ± 0.25 |
| G-NonP | 3.9 ± 0.56 | 1.31 ± 0.6 | 5.8 ± 0.15 |
| G-Phos | 3.6 ± 0.53 | 1.20 ± 0.7 | 5.5 ± 0.42 |
| G-Perm | 4.0 ± 0.21 | 1.15 ± 0.3 | 6.0 ± 0.19 |
Electrophysiological responses to the perforant path stimulus prior to LTP were not different among the three transgenic mouse lines (G-Phos, G-NonP, and G-Perm), nor was there a difference between transgenic mice and nontransgenic (WT) controls. Among the four groups, there were thus no differences in: (i) baseline voltage, the voltage level used to stimulate perforant path and elicit the criterion baseline response, a population spike having one-third the amplitude of the population EPSP; (ii) baseline spike response, population spike amplitude prior to LTP; and (iii) tetanus voltage, the minimum stimulation level required to elicit the maximum population spike. The data represent mean values ± SEM.
There were also no detectable distortions in the cellular arrays or lamination patterns of the hippocampus (compare Fig. 3 A and B), such as has been seen in other mouse mutants (e.g., ref. 38). Electrophysiological analysis of laminar profiles, performed as described previously (35), was essentially identical in transgenic and nontransgenic animals. There was also no apparent alteration in immunohistochemical staining of the endogenous GAP-43, nor were alterations detected in calretinin and γPKC distribution in hippocampal cell fields among the three transgenic strains compared with WT controls. An initial count of the number of synapses in WT and G-Perm mice by using serial electron microscopy (EM) sections revealed no difference between WT controls and transgenic G-Perm mice in the hippocampal molecular layer. This procedure was used in two separate synaptic counts of different tissue blocks performed blind within both the inner and middle molecular layer of the dentate gyrus. Nor were there any apparent differences among the transgenic mice and WT controls in location or silver staining intensity of over 200 non-GAP-43 proteins detected in two-dimensional gels (data not shown).
Discussion
It is concluded from the present behavioral and electrophysiological studies that both learning and synaptic plasticity were enhanced by overexpression of a brain growth protein. Moreover, this regulation occurred through the PKC phosphorylation site of GAP-43. Prior studies demonstrating both increased GAP-43 phosphorylation after learning and highly correlated increases in GAP-43 phosphorylation with LTP-induced change converge with the present results to indicate that this phosphorylation event is a critical one for learning and subsequent retention. Time-dependent blockade of memory and LTP by PKC inhibitors that prevent LTP-induced increases in GAP-43 phosphorylation support this conclusion (11, 14–16).
Overexpression of GAP-43 is likely to enhance learning and LTP in two ways. First, input-dependent alterations in synaptic connectivity during postnatal development may occur. Because there is more GAP-43 in the synaptic terminal, normal inputs experienced by the mice would amplify these subtle synaptic modifications. This synaptic change could then lead to an elaborated synaptic network that would enhance learning and LTP. Indeed, the mossy fiber system, in some cases, can sprout into the stratum oriens from stratum lucidum (20). Because only some transgenic mice showed this pattern, the possibility exists that this is an input-dependent function interacting with GAP-43 overexpression. This proposal is supported by recent work showing that growth of mossy fibers on postnatal days 4–8 can be regulated through the NMDA receptor (39). Moreover, NMDA receptor antagonists reduce the GAP-43 mRNA of the cells of origin of the affected axons and significantly reduce this growth. It is interesting that the transgenic protein is driven by the Thy-1 promoter, which is activated postnatally beginning on day 7.
A second way in which increased levels of GAP-43 in the presynaptic terminal could enhance learning and LTP is at the time of the experiment itself. In addition to synaptic growth, GAP-43 may also facilitate synaptic function via enhanced transmitter release (6). The schematic diagram of Fig. 4 depicts how each GAP-43 transgene, except G-NonP, might combine with endogenous GAP-43 to regulate transmitter release.
Figure 4.

Proposed mechanism to explain enhanced LTP in transgenic animals overexpressing GAP-43. In nontransgenic animals, presynaptic PKC is activated by an NMDA-dependent postsynaptic retrograde signal. Phosphorylated endogenous GAP-43 (black circle) interacts only with calcium-sensor proteins of the exocytotic protein machine (EPM) (see ref. 49) to enhance release when intraterminal calcium is raised sufficiently. Because low frequency activity does not raise intraterminal calcium to activate EPM sufficiently, phosphorylated GAP-43 alone would be insufficient to induce LTP. Once PKC phosphorylates both endogenous and transgenic G-Phos (red circle), the terminal is “primed” to release more transmitter upon subsequent depolarization of the presynaptic terminal. Because the G-Perm variant of GAP-43 (blue square) can bind to activated EPM without PKC phosphorylation, this mutated form of GAP-43 does not require the influence of NMDA receptor activation as shown in Fig. 2 D and E. Note that either G-Phos or G-Perm transgenic GAP-43, but not the G-NonP variant (green circle), can sum with endogenous GAP-43 to enhance exocytosis.
Fig. 4 may be applied to the developmental mechanism just discussed because NMDA receptor activation would regulate postnatal developmental growth through its retrograde influence on PKC phosphorylation of GAP-43. This pathway may act through N-CAM-mediated activation of fibroblast growth factor (40). There is considerable debate concerning the identity of the retrograde factor. One attractive possibility is an unsaturated fatty acid such as arachidonate, which has been shown to be released after LTP (41), to activate PKC (42, 43), and to increase GAP-43 phosphorylation (37).
The second mechanism of enhancement proposes that the presence in the presynaptic terminal of increased GAP-43 at the time of the learning or LTP induction is critically important. The presence of the overexpressed G-Phos or G-Perm sums with endogenous GAP-43 and by complexing with exocytotic protein machinery (EPM) facilitates synaptic transmission (Fig. 4). GAP-43 exists within a presynaptic lattice of dynamic protein–protein interactions. Phosphorylated GAP-43 complexes with exocytotic proteins such as synaptotagmin (44, 45) to facilitate release and with endocytotic proteins such as rabaptin-5 (46) to facilitate reuptake. GAP-43 overexpression would likely alter the distribution of the membrane components to which it binds, thereby giving rise to the altered morphological changes at the synapse (47, 48) often subsumed under the rubric of “synaptic growth.” Moreover, GAP-43 also binds cytoskeletal elements such as actin (17, 1) and brain spectrin or fodrin (18).
Perturbation of the presynaptic protein lattice by mutation of the GAP-43 phosphorylation site could thus enhance both transmitter release and presynaptic terminal growth necessary for enhanced LTP. This latticial adjustment within individual synapses would in turn alter synaptic strength in the circuits that underlie the enhanced learning observed.
The striking difference in persistence of LTP enhancement between G-Perm vs. G-Phos mice may be related to the fact that the GAP-43 in G-Perm mice cannot be dephosphorylated, hence, their persistence of enhanced potentiation (Fig. 2C). The enhancement of learning in G-Phos mice in contrast to G-Perm animals strongly suggests a critical role for the GAP-43 phosphorylation–dephosphorylation cycle. The absence of such cycles in G-Perm mice would not hinder expression in the single-trial LTP paradigm. Such cycles do not occur with the mutated transgenic protein in either poor learning G-NonP or G-Perm mice. Because it has been shown that learning and retention selectively increase GAP-43 phosphorylation (9), it is attractive to think that learning requires a GAP-43 that can fine-tune its level of phosphorylation.
The model of Fig. 4 helps explain both why LTP is enhanced even though GAP-43 is already phosphorylated in G-Perm mice and why they do not show enhancement with low frequency stimulation. Concerning the enhancement, the model indicates that G-Perm combines with Ca2+-activated EPM to enhance transmitter release. Thus, under physiological conditions, the increment of phosphorylated GAP-43 occurs shortly after tetanus; in the transgenic mouse the elevated level of phosphorylated GAP-43, while present before tetanus, combines with EPM after the tetanus. In G-Perm, moreover, both the physiological and transgenic phosphorylated GAP-43 combine as shown in Fig. 4 to produce the enhanced LTP. Thus, it is not the process of phosphorylation per se, but the presence of phosphorylated GAP-43 in combination with Ca2+-activated EPM that is critical. This protein–protein interaction relates to the second issue of the absence of low frequency enhancement in G-Perm mice. This can be understood in Hebbian terms (49), given that enhancement occurs only with co-occurrence, here satisfied by the conjunction of a postsynaptic release of retrograde factor combining with presynaptic activation of EPM. Thus, there is no enhancement with low frequency stimulation because the conjunction does not take place.
Perusal of Fig. 4 suggests a unique prediction. Because the retrograde influence of NMDA receptor activation on GAP-43 phosphorylation would be present for G-Phos but not G-Perm mice, NMDA receptor blockade should not influence LTP in G-Perm mice to the same extent as in G-Phos animals. As Fig. 2 D and E shows, this prediction was confirmed. These data make the essential point that LTP can be rescued from NMDA receptor blockade effects when permanently phosphorylated GAP-43 is present in the presynaptic terminal. This result adds weight to the proposal that GAP-43 phosphorylation is downstream from NMDA receptor activation (35–37). Overexpression of the permanently phosphorylated form of GAP-43 could alter postsynaptic α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) and/or glutamate metabotropic receptors in such a way that initiation of synaptic potentiation would bypass the NMDA receptor (50).
It was recently reported that overexpression of the NMDA receptor subunit NR2B led to a genetically induced enhancement of learning and of LTP (33) not dissimilar from that reported here. Such a result lends support to the present proposed schema (Fig. 4) that emphasizes the linkage between activation of the NMDA receptor postsynaptically and GAP-43 phosphorylation presynaptically. Because blockade of the NMDA receptor in fact blocks LTP-induced increases in GAP-43 phosphorylation (35–37), overexpression of the NMDA receptor, by producing a greater retrograde signal, would be predicted by the model to increase GAP-43 phosphorylation and thereby enhance learning.
Linking different genes whose overexpression yields similar phenotypic enhancements of learning suggests a genetic approach to specifying the particular signaling pathways of synaptic plasticity. The fact that G-NonP animals demonstrated “normal” LTP no different from controls indicates that their inability to learn win-shift rules was not due to a gross deficiency in capacity for synaptic modification. On the other side of the coin, the superior learning performance of G-Phos mice suggests novel therapeutic approaches that seek to mimic the learning–enhancing chemistries produced here.
Acknowledgments
We thank Dr. Pico Caroni at the Friedrich-Miescher Institute, Basel, Switzerland, for providing the transgenic mice. Gratitude is also expressed to him and Dr. James L. McGaugh for their valuable comments on earlier drafts of the manuscript. Thanks to Dr. Yuri Geinisman for his assistance with the electron microscopy sections, Dr. Laurence Baizer for chick GAP-43 cDNA and Dr. Karina Meiri for the GAP-43 Abs. This work was supported by the Whitehall Foundation, P. Randall, National Science Foundation Grant IBN-9811592 (to A.R.), Fundacion Barcalo (to I.C.), and a National Science Foundation Postdoctoral Fellowship (to P.S.).
Abbreviations
- GAP-43
growth-associated protein of 43 kDa
- PKC
protein kinase C
- WT
wild-type
- LTP
long-term potentiation
- NMDA
N-methyl-d-aspartate
- EPM
exocytotic protein machinery
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Regression lines for each group were calculated, plotting errors over days until chance performance was reached. The slopes of the lines were determined by regression coefficients: G-Phos, −0.50 (0.17) F(1,42) = 8.42, P < 0.007; G-Perm, −0.26 (0.09), F(1,52) = 7.62, P < 0.01; and WT, −1.09 (0.31), F(1,26) = 11.99, P < 0.003.
References
- 1.Fischer M, Kaech S, Knutti D, Matus A. Neuron. 1998;20:847–854. doi: 10.1016/s0896-6273(00)80467-5. [DOI] [PubMed] [Google Scholar]
- 2.Engert F, Bonhoeffer T. Nature (London) 1999;399:66–70. doi: 10.1038/19978. [DOI] [PubMed] [Google Scholar]
- 3.Maletic-Savatic M, Malinow R, Svoboda K. Science. 1999;283:1923–1927. doi: 10.1126/science.283.5409.1923. [DOI] [PubMed] [Google Scholar]
- 4.Ramón y Cajal S. Histologie du Système Nerveux de L'homme et des Vertébrés (Consejo Superior de Investigaciones Científicas, Madrid) trans. Azoulay, L. (1909) Textura del Sistema Nervioso del Hombre y de los Vertebrados. 1952. [Google Scholar]
- 5.Hebb D O. Organization of Behavior. New York: Wiley; 1949. [Google Scholar]
- 6.Benowitz L I, Routtenberg A. Trends Neurosci. 1997;20:84–91. doi: 10.1016/s0166-2236(96)10072-2. [DOI] [PubMed] [Google Scholar]
- 7.Nishizuka Y. FASEB J. 1995;9:484–496. [PubMed] [Google Scholar]
- 8.Mellor H, Parker P J. Biochem J. 1998;332:281–292. doi: 10.1042/bj3320281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Linden D J, Routtenberg A. Brain Res Rev. 1989;14:279–296. doi: 10.1016/0165-0173(89)90004-0. [DOI] [PubMed] [Google Scholar]
- 10.Sheu F S, McCabe B J, Horn G, Routtenberg A. Proc Natl Acad Sci USA. 1993;90:2705–2709. doi: 10.1073/pnas.90.7.2705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jerusalinsky D, Quillfeldt J A, Walz R, Da Silva R C, Medina J H, Izquierdo I. Behav Neural Biol. 1994;61:107–109. doi: 10.1016/s0163-1047(05)80063-9. [DOI] [PubMed] [Google Scholar]
- 12.Ehrlich Y H, Rabjohns R R, Routtenberg A. Pharm Biochem Behav. 1977;6:169–174. doi: 10.1016/0091-3057(77)90068-5. [DOI] [PubMed] [Google Scholar]
- 13.Cammarota M, Paratcha G, Stein M, Bernebeau R, Izquierdo I, Medina J H. Neurochem Res. 1997;22:499–505. doi: 10.1023/a:1027324214060. [DOI] [PubMed] [Google Scholar]
- 14.Lovinger D, Akers R, Colley P, Linden D, Routtenberg A. Brain Res. 1986;399:205–211. doi: 10.1016/0006-8993(86)91510-6. [DOI] [PubMed] [Google Scholar]
- 15.Lovinger D M, Wong K L, Murakami K, Routtenberg A. Brain Res. 1987;436:177–183. doi: 10.1016/0006-8993(87)91573-3. [DOI] [PubMed] [Google Scholar]
- 16.Colley P A, Sheu F-S, Routtenberg A J. J Neurosci. 1990;10:3353–3360. doi: 10.1523/JNEUROSCI.10-10-03353.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.He Q, Dent E W, Meiri K F. J Neurosci. 1997;17:3515–3524. doi: 10.1523/JNEUROSCI.17-10-03515.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Riederer B, Routtenberg A. Mol Brain Res. 1999;71:345–348. doi: 10.1016/s0169-328x(99)00179-5. [DOI] [PubMed] [Google Scholar]
- 19.Strittmatter S M, Fankhauser C, Huang P L, Mashimo H, Fishman M C. Cell. 1995;80:445–452. doi: 10.1016/0092-8674(95)90495-6. [DOI] [PubMed] [Google Scholar]
- 20.Aigner L, Arber S, Kapfhammer J P, Laux T, Schneider C, Botteri F, Brenner H-R, Caroni P. Cell. 1995;83:269–278. doi: 10.1016/0092-8674(95)90168-x. [DOI] [PubMed] [Google Scholar]
- 21.Sambrook J, Fritsch E F, Maniatis T. Molecular Cloning: A Laboratory Manual. Plainview, NY: Cold Spring Harbor Lab. Press; 1989. [Google Scholar]
- 22.Baizer L, Alkan S, Stocker K, Ciment G. Mol Brain Res. 1990;7:61–68. doi: 10.1016/0169-328x(90)90074-n. [DOI] [PubMed] [Google Scholar]
- 23.Cimler B M, Giebelhaus D H, Wakim B T, Storm D R, Moon R T. J Biol Chem. 1987;262:12158–12163. [PubMed] [Google Scholar]
- 24.Meberg P J, Routtenberg A. Neuroscience. 1991;45:721–733. doi: 10.1016/0306-4522(91)90284-u. [DOI] [PubMed] [Google Scholar]
- 25.Cantallops I, Routtenberg A. J Comp Neurol. 1996;366:303–319. doi: 10.1002/(SICI)1096-9861(19960304)366:2<303::AID-CNE9>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 26.Meiri K F, Bickerstaff L E, Schwob J E. J Cell Biol. 1991;112:991–1005. doi: 10.1083/jcb.112.5.991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Collier T J, Quirk G S, Routtenberg A. Brain Res. 1987;409:316–328. doi: 10.1016/0006-8993(87)90717-7. [DOI] [PubMed] [Google Scholar]
- 28.Namgung U, Valcourt E, Routtenberg A. Brain Res. 1995;689:85–92. doi: 10.1016/0006-8993(95)00531-t. [DOI] [PubMed] [Google Scholar]
- 29.Namgung U, Matsuyama S, Routtenberg A. Proc Natl Acad Sci USA. 1997;94:11675–11680. doi: 10.1073/pnas.94.21.11675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McNamara R K, Namgung U, Routtenberg A. Mol Brain Res. 1996;40:177–187. doi: 10.1016/0169-328x(96)00048-4. [DOI] [PubMed] [Google Scholar]
- 31.Bliss T V P, Collingridge G L. Nature (London) 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- 32.Shors T J, Matzel L D. Behav Brain Sci. 1997;20:597–614. doi: 10.1017/s0140525x97001593. [DOI] [PubMed] [Google Scholar]
- 33.Tang Y-P, Shimizu E, Dube G R, Rampon C, Kerchner G A, Zhuo M, Liu G, Tsien J. Nature (London) 1999;401:63–69. doi: 10.1038/43432. [DOI] [PubMed] [Google Scholar]
- 34.Migaud M, Charlesworth P, Dempster M, Webster L C, Watabe A M, Makhinson M, He Y, Ramsay M F, Morris R G, Morrison J H, O'Dell T J, Grant S G. Nature (London) 1998;396:433–439. doi: 10.1038/24790. [DOI] [PubMed] [Google Scholar]
- 35.Linden D J, Wong K L, Sheu F-S, Routtenberg A. Brain Res. 1988;458:142–146. doi: 10.1016/0006-8993(88)90506-9. [DOI] [PubMed] [Google Scholar]
- 36.Leahy J C, Luo Y, Kent C S, Meiri K F, Vallano M L. Neuroscience. 1993;52:563–574. doi: 10.1016/0306-4522(93)90406-6. [DOI] [PubMed] [Google Scholar]
- 37.Luo Y, Vallano J. Neurochemistry. 1995;64:1808–1818. doi: 10.1046/j.1471-4159.1995.64041808.x. [DOI] [PubMed] [Google Scholar]
- 38.Grant S G, O'Dell T J, Karl K A, Stein P L, Soriano P, Kandel E R. Science. 1992;258:1903–1910. doi: 10.1126/science.1361685. [DOI] [PubMed] [Google Scholar]
- 39.Cantallops I, Routtenberg A. J Neurobiol. 1999;41:208–220. [PubMed] [Google Scholar]
- 40.Meiri K F, Saffell J L, Walsh F S, Doherty P J. J Neurosci. 1998;15:10429–10437. doi: 10.1523/JNEUROSCI.18-24-10429.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Clements M P, Bliss T V, Lynch M A. Neuroscience. 1991;45:379–389. doi: 10.1016/0306-4522(91)90235-g. [DOI] [PubMed] [Google Scholar]
- 42.Murakami K, Routtenberg A. FEBS Lett. 1985;192:189–193. doi: 10.1016/0014-5793(85)80105-8. [DOI] [PubMed] [Google Scholar]
- 43.Murakami K, Chan S Y, Routtenberg A. J Biol Chem. 1986;261:15424–15429. [PubMed] [Google Scholar]
- 44.Haruta T, Takami N, Ohmura M, Misumi Y, Ikehara Y. Biochem J. 1997;325:455–463. doi: 10.1042/bj3250455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sudhof T C. Nature (London) 1995;375:645–653. doi: 10.1038/375645a0. [DOI] [PubMed] [Google Scholar]
- 46.Neve R L, Coopersmith R, McPhie D L, Santeufemio C, Pratt K G, Murphy C J, Lynn S D. J Neurosci. 1998;18:7757–7767. doi: 10.1523/JNEUROSCI.18-19-07757.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tarrant S, Routtenberg A. Tissue Cell. 1977;9:461–473. doi: 10.1016/0040-8166(77)90006-4. [DOI] [PubMed] [Google Scholar]
- 48.Geinisman Y. Hippocampus. 1994;3:143–157. [Google Scholar]
- 49.Routtenberg A. Trends Neurosci. 1999;22:255–256. doi: 10.1016/s0166-2236(99)01412-5. [DOI] [PubMed] [Google Scholar]
- 50.Manahan-Vaughan D, Reiser M, Pin J P, Wilsch V, Bockaert J, Reymann K G, Riedel G. Neuroscience. 1996;72:999–1008. doi: 10.1016/0306-4522(95)00594-3. [DOI] [PubMed] [Google Scholar]