

Abstract
HIV drug therapy often fails because of the appearance of multidrug-resistant virus. There are two possible scenarios for the outgrowth of multidrug-resistant virus in response to therapy. Resistant virus may preexist at low frequencies in drug-naïve patients and is rapidly selected in the presence of drugs. Alternatively, resistant virus is absent at the start of therapy but is generated by residual viral replication during therapy. Currently available experimental methods are generally too insensitive to distinguish between these two scenarios. Here we use deterministic and stochastic models to investigate the origin of multidrug resistance. We quantify the probabilities that resistant mutants preexist, and that resistant mutants are generated during therapy. The models suggest that under a wide range of conditions, treatment failure is most likely caused by the preexistence of resistant mutants.
In recent years, management of HIV infection has greatly improved because of the development of new treatment protocols, involving the combination of highly potent drugs (1–5). However, combination therapy is not effective in all patients and may fail because of severe side effects, nonadherence to therapy protocol, lack of potency of drugs, or emergence of resistant virus (refs. 2 and 6; See www.hivatis.org/trtgdlns.html and refs. therein).
Generally, there are two main processes leading to resistance-related treatment failure: preexisting resistant strains may be selected by the drugs used, or resistant mutants are generated de novo by residual virus replication during treatment. It is important to distinguish between these processes, because they require different actions to improve therapy. If treatment fails because of preexisting resistant virus, than increasing the efficacy of the drugs (for example by increasing the dosage) may not suffice to control virus replication. Rather, several drugs with different resistance profiles need to be combined, reducing the likelihood that strains resistant to combination therapy are present in the first place. On the other hand, if resistance arises de novo during treatment, then increasing the dosage of the drug may lead to a more effective treatment. In this case, the objective would be to minimize any residual replication of the sensitive virus during therapy, because this would reduce the probability of producing a resistant mutant.
Thus, determination of which of the two causes for treatment failure is more likely may be helpful to find the best therapy regimen. Laboratory testing for the presence of resistant strains in a patient has been proposed (7–12). However, current methods either are not sensitive enough to detect mutants at very low frequencies or are too laborious to be used in clinical practice (7). In view of these difficulties, this question has been addressed by using population dynamical models (13–15). However, so far, these theoretical approaches have underestimated the probability of treatment failure attributed to de novo generation of resistance by the residual virus replication during treatment. Here, we use both deterministic and stochastic approaches to investigate the origin of drug-resistant mutants and derive an upper limit to the probability of emergence of resistant virus during therapy. On the basis of quantifiable parameters such as the viral load and the viral mutation rate, we estimate the likelihood of preexistence of resistant strains in comparison to the likelihood of emergence of resistant virus during therapy. We emphasize at the outset that we are not concerned with drug resistance in patients who were infected by resistant carriers. Although a major concern for the future, to date the spread of resistant strains seems to account only for a minority of treatment failures caused by resistance (16–18). Instead, we focus on the emergence of resistance in drug-naïve patients who were infected with sensitive virus, but who may develop resistant mutants at low frequency in a mutation-selection equilibrium.
Definition of the Model
We begin with the basic model of HIV dynamics (13, 14, 19–22):
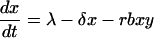 |
1 |
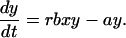 |
For a detailed description of this model, see ref. 23. Here the variables x and y denote the population densities of susceptible and infected cells, respectively. The model has five parameters: λ, the rate of immigration of susceptible cells from a pool of precursor cells; δ, the death rate of susceptible cells; b, the infectivity rate; a, the per capita death rate of infected cells; and r, the inhibitory effect of drug therapy on virus replication, which is between 0 and 1, with r = 1 corresponding to no treatment.
This model has two equilibria corresponding to the uninfected and infected steady states. The evolution of the system to one or the other steady state is determined by the basic reproductive ratio, R0 (24–26). In the present context, R0 is defined as the average number of secondary infected cells produced by the first infected cell introduced in a wholly susceptible population. For the above model, the basic reproductive rate before the start of treatment is given by Rb = λb/(δa). If Rb < 1, then on average one infected cell produces less than one secondary infected cell, and hence the system goes to the uninfected steady state given by xU = λ/δ and yU = 0. Conversely, if Rb > 1, the system goes to the infected steady state given by xI = a/b and yI = λ/a − δ/b.
In the absence of treatment (r = 1), the virus is expected to have a basic reproductive ratio larger than 1, because otherwise it is unable to cause or sustain an infection. During treatment (r < 1), the new basic reproductive ratio, Rd = rλb/(δa), can be smaller or larger than 1, depending on whether the virus population is sensitive or resistant to treatment. In the following, we define resistant viruses as those viruses with a basic reproductive ratio during treatment larger than one, Rd > 1. This defines drug resistance not in terms of an in vitro assay, but as a combined property of host, virus, and drug. Note, however, that a basic reproductive ratio smaller than one does not imply that there is no replication. The de novo production of a resistant virus during therapy is possible so long as the basic reproductive ratio is larger than zero.
Analysis of the Model
We use the above model to distinguish between the two alternative hypotheses for the cause of treatment failure caused by resistance: (i) The preexistence hypothesis, according to which treatment failure is caused by the existence of resistant mutants in the patient's virus population before the start of therapy; and (ii) the emergence hypothesis, according to which resistant variants are absent before therapy, but treatment failure occurs because resistant mutants are produced from the sensitive virus population as it declines during therapy.
The likelihood of emergence of resistance during therapy depends on the number of cells that become newly infected as the sensitive virus population declines during therapy. This number is given by rb ∫t=0t=∞ xydt, where t = 0 is the start of therapy, and r < 1. Unfortunately, this integral cannot be calculated in closed form, because we do not have analytical solutions for x(t) and y(t). However, we can approximate Eq. 1 by neglecting the term rbxy in the first equation for x(t). The simplified model is:
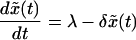 |
2 |
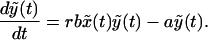 |
Because the simplified model has no nonvanishing steady state for the infected cell population, we use the infected steady state of Eq. 1 as the initial condition. Because Eq. 2 neglects the loss rate of susceptible cells because of infection, x̃(t) overestimates x(t), as given by the full model (Eq. 1). As a consequence, ỹ(t) also overestimates y(t), because the per capita rate of production of new infected cells, rbx̃(t), overestimates rbx(t). For a numerical comparison of the models, see Fig. 1. For the simplified model, we can obtain analytical solutions for x̃(t) and ỹ(t) and thus can compute rb ∫0∞x̃ỹdt, which gives an upper limit to rb ∫0∞ xydt.
Figure 1.

Graphical comparison of the full (solid line) and simplified (dashed line) models under drug therapy (see text). In A, we compare the increase in the number of susceptible cells and in B, the decrease in the infected cell population. Notice that in both cases, the simplified model overestimates the full model. Parameters are as follows: λ = 106, b = 5 × 10−8, bd = rb = 2.25 × 10−8, a = 0.5, δ = 0.05. Thus, Rb = 2 and Rd = 0.9.
In equilibrium, the probability of preexistence of resistant virus depends on the total number of infected cells present at the start of therapy, which is given by yI. We thus calculate the ratio, Θ, of the number of cells infected during therapy (given by rb ∫0∞x̃ỹdt) and the number of infected cells present at the start of therapy (given by yI). A detailed derivation in the Appendix yields:
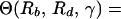 |
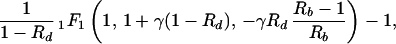 |
3 |
where 1F1 is the generalized hypergeometric function, γ = a/δ is the ratio of the death rates of infected and uninfected cells, and Rb and Rd are the basic reproductive ratios of the sensitive virus before and during treatment, respectively. Note also that the ratio, Θ, depends only on three parameters, γ, Rb, and Rd.
As the sensitive virus is able to maintain an infection in the absence but not in the presence of treatment, we have Rb > 1 and Rd < 1. Furthermore, it is reasonable to assume that the death rate of infected cells exceeds that of uninfected cells, i.e. γ > 1. Fig. 2 shows the ratio Θ as a function of Rb and Rd for a conservative minimal value of γ = 10 (21, 27, 28). [Generally, Θ(Rb, Rd, γ) is a decreasing function of γ.] Interestingly, the ratio Θ is smaller than 1 for most of the parameter region, implying that the number of infected cells present at the start of therapy is larger than the number of cells infected during therapy. Θ is larger than 1 only if Rd is close to 1. However, this is also the region where Θ greatly overestimates the corresponding ratio (rb/yI ∫0∞ xydt) of the full model defined by the system of Eq. 1.
Figure 2.

Behavior of Θ (the ratio between the likelihood of emergence of resistant mutants emergence during therapy and the likelihood of preexistence of resistant mutants) with Rd and Rb, for γ = 10. For most of the parameter region, the ratio is smaller than one. The ratio Θ becomes close to one only when Rd, the basic reproductive ratio during therapy, is also close to one.
Thus, typically the number of infected cells present at the start of therapy exceeds the number of infected cells produced during therapy, which suggests that the probability of preexistence of resistance is larger than the probability of production of resistance during therapy. Exceptions to this rule may occur in a narrow region, where the basic reproductive ratio of the sensitive virus during therapy, Rd, is very close to but below 1. Nonetheless, numerical simulations of the full model, given by Eq. 1, suggest that this region is actually smaller than that shown in Fig. 2.
Multistrain Model
Although the comparison of the number of infected cells present at the start of therapy with the number of cells infected during therapy provides an intuitive measure of the relative likelihood of the preexistence and emergence hypothesis, this is only partially satisfactory, because it ignores the full complexity of a heterogeneous virus population before and during selection pressures imposed by drug treatment. Therefore, we extend the above model and subdivide the population of infected cells into l populations, yi, each infected with a different virus mutant i. Again we calculate the ratio of expected production during therapy and expected frequency before therapy, but this time we calculate this ratio for each mutant, as a function of the number of point mutation differences between the mutant and the predominant wild type. The modified dynamical equations are:
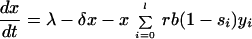 |
4 |
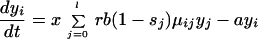 |
Here the infectivity parameter b is multiplied by a factor 1 − si, which accounts for the selective disadvantage of mutant i in comparison to the wild type. Hence, s0 = 0 for the wild type (mutant 0), and 0 < si ≤ 1 for all other mutants. The matrix μij describes the probability of mutation of strain j into strain i during reverse transcription. Hence, cells infected by mutant i are produced either by infection of a susceptible cell with mutant i or by mutation of strain j into strain i during infection.
We consider only the n nucleotide sites in conferring resistance to a particular drug regimen. That is, we group all possible strains in the viral population in classes according to their status at the sites that confer resistance. At these n sites, each strain either has the nucleotide necessary for resistance or not, which we call 1 and 0, respectively. This represents a binary model for n nucleotides. The resistant mutant is n-point mutations away from the wild type, and for each class of k-point mutants, there are (kn) strains. For instance, if we consider n = 3, then there are three one-point mutants (001, 010, 100) and three two-point mutants (011, 101, 110). The total number of strains, l, in Eq. 4 thus equals 2n.
For simplicity, we assume that the point mutation rate per replication cycle is the same at all sites, μ, such that μij = μ|i−j| for i ≠ j, and μii = 1 − ∑i≠j μij = (1 − μ)n (where |i − j| is the number of sites at which mutant i and j differ). Assume further that all mutants have the same selective disadvantage in comparison to the wild type (si = s ≪ 1 for 1 ≤ i ≤ l). In this case, a simple expression can be obtained for the equilibrium frequency of a k-point mutant (29):
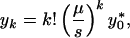 |
5 |
where y*0 stands for the equilibrium frequency of the wild type.
For the resistant n-point mutant, we calculate the ratio between the number of cells newly infected during therapy and the number of cells infected with that strain present at the start of therapy. To this end, we calculate the total production of the n-point mutant during therapy, provided it did not exist before therapy, but all 0 to n − 1 point mutants were in the mutation-selection equilibrium given by expression 5. Again, at t = 0, we initiate therapy which reduces the basic reproductive ratio of all strains present to 0 < Rd < 1. The total production of n-point mutants by a strain k(0 ≤ k < n) during therapy is given by:
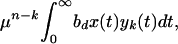 |
6 |
where bd = rb(1 − s) is the infectivity during therapy. Hence, the total production of n-point mutants by all other strains k(0 ≤ k ≤ n − 1) can be approximated as:
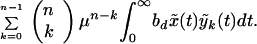 |
7 |
This expression represents the sum of the contribution of all preexisting strains according to Eq. 6, taking into account that for each k there are (kn) strains, as explained above.
Evaluating the sum in Eq. 7 (see Appendix) and dividing by expression 5 (with k = n), we obtain for the ratio of the production of n-point mutants during therapy and their frequency at the start of therapy:
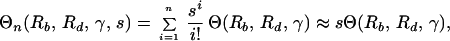 |
8 |
where Θ(Rb, Rd, γ) is given by Eq. 3. The approximation is valid for s < 0.2 and n > 3 (see Appendix).
Θn has a number of surprising properties. First, it is independent of the mutation rate, μ. Thus, counterintuitively, the relative likelihood of a mutant being produced during therapy and being present at the start of therapy does not depend on the mutation rate. Second, also contrary to expectation, Θn increases with increasing selective disadvantage of the mutants, s (Fig. 3). This result arises because when s increases, the probability of preexistence decreases disproportionately in relation to the decrease in the likelihood of emergence during therapy. Third, Θn depends only very weakly on n. Hence, the relative likelihood of emergence and preexistence is approximately the same regardless of the number of point mutations by which the resistant differs from the wild type (at least for n ≥ 3). Finally, Θn is typically smaller than Θ, because for small-to-moderate selective disadvantages (s < 0.6), we have ∑i=1n si/i! ≈ (es − 1) < 1. This implies that mutants are less likely to be produced (for the first time) during therapy than to preexist when therapy is started. This conclusion holds so long as the basic reproductive ratio of the sensitive virus during therapy is not very close to one, and the selective disadvantages involved are small.
Figure 3.

Behavior of Θn with (A) increasing selective disadvantage and (B) larger number of point mutations separating the wild type and the resistant mutant. Notice in A that the ratio Θn is bigger than one only for large values of the selective disadvantage. Parameters are as in Fig. 1 except for the ones indicated; in A, n = 3 and in B, s = 0.1.
Stochastic Simulations
The results described so far were obtained by using deterministic models to calculate the number of infected cells produced during therapy and present at the start of therapy. In a deterministic model, the frequency of a mutant will never go to zero, provided its basic reproductive ratio is larger than one. However, this may not be realistic. A particular strain might be created by mutation, but there is some probability that subsequently it will be lost again because of stochastic effects, even if its basic reproductive ratio is larger than one, R0 > 1. This is an important consideration, because the frequencies of mutants are often very small and therefore subject to stochastic fluctuations. In this section, we develop a stochastic approach to the problems addressed above. Instead of the number of infected cells produced, we consider the actual probabilities that a specific mutant exists in the population before and during treatment. Although an analytic approximation for this stochastic model is possible (30), here, in the interest of space, we show only results of simulations.
In the stochastic framework, each event (production and death of susceptible and infected cells) is assigned a probability as opposed to a deterministic rate. The probability of each event occurring is related to the rates in the system of Eq. 1. Table 1 shows the structure of the stochastic model with its events and associated probabilities.
Table 1.
Structure of the stochastic process
| Event | Population at t | Population at t + Δt | Probability | |
|---|---|---|---|---|
| Production of uninfected cell | x | → | x + 1 | λ Δt |
| Death of uninfected cell | x | → | x − 1 | δxΔt |
| Production of infected cell, yi | {yix | → | {yi + 1x − 1} | x ∑j bjyjμijΔt |
| Death of infected cell, yi | yi | → | yi − 1 | ayi Δt |
| None of the above events happen | {yix | → | {yix} | 1 − ΦΔt |
Structure of the stochastic process used in simulations and associated probabilities. Φ = (λ + δx + x ∑ biyiμi + ∑ ayi), and n is the number of sites. Thus, indices i, j run from 0 to 2n. The parameters bj = (1 − sj)bwt and μij are the same as in the deterministic model.
For the following simulations, we assume a scenario where three particular point mutations confer drug resistance. Hence we consider 8(=23) different mutants, corresponding to all possible one- and two-point mutants at the three relevant sites, as well as the wild type (000) and the strain (111) with all the required mutations for resistance. For simplicity, we assume, as in the previous section, that all intermediate one- and two-point mutants and the resistant three-point mutant have the same selective disadvantage, s. At the start of the simulation, all strains except the resistant strain are present at their respective equilibrium frequencies, given by Eq. 5. The simulation is run for a period that allows the generation of stochastic diversity around the equilibrium. Then treatment is started. If the resistant mutant is present, the simulation is stopped. Otherwise, the simulation is continued either until the virus population is eradicated or until a resistant mutant emerges.
We simulated all permutations of the following set of parameters: selective disadvantage s = 0.002, 0.005, 0.009; ratio of infected to uninfected cell death rates γ = 5, 10; basic reproductive ratio before therapy Rb = 2, 10; and basic reproductive ratio during therapy Rd = 0.9, 0.98. This makes a total of 24 sets of simulations. For each set of parameters, the simulation was run 100 times to calculate the average probability of emergence of a resistant mutant and 500 times for the average probability of resistance emerging during treatment. We calculated the probability of emergence of the resistant strain as the proportion of runs the resistant strain was present at the end of a sufficiently long time, with or without treatment. We also calculated the fraction of the time that a resistant mutant is present in an untreated patient. Because these results are equivalent to the number of times a resistant strain is present at the end of a sufficiently long run, these results are not shown here.
In support of the conclusions drawn from the deterministic models, the probability of the resistant strain emerging after treatment was in no case higher than the probability of the resistant virus already being present before treatment (data not shown). Even when Rd(=0.98) and Rb(=2) are close to 1, for which the analytical calculations are most inaccurate, the relevant ratio is still smaller than 1.
To study the effect of the different parameters on the ratio of probabilities, the results of further simulations are presented in Table 2. In the top of the table, we find that for increasing Rd, the ratio increases, as expected from the deterministic theory. This increase in the ratio is because of an increase in the probability of the resistant strain emerging after the start of therapy, because, as expected, the corresponding probability before therapy stays roughly constant. Although the ratio increases with Rd, even for Rd = 0.99, the ratio of probabilities is small.
Table 2.
Ratio of probabilities obtained in the stochastic simulations
| Parameters | Probability before | Probability after | Ratio | Estimated ratio | Standard deviation of ratio |
|---|---|---|---|---|---|
| Rd* | |||||
| 0.8 | 0.32 | 0.026 | 0.081 | 0.083 | 0.026 |
| 0.9 | 0.35 | 0.034 | 0.097 | 0.099 | 0.027 |
| 0.98 | 0.31 | 0.052 | 0.168 | 0.172 | 0.042 |
| 0.99 | 0.35 | 0.056 | 0.160 | 0.163 | 0.037 |
| γ† | |||||
| 1 | 0.35 | 0.232 | 0.663 | 0.675 | 0.107 |
| 2 | 0.32 | 0.132 | 0.413 | 0.421 | 0.078 |
| 5 | 0.31 | 0.052 | 0.168 | 0.172 | 0.042 |
| 10 | 0.37 | 0.016 | 0.043 | 0.044 | 0.016 |
| s‡ | |||||
| 0.0015 | 0.54 | 0.100 | 0.185 | 0.187 | 0.030 |
| 0.002 | 0.31 | 0.052 | 0.168 | 0.172 | 0.042 |
| 0.003 | 0.10 | 0.030 | 0.300 | 0.327 | 0.129 |
| 0.005 | 0.03 | 0.004 | 0.133 | 0.176 | 0.160 |
| 0.009 | 0.01 | 0.004 | 0.400 | 0.796 | 0.971 |
Simulation results for the probability of emergence of a resistant mutant before and during therapy. The following parameters are the same in all simulations: λ = 106, a = 0.5, δ = 0.1, n = 3, μ = 3 × 10−5, bwt = Rd × (aδ/λ), and bi = (1 − s)bwt. The simulations were run at least 100 times for the estimation of probability before therapy and 500 times for the corresponding probability during therapy. The estimated ratio and standard deviation of the ratio are calculated according to the δ method (36). Note that the large standard deviation for very small values of s is because of the small values of the probability of emergence before and during treatment.
Parameters of the simulation are: Rb = 10, γ = 5 and s = 0.002.
Parameters of the simulation are: Rd = 0.98, Rb = 10, and s = 0.002.
Parameters of the simulation are: Rd = 0.98, Rb = 10, and γ = 5.
In the middle of the table, the effect of γ is shown. Again, the probability of the resistant mutant being present before therapy stays roughly constant, suggesting that this probability is independent of γ. On the other hand, for smaller γ, the probability of producing a mutant during therapy is higher. Thus, the ratio of probabilities increases for smaller γ, but remains below one even for an unrealistic value of γ = 1 (implying that uninfected and infected cells have the same life span). The bottom of the table shows the effect of s. The selective disadvantage affects both probabilities before and after treatment in the same way: the probabilities decrease for larger values of s. This double effect makes it difficult to discern a clear trend for the ratio of probabilities. However, it seems that the ratio of probabilities increases with s, because the probability of the resistant strain being present before therapy decreases disproportionately in relation to the decrease in the corresponding probability after treatment. In any case, also in these simulations, the ratio of probabilities is always smaller than one.
In summary, the stochastic simulations are in excellent agreement with our analytical calculations. They strongly support the hypothesis that the more likely cause of resistance-related treatment failure is the presence of resistant strains before therapy. Moreover, even for unrealistic sets of parameters, corresponding to cases where the theoretical value of Θn is close to or above one, the probability of producing a new resistant mutant is smaller than the probability of this mutant preexisting in the population.
So far, we have assumed that after therapy, all sensitive strains have the same basic reproductive ratio, Rd. If, on the other hand, therapy reduces Rd of the wild type more than it reduces Rd of the other sensitive strains, such that Rd(wt) < Rd(other strains), the results obtained can be quite different. In Table 3, we show some results for the limiting case of that inequality, i.e., the wild type does not replicate at all after therapy, Rd(wt) = 0, and Rd(other strains) is close to one. In this limiting case, the probability of producing a resistant mutant is higher than that observed when the wild type also replicates.
Table 3.
Ratio of probabilities when the wild type does not replicate
| Probability before | Probability after | Probability after (βwt = 0) | Ratio | Ratio (βwt = 0) | |
|---|---|---|---|---|---|
| i | 0.01 | <0.002 | 0.010 | – | 1.000 |
| ii | 0.02 | 0.008 | 0.006 | 0.400 | 0.300 |
| iii | 0.13 | 0.042 | 0.072 | 0.323 | 0.554 |
| iv | 0.32 | 0.132 | 0.396 | 0.413 | 1.238 |
| v | 0.035 | 0.232 | 0.632 | 0.663 | 1.806 |
Results of the simulations showing the effect of total suppression of wild type replication on the probability of emergence of resistance during therapy. The simulations were run at least 100 times for the estimation of probability before therapy and 500 times for the corresponding probability during therapy. Parameters of the simulations are: for i, ii, and iii, γ = 5, Rd = 0.98, Rb = 2, and s = 0.009, s = 0.005, and s = 0.002, respectively; for iv and v, Rd = 0.98, Rb = 10, s = 0.002, and γ = 2, and γ = 1, respectively. Other parameters are in all cases: λ = 106, a = 0.5, δ = a/γ, μ = 3 × 10−5, n = 3, βwt = Rd × (aδ/λ), and βi = (1 − s)βwt.
When therapy is more efficient in relation to the wild type than in relation to the other preexisting sensitive strains, the probability of production of a resistant mutant during therapy may sometimes be higher than the probability of that mutant already existing before therapy.
Discussion
The appearance of HIV strains resistant to a particular drug regimen is the main problem during treatment of infected individuals (refs. 2 and 7; www.hivatis.org/trtgdlns.html). In principle, there are two ways in which resistance can emerge in response to therapy (13, 14): (i) resistant strains may already exist when therapy is started; or (ii) all preexisting strains are sensitive, but the drug regimen is not 100% effective, and the resistant mutant is created de novo during treatment. Using various deterministic and stochastic models, we have shown that almost universally treatment fails because of the preexistence of resistant strains in the drug naïve viral population. It is generally less likely that resistant mutants are generated for the first time during treatment. This suggests that efforts to reduce the risk of treatment failure need to concentrate on the combination of drugs with different resistance profiles in order to minimize the risk that multidrug-resistant strains preexist in a drug-naïve viral population. Increasing the efficacy of replication inhibition is only of secondary concern.
It is important to emphasize at this point that this paper has not dealt with the evolution of resistance caused by poor adherence to the drug regimen. Clearly, if patients at certain periods take only a subset of the prescribed drugs, then resistance may evolve successively to each of the drugs used. Similarly, spatial heterogeneity in the distribution of the drugs used may facilitate the evolution of resistance as the virus may locally be controlled only by a single drug (31). Furthermore, this paper also does not consider the contribution of viral replication at sanctuary sites to the evolution of resistance. In the narrow sense of our definition of drug resistance, a virus capable of persisting at a sanctuary site is resistant, because its basic reproductive ratio is larger than one during treatment. However, the question is whether the virus replicating at these sanctuary sites is actually likely to produce a fully resistant virus that is capable of recolonizing the main sites of infection in the presence of treatment. Clearly, continuous replication of the virus at sanctuary sites increases the risk that a fully resistant mutant is produced during treatment. However, if such a fully resistant mutant is unlikely to be present at the start of therapy, then we expect that its production during treatment should take a long time, because the viral population replicating during treatment is typically orders of magnitude smaller than the viral population at the start of therapy.
Although our results show, as expected, that the likelihood of creating a resistant mutant during therapy decreases with more effective inhibition of replication (i.e., with smaller Rd), this likelihood is generally smaller than the likelihood that resistant viruses preexist. Furthermore, it is interesting that the ratio of these probabilities increases for higher selective disadvantages of the sensitive strains present at the start of therapy. We must caution, however, that this is true only for the relative risk. In absolute terms, the risk decreases with increasing s (Table 2). Most surprisingly, the relative risk is independent of both the mutation rate and the number of point mutations necessary for resistance.
Although much is now known about HIV genotypic and phenotypic resistance (11), testing for the presence of resistant strains in the context of drug therapy is still not very effective. This is because of the lack of sensitive and reliable tests, which can rapidly track resistance mutations (7–10). However, results of this paper suggest a new reason why great care must be taken in the choice of therapies. If the drugs are specifically targeted to be more efficient against the wild type but are less effective against other sensitive strains present in the viral quasispecies, the risk of producing a mutant during therapy increases relative to that of preexistence of resistance. The reason is that intermediate mutants contribute disproportionately to the production of the resistant strain, even though they are present at very low frequencies. If drugs control the wild type selectively, the other sensitive strains present may have more target cells available for infection (32–35), as they decline under treatment. The increased opportunities to infect cells, in turn, increase the risk of mutation into the resistant virus.
In summary, under very general conditions, analytical and numerical analysis of the viral quasispecies' response to selection pressures imposed by drug therapy argues strongly that the resistant mutants that appear in patients who failed on therapy are most likely present already at the start of therapy. Even those patients who fail on triple combination therapy but were fully compliant most likely already harbored resistant virus when therapy was started. Thus the key to drug resistance lies in the diversity of the viral population at the start of therapy.
Acknowledgments
Support by the Novartis Research Foundation is gratefully acknowledged (S.B.). R.M.R. is supported by the PRAXIS XXI program of Fundação para a Ciência e Técnologia.
Appendix
Here we present in detail the analytical expressions for the production of infected cells during treatment in the basic and multistrain models.
Basic Model.
The solution of the first equation in system 2 is
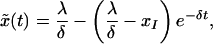 |
9 |
where x̃(0) = xI = a/b corresponds to the infected steady state of the full model at the start of therapy. Substituting solution 9 into the second equation of system 2 and solving for ỹ, we obtain
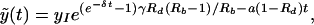 |
10 |
where ỹ(0) = yI = λ/a − δ/b is the steady-state frequency of infected cells at the start of therapy for the full model. Rb = λb/(δa) and Rd = rRb are the basic reproductive ratios of the virus before and during treatment, and γ = a/δ is the ratio of the life spans of infected and uninfected cells. Using these definitions, we can transform the integral rb ∫0∞x̃(t)ỹ(t)dt as follows:
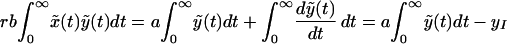 |
11 |
Substituting z = e−δt, ρ = γRd(Rb − 1)/Rb and φ = a(1 − Rd), and using Eq. 10, we obtain:
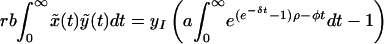 |
12 |
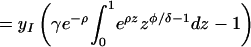 |
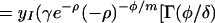 |
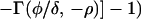 |
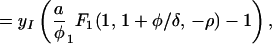 |
where Γ(φ/δ) is the gamma function, Γ(φ/δ, −ρ) is the incomplete gamma function, and 1F1(1, 1 + φ/δ, −ρ) is the generalized hypergeometric function. The integrals and functional relationships used in the calculation can be found in ref. 37. Expressed in terms of basic reproductive ratios, we obtain, by back-substituting z, ρ, and φ,
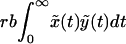 |
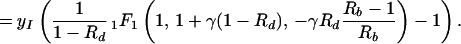 |
13 |
The ratio of the number of infected cells produced during therapy (Eq. 13) and the number of infected cells before therapy (yI) is:
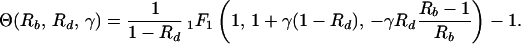 |
14 |
Multistrain Model.
The total production of cells infected with the resistant n-point mutant during therapy is given by Eq. 7. Using Eqs. 7 and 5 and substituting yk(0) = k!(μ/s)ky*0, we obtain:
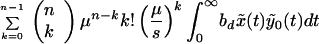 |
15 |
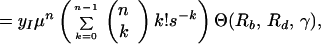 |
where y0(0) = yI. The ratio of production of n-point mutants during therapy and their frequency of preexistence is obtained by dividing expression 15 by expression 5, with k replaced by n:
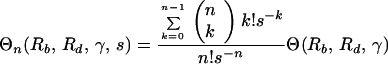 |
16 |
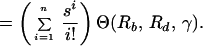 |
For s < 0.2 and n > 3, we can simplify further (with an error of less than about 10%):
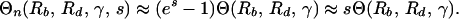 |
17 |
References
- 1.BHIVA Guidelines Coordinating Committee. Lancet. 1997;349:1086–1092. [PubMed] [Google Scholar]
- 2.Gazzard B, Moyle G. Lancet. 1998;352:314–316. doi: 10.1016/s0140-6736(98)04084-7. [DOI] [PubMed] [Google Scholar]
- 3.Rachlis A R, Zarowny D P. Can Med Assoc J. 1998;158:496–505. [PMC free article] [PubMed] [Google Scholar]
- 4.Carpenter C C, Fischl M A, Hammer S M, Hirsch M S, Jacobsen D M, Katzenstein D A, Montaner J, Richman D D, Saag M S, Schooley R T, et al. J Am Med Assoc. 1997;277:1962–1969. [PubMed] [Google Scholar]
- 5.NIH Panel to Define Principles of Therapy of HIV Infection. Ann Intern Med. 1998;128:1057–1078. doi: 10.7326/0003-4819-128-12_part_2-199806151-00002. [DOI] [PubMed] [Google Scholar]
- 6.Gazzard B. Int J Clin Pract. 1999;S103:45–48. [PubMed] [Google Scholar]
- 7.Hirsch M S, Conway B, D'Aquila R T, Johnson V A, Brun-Vezinet F, Clotet Demeter L M, Hammer S M, Jacobsen D M, Kuritzkes D R, Loveday C, et al. J Am Med Assoc. 1998;279:1984–1991. doi: 10.1001/jama.279.24.1984. [DOI] [PubMed] [Google Scholar]
- 8.Perrin L, Telenti A. Science. 1998;280:1871–1873. doi: 10.1126/science.280.5371.1871. [DOI] [PubMed] [Google Scholar]
- 9.Loveday C, Dunn D, McCormack S, Babiker A. Sex Transm Infect. 1999;75:140–141. [PubMed] [Google Scholar]
- 10.Gazzard B G, Moyle G. Sex Transm Infect. 1999;75:141–142. [PubMed] [Google Scholar]
- 11.Vandamme A M, van Laethem K, de Clercq E. Drugs. 1999;57:337–361. doi: 10.2165/00003495-199957030-00006. [DOI] [PubMed] [Google Scholar]
- 12.Gomez-Cano M, Rubio A, Ruiz L, Perez-Olmeda M, Leal M, Clotet B, Soriano V. Antiviral Ther. 1999;4:123–124. [PubMed] [Google Scholar]
- 13.Bonhoeffer S, Nowak M A. Proc R Soc London B. 1997;264:631–637. doi: 10.1098/rspb.1997.0089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bonhoeffer S, May R M, Shaw G M, Nowak M A. Proc Natl Acad Sci USA. 1997;94:6971–6976. doi: 10.1073/pnas.94.13.6971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lipsitch M, Levin B R. Antimicrob Agents Chemother. 1997;41:363–373. doi: 10.1128/aac.41.2.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Birk M, Sonnerborg A. AIDS. 1998;12:2369–2375. doi: 10.1097/00002030-199818000-00005. [DOI] [PubMed] [Google Scholar]
- 17.Gomez-Cano M, Rubio A, Puig T, Perez-Olmeda M, Ruiz L, Soriano V, Pineda J A, Zamora L, Xaus N, Clotet B, et al. AIDS. 1998;12:1015–1020. [PubMed] [Google Scholar]
- 18.Wainberg M A, Friedland G. J Am Med Assoc. 1998;279:1977–1983. doi: 10.1001/jama.279.24.1977. [DOI] [PubMed] [Google Scholar]
- 19.Nowak M A, Nowak M A, Anderson R M, McLean A R, Wolfs T F, Goudsmit J, May R M. Science. 1991;254:963–969. doi: 10.1126/science.1683006. [DOI] [PubMed] [Google Scholar]
- 20.Frost S D W, McLean A R. AIDS. 1994;8:323–332. doi: 10.1097/00002030-199403000-00005. [DOI] [PubMed] [Google Scholar]
- 21.Wei X, Ghosh S K, Taylor M E, Johnson V A, Emini E A, Deutsch P, Lifson J D, Bonhoeffer S, Nowak M A, Hahn B H, et al. Nature (London) 1995;373:117–122. doi: 10.1038/373117a0. [DOI] [PubMed] [Google Scholar]
- 22.Nowak M A, Bonhoeffer S, Shaw G M, May R M. J Theor Biol. 1997;184:203–221. doi: 10.1006/jtbi.1996.0307. [DOI] [PubMed] [Google Scholar]
- 23.Bonhoeffer S. Aids Patient Care STDs. 1998;12:769–774. doi: 10.1089/apc.1998.12.769. [DOI] [PubMed] [Google Scholar]
- 24.May R M, Anderson R M. Nature (London) 1979;280:455–461. doi: 10.1038/280455a0. [DOI] [PubMed] [Google Scholar]
- 25.Anderson R M, May R M. Infectious Dis. Hum. Oxford: Oxford Univ. Press; 1991. [Google Scholar]
- 26.Heesterbeek J A P, Dietz K. Stat Neerlandica. 1996;50:89–110. [Google Scholar]
- 27.Ho D D, Neumann A U, Perelson A S, Chen W, Leonard J M, Markowitz M. Nature (London) 1995;373:123–126. doi: 10.1038/373123a0. [DOI] [PubMed] [Google Scholar]
- 28.McLean A R, Michie C A. Proc Natl Acad Sci USA. 1995;92:3707–3711. doi: 10.1073/pnas.92.9.3707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ribeiro R M, Bonhoeffer S, Nowak M A. AIDS. 1998;12:461–465. doi: 10.1097/00002030-199805000-00006. [DOI] [PubMed] [Google Scholar]
- 30.Ribeiro R M. Ph.D. thesis. Oxford: Univ. of Oxford; 1999. [Google Scholar]
- 31.Kepler T B, Perelson A S. Proc Natl Acad Sci USA. 1998;95:11514–11519. doi: 10.1073/pnas.95.20.11514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McLean A R, Emery V C, Webster A, Griffiths P D. AIDS. 1991;5:485–489. doi: 10.1097/00002030-199105000-00002. [DOI] [PubMed] [Google Scholar]
- 33.McLean A R, Nowak M A. AIDS. 1992;6:71–79. doi: 10.1097/00002030-199201000-00009. [DOI] [PubMed] [Google Scholar]
- 34.De Boer R J, Boucher C A B. Proc R Soc London Ser B. 1996;263:899–905. doi: 10.1098/rspb.1996.0133. [DOI] [PubMed] [Google Scholar]
- 35.De Boer R J, Perelson A S. J Theor Biol. 1998;190:201–214. doi: 10.1006/jtbi.1997.0548. [DOI] [PubMed] [Google Scholar]
- 36.Kotz S, Johnson N L. The Encyclopaedia of Statistical Sciences. Vol. 8. New York: Wiley; 1989. [Google Scholar]
- 37.Gradsteyn I S, Ryzhik I M. Tables of Integrals, Series, and Products, Corrected and Enlarged. 4th Ed. San Diego: Academic; 1980. [Google Scholar]