

Summary
Acireductone dioxygenase (ARD) from Klebsiella ATCC 8724 is a metalloenzyme that is capable of catalyzing different reactions with the same substrates (acireductone and O2) depending upon the metal bound in the active site. A model for the solution structure of the paramagnetic Ni2+-containing ARD has been refined using residual dipolar couplings (RDCs) measured in two media. Additional dihedral restraints based on chemical shift (TALOS) were included in the refinement, and backbone structure in the vicinity of the active site was modeled from a crystallographic structure of the mouse homolog of ARD. The incorporation of residual dipolar couplings into the structural refinement alters the relative orientations of several structural features significantly, and improves local secondary structure determination. Comparisons between the solution structures obtained with and without RDCs are made, and structural similarities and differences between mouse and bacterial enzymes are described. Finally, the biological significance of these differences is considered.
Keywords: metalloenzyme, magnetic alignment, nickel enzyme, paramagnetic, homology modeling
Introduction
Acireductone dioxygenase (ARD), a metalloenzyme from the methionine salvage pathway of the bacterium Klebsiella ATCC 8724, is unusual in that it catalyzes different dioxygenase reactions with identical substrates depending upon the metal bound in the active site (Dai et al., 1999; Dai et al., 2001). The methionine salvage pathway returns the γ-thiomethyl group of methylthioadenosine (MTA) to methionine (Kestell, 1996). MTA is derived from S-adenosylmethionine (SAM, AdoMet), that in turn is formed by S-alkylation of methionine at the 5′-ribose carbon of ATP. The pentultimate intermediate in the methionine salvage pathway is 1,2-dihydroxy-3-oxo-5-(methylthio)pent-1-ene, commonly termed acireductone, the substrate for ARD (Wray and Abeles, 1993). ARD is a branch point in the methionine salvage pathway. If Fe2+ is bound in the active site (FeARD), the substrate acireductone reacts with O2 to yield the ketoacid precursor of methionine, 2-oxo-4-(methylthio)butyrate, and formate. The Ni2+-containing NiARD catalyzes an off-pathway oxidation of acireductone with formation of carbon monoxide, formate and methylthiopropionate. The two ARD isoforms are chromatographically separable on hydrophobic interaction and strong anion exchange liquid chromatography phases, suggesting significant structural differences between them.
We previously reported 1H, 15N and 13C resonance assignments for NiARD (Mo et al., 1999) and described a structural model of NiARD using NOE and J-coupling restraints in regions of the enzyme unaffected by paramagnetism of the single Ni2+ ion bound in the active site (Pochapsky et al., 2002). Paramagnetism broadens 1H resonance lines within ~9 Å from the metal center (approximately 15% of the 1H resonances in the protein) beyond detection by standard multidimensional NMR methods. In the absence of sequence-specific assignments for this region of the protein, and lacking a crystallographic structure for any near homologue of ARD, we used conserved domain homology modeling based on the structure of jack bean canavalin (Ko et al., 2000) to propose a structure for the metal binding site of NiARD. To support the modeling, we employed X-ray absorption spectroscopy (XAS) and paramagnetic NMR to characterize the first- and second coordination sphere of the nickel ion (Al-Mjeni et al., 2002).
The resulting active site structure is consistent with the known enzymatic activity of NiARD and is similar to a number of other metalloenzymes, in particular, the Mn2+-containing oxalate decarboxylase (OxD) (Anand et al., 2002) and oxalate oxidase (OxOx) (Woo et al., 2000). ARD, canavalin, OxD and OxOx are all members of the cupin superfamily that is characterized by a conserved small β-barrel fold (sometimes referred to as a “jelly roll”) from which the name of the superfamily is derived, although “β-sandwich” is probably a more accurate description of the fold (Dunwell et al., 2001). Recently, a crystallographic structure of an ARD homologue from Mus musculus (house mouse), MmARD, was deposited in the PDB database (entry 1VR3, (JCSG, 2005). The overall homology between the two proteins is low (23% identity). Still, the MmARD structure supports our proposed ligation geometry for the active site, although the nature of the bound metal and the enzymatic activity of the mouse ARD homologue have yet to be reported.
We now describe a refinement of the NiARD structural model that employs 1H-15N RDCs measured in two alignment media, 1-hexanol/ nonsymmetric polyol HO-(CH2O)6-(CH2)11CH3 (C12E5) (Ruckert and Otting, 2000) and filamentous phage (fd) (Hansen et al., 1998). The use of residual dipolar couplings (RDCs) has revolutionized the determination of solution structures of proteins by NMR (Prestegard et al., 2004; Tjandra et al., 1997). Unlike NOEs and J-coupling that are local constraints and not related to a single frame of reference, RDCs provide an independent structural parameter that relate to a single frame of reference provided by the order tensor. If multiple sets of RDCs can be measured in different aligned media with independent order tensors, elimination of ambiguity in the direction of individual dipole vectors is possible (Prestegard et al., 2004). The current structural refinement provides structures that satisfy both sets of RDC restraints as well as NOE, chemical shift and dihedral angle restraints. In the current refinement, we made use a greater number of dihedral restraints generated from chemical shift database correlations (TALOS) than in the previous calculations (Cornilescu, Delaglio et al., 1999).
Improved 2D methods for sequential assignment of paramagnetically relaxed 13C and 15N resonances have permitted us to make some assignments in the region of the NiARD active site (Kostic et al., 2002). However, these assignments are not yet sufficiently complete to forgo modeling in the current structural refinement. In the current calculations, we have replaced the coordinates from canavalin used in the original structure calculations with the coordinates from MmARD (vide supra). We note, however, that the backbone structure in the vicinity of the active site in the current ensemble of structures is little different from our original modeling using the canavalin coordinates (<0.8 Å RMSD for modeled coordinates), indicating a remarkable degree of structural homology in this region across proteins of very different structures and functions.
Materials and Methods
Polyether/alcohol aligned medium
The asymmetric polyol HO-(CH2O)6-(CH2)11CH3 (C12E5) (Aldrich) was prepared as a 10% w/w solution in standard 50 mM KPi 90/10 H2O/D2O pH 7.4. 200 μL of this solution was then added to an equal volume of 3 mM 15N-labeled NiARD prepared as described previously (Mo et al., 1999) in the same buffer and gently mixed. Aliquots of 1-hexanol (anhydrous, Sigma) was added to a final mol/mol ratio C12E5/hexanol of 0.96 (~ 9 μL), the mixture gently vortexed and centrifuged to remove bubbles. The sample had a slightly opalescent appearance. The sample was placed in a Shigemi tube, immediately transferred to the spectrometer and allowed to equilibrate at 303 K. Formation of an aligned phase was monitored by splitting of the 2H water signal with a splitting of ~ 22 Hz observed within 1 hour.
Growth and purification of fd bacteriophage
A single colony from a streak plate of XL1-Blue (Stratagene) cells was used to inoculate an LB culture supplemented with 50 μg/mL tetracycline. The culture was grown until the mixture was cloudy, but halted prior to saturation to prevent loss of the F′ episome (~10 hours at 37° C). A stock solution of fd phage was serially diluted in LB. Mixtures of the bacterial culture (150 μl), phage titrations (10 μL), and melted top agar (3 mL) were plated on LB supplemented with tetracycline. The plates were grown overnight at 37°C. Isolated plaques were selected from the plate with the 10−9 phage dilution and used to inoculate two 25 ml LB cultures. The cultures were grown overnight at 37° C. The bacterial cells were pelleted by centrifugation (3,000 x g, 40 min), and the supernatant was retained. This is the “infecting phage” used for large-scale preparations.
A 5600 mL LB culture supplemented with tetracycline was inoculated with 700 μL of bacterial culture and incubated at 37°C for ~4 hours. The culture was then infected with 300 μL of the infecting phage and incubated at 37°C overnight. The bacterial cells were pelleted by centrifugation (3,000 x g, 40 min). The bacteriophage was precipitated from the supernatant by the addition of PEG 8000 (100 g) and NaCl (100 g), and collected by centrifugation (3,000 x g, 40 min). The pellet was resuspended in distilled water and clarified by centrifugation (20,000 x g, 1 hr) twice. The supernatant was retained and the pellet discarded both times. The presence of bacteriophage in the supernatant was confirmed by spectrophotometry (OD269 = 3.84 for 1 cm path length x 1 mg/ml fd phage). The yield at this stage was 147 mg. The bacteriophage was ultracentrifuged (~250,000 x g, 3 hrs) and the pellet resuspended in water. The virus solution was dialyzed three times against 1000 ml 10 mM Tris-Cl pH 7.4 (membrane cutoff = 12–14 kD). The yield after dialysis was 95 mg. The virus solution was ultracentrifuged (~250,000 x g, 3 hrs), and the pellet resuspended in distilled water to a concentration of 46.8 mg/ml.
Samples of 15N-labeled NiARD were mixed gently with phage to prevent introduction of bubbles to a final phage concentration of 20 mg/mL. We note that, unlike the polyether-aligned medium, little quadrupolar splitting of the D2O signal was observed (only 3.6 Hz) after alignment was complete. Alignment was confirmed by the use of crossed polarizers and the observation of changes in 1JNH splittings consistent with alignment.
NMR data acquisition and analysis
The NMR experiments and methodology used for sequential assignment and tertiary structural determination of Ni-ARD have been described previously (Mo et al., 1999; Pochapsky et al., 2002). Determination of residual dipolar couplings was accomplished using 1H, 15N HSQC spectra obtained without 1H decoupling in the indirect dimension on a Varian INOVA 14 T (600 MHz 1H) NMR spectrometer, Quadrature detection in the indirect dimension was obtained using the Rance-Kay method (Muhandiram and Kay, 1994). 512 increments were obtained in the indirect (15N) dimension for each spectrum. Reference spectra were acquired at the appropriate temperatures without alignment, followed immediately by acquisition of identical spectra under aligned conditions. NMR data was processed using XWinNMR (Bruker Biospin GMBH). Spectra were zero-filled to an apparent resolution of 1 Hz/pt in the 15N dimension. The data was analyzed using XEASY (Bartels et al., 1995). Residual dipolar couplings were obtained by comparison between the reference spectrum and those obtained in aligned media. Estimated error in measurement is +/− 2 Hz. Overall spectral quality in the presence of phage was significantly poorer than that observed in the C12E5-aligned phase, so that only 30 well-resolved RDC values were obtained with the phage alignment, as opposed to the 102 RDC values obtained with the C12E5 phase.
Initial values of anisotropy Da and rhombicity R=Dr/Da for each alignment medium were obtained using a subset of the experimental dipolar coupling values fitted to the original structure of NiARD (1M40) using the SSIA program (http://spin.niddk.nih.gov/bax/software/) (Zweckstetter and Bax, 2000). Outliers were removed to improve the fit, and the values for Da and R thus obtained were used as starting points for XPLOR calculations. For the most part, the NH couplings in the initial set corresponded to amide bonds well defined by regular secondary structural features, especially within the β-sandwich. As the refinement progressed, values for Da and R were re-calculated with larger numbers of experimental RDCs to improve the estimates of Da and R.
Structural calculations
Structural calculations were performed on an MB computer with an Intel 386 processor running a RedHat Linux operating system. Restrained molecular dynamics/simulated annealing using torsional dynamics were performed with the NIH implementation of XPLOR version 2.10. The NIH implementation of XPLOR includes database potentials for improving local geometry (backbone and sidechain torsional angles) while maintaining the primacy of experimental restraints (Kuszewski et al., 1996; Kuszewski et al., 1997; Kuszewski and Clore, 2000). Energies were calculated using bond, angle, van der Waals, improper, NOE, dihedral angle, J-coupling, residual dipolar coupling (SANI), 13C chemical shift, hydrogen bonding and database torsional (RAMA) terms. Electrostatic terms were not included in the refinement. Experimental and modeling restraints used in the calculations are summarized in Table 1. Initial estimates of Da and R for use in the calculations were obtained from a subset of RDCs from each dataset (fd and C12E5) as described above. As the refinement progressed, this calculation was repeated with larger numbers of RDCs with refined structures to improve the best-fit value of Da and R for each of the two phases. Final fits were obtained using complete backbone RDC restraint sets. The final alignment tensors for NiARD in the two phases when superimposed showed approximately the same z-axis orientation (+8° tilt of fd relative to C12E5) and nearly opposite directions for x and y (+176° for y and +172° for x of fd relative to C12E5). The positions of the reference pseudoatoms used by the SANI subroutine for fitting RDCs to the alignment tensor for all accepted structures were consistently in the same octant of Cartesian space relative to the structure for each alignment, but unique for each alignment medium. That is, the pseudoatoms used to fit fd always appeared in the same octant relative to the structure, and those used to fit C12E5 always appeared in the same (different than the fd pseudoatoms) octant.
Table 1.
| Experimental restraints | |
| long range NOEs (i - j > 10)1 | 459 |
| short range interresidue NOEs (i - j < 10) | 566 |
| restrictive intraresidue NOEs | 210 |
| paramagnetic restraints2 | 27 |
| 13C chemical shift restraints | 271 |
| Residual dipolar couplings (C12E5, Da=20 Hz, R = 0.283) | 102 |
| Residual dipolar couplings (fd, Da=13.1 Hz, R = 0.309) | 32 |
| J-coupling restraints (periodic) | 64 |
| dihedral angle restraints3 | 333 |
| total number of experimental restraints | 2,054 |
| Modeling restraints | |
| hydrogen bonds in regular secondary structure4 | 59 |
| harmonic and modeling restraints5 | 71 |
| total number of modeling restraints | 125 |
| Structural statistics over 17 models | |
| no. of distance violations > 0.5 Å | none |
| Goodness of fit of RDC restraints to structures | |
| C12E5 | R=1.00, Q=0.05 |
| fd | R=0.99, Q=0.12 |
| Ramachandran analysis7 | |
| residues in most favored regions | 80.9% |
| residues in additional allowed regions | 16.8% |
| residues in generously allowed regions | 2.3% |
| residues in dissallowed regions | 0.0% |
| Atypical χ1 rotamers8 | 1.2% |
| Side-chain steric interactions < 0.8 Å9 | <1% |
| backbone RMSD (Å) for 17 models | 0.62 |
i and j refer to residue numbers.
Paramagnetically broadened resonances were constrained to be ≤ 10 Å from the nickel.
Dihedral angle restraints were obtained by chemical shift analysis using TALOS (2004 edition) (Cornilescu et al., 1999) (see text).
Hydrogen bonds within regular secondary structure were determined by repeated occurrence in unrestrained structures.
Harmonic restraints and three hydrogen bonds were applied to model the paramagnetic region as described in text.
Ramachandran statistics were calculated using PROCHECK_NMR (Laskowski et al., 1996) and excludes prolines, glycines and terminal residues.
Sidechain dihedral angles (χ1) noted as low frequency by WHAT_CHECK (Hooft et al., 1996). No “bad” scores (lower than 0.30) were found in any structure. In eight of the 17 structures, residue 38 was identified as a rare conformer. In seven of the structures, residue 37 was identified as a rare conformer. Residues 7, 77 and 133 occur as rare conformers in more than one structure.
Steric interactions occurred in ten structures between the side chain atoms Cɛ2 of Trp 16 and Hɛ22 of Gln 27, and between Cζ2 of Trp 16 and Hɛ22 of Gln 27 in seven structures. Interactions between Hɛ21 of Gln 58 and Cγ1 Ile 61 occur in nine structures. Steric interactions were detected using the van der Waals subroutine of MOLMOL (Koradi et al., 1996).
After an initial (5000 step) energy minimization using bond, angle and harmonic restraints with reduced van der Waals repulsion terms, structures were heated to 3000 K for 5,000 steps of torsional dynamics using a 2 fs step size. The simulation temperature was reduced gradually during the annealing phase (12.5 K temperature steps of 50 steps each, 2 fs step size, final temperature = 25 K), while van der Waals repulsive terms, experimental, database and harmonic restraints were gradually increased to their full values. 500 steps of minimization were used to complete the calculation. Accepted structures gave no NOE violations, good fits of RDCs to calculated order tensors (R=1.00 for C12E5 and R=0.99 for phage) and reasonable Ramachandran plots. Structural statistics are summarized in Table 1.
Modeling restraints on the metal cluster environment
The modeling of the metal binding site for structural calculations proceeded as follows. The polypeptide segments that form the metal binding site, including all backbone heavy atoms (N, Cα, C and O) of residues 96–102 and 136–143, backbone N atoms of residues 103 and 144, and backbone CO groups of residues 95 and 135, were harmonically restrained to the coordinates obtained for the corresponding residues in MmARD (1VR3). Harmonic restraints were applied to backbone heavy atoms as well as to the Cβ atoms of the proposed protein-based ligands (His 96, His 98, Glu 102 and His 140). Appropriate bond lengths as established by XAS and bond angles based on expected octahedral geometry and standard metal ligation geometries for His, Glu and TIP3 waters were used to restrain ligands and metal. The average Ni-N (His) bond length of 2.09 Å was obtained in the best fit of XAS data, with an overall average Ni-X bond length for all six ligands is 2.06 Å (Al-Mjeni et al., 2002). Co-planarity of atoms bound to the nickel within the pseudosymmetric reflection planes dictated by the octahedral ligand arrangement was enforced by application of improper angle restraints.
Based upon observations from the crystal structure of MmARD (vide inftra), two hydrogen bond restraints were imposed between the backbone amides of Ile 163 and Thr 97 to place those residues in an extended antiparallel arrangement. The extension of helix G to include residues 93 and 94 is observed in a metal-free mutant of ARD (Tingting Ju, unpublished results), and was modeled by inclusion of a hydrogen bond between the NH of Asn 94 and the carbonyl of Glu 90.
Results and Discussion
Global fold and tertiary structure of NiARD
The refined model for the solution structure of NiARD is detailed in Figure 1. The superposition of the family of 17 accepted structures deposited in the PDB database as 1ZRR is shown in Figure 2. The topology of the refined structure is unchanged from our original published structure, 1M4O (Pochapsky et al., 2002). The structure is lettered slightly differently in the current presentation to include a short β-strand (D) that was not labeled in the previous scheme. The structure is defined primarily by the β-sandwich formed in part by a right-handed β-helix that contains the active site of the enzyme, including the nickel binding site (see Figure 2). The β-helix originates at a turn formed by residues 117–121 between strands J and K, continues through one complete helical turn that forms the active site of the enzyme, including antiparallel strands I:L and H:M. The β-helix is interrupted at this point by α-helix G near the opening of the active site, although the antiparallel arrangement of the polypeptide continues, and the β-sandwich structure resumes as antiparallel β-strands F:N. Strand N diverges from strand I after the Ile 153:Phe 105 hydrogen bond pair. The N-terminal strands A and B also form an extension of the β-sandwich, with strand A arranged in an antiparallel fashion between strands K and B. α-helix C closes one end of the β-sandwich, with a short strand D forming an extension of the β-sandwich antiparallel to strand L.
Figure 1.

Ribbon structure of NiARD. Letters reference to the ARD sequence as follows: A (Ala 2-Phe 6), B (Leu 15-Ser 18), C (Glu 23-Lys 31), D (Val 33-Glu 36), E (Thr 52-Tyr 57), E′ (Ile 61-Lys 68), F (Ser 72-Leu 78), G (Lys 85-Glu 90), H (Phe 92-Glu 95), I (Arg 104-Val 107), J (Gly 111-Ile 117), K (Glu 120-Leu 125), L (Asn 129-Ile 132), M (His 140-Met 144), N (Phe 150-Phe 156), O (Trp 162-Phe 166), P (Ile 171-Ala 174). Position of Ni2+ is shown as a green sphere. Figure generated with MOLSCRIPT (Kraulis, 1991).
Figure 2.

(A) Stereoview of the superposition of the ensemble of 17 accepted structures of NiARD from the current refinement. Orientation is approximately the same as in Figure 1. RMSD for backbone heavy atoms is 0.65 Å. RMSD for all heavy atoms is 1.28 Å. Figure generated using MOLMOL (Koradi et al., 1996). (B) Heavy atom positions from a representative member of the ensemble in A, shown in approximately the same orientation as in A. Paramagnetically affected residues are shown in bold. Color key: red, Trp 162-Phe 166; purple, Asn 94-Val 103; yellow, Val 134-Asp 143. Position of Ni2+ indicated by a green sphere. (C) Metal binding site of NiARD based on modeling as described in the text. Position of Ni2+ indicated by a green sphere. View is from the opening to the active site (over strand N, with helix G to the upper left).
Helices E′ and P approximately bisect the β-sandwich on the top and bottom faces as viewed in Figure 1. The side chains of residues Ala 60 and Ile 61 on helix E′ contact two tryptophan side chains, Trp 38 and Trp 73, that occupy the cleft below the β-sandwich where helices E and E′ are situated. The long axes of helices E and E′ lie at about a 135° angle to each other. Helix P is a short (two-turn) 3,10 helix formed by residues 170–175 near the C-terminal. The terminal residues L178 and A179 interact with the loop between strands M and N (residues 145 and 146).
Comparison of 1M4O structures and current refinement
A comparison between the original NMR-derived structure for ARD (1M4O) and the current refinement is shown in Figure 3. As can be seen, the structures differ considerably, despite the fact that the same NOE-determined tertiary contacts are present in both structures. In particular, the E and E′ helices are displaced from their positions in 1M4O, if backbone positions in the β-sandwich are superimposed (Fig. 3). There are a number of reasons for this displacement, including the increased global restraints provided by RDCs on residues in strand N (Phe 150-Phe 156) and in a tight turn (Asn 158-Pro 159-Glu 160-Gly 161) immediately preceding the paramagnetically broadened Trp 162-Phe 166. The conformation and positions of these residues strongly affect the position of the C-terminal of helix E′. This is due to NOEs that are observed between the side chains of Tyr 70 and Ile 155, and between the Tyr 70 side chain and backbone resonances of Phe 156 and Asn 158 (strand N and turn-loop O). These NOEs were critical in determining the orientation of helix E′ in the 1M4O structures. As a result of the imposition of RDC restraints (and importantly, the modeled restraints on residue 163 relative to the metal binding site, vide infra), the conformation and position of this region is now better defined with respect to the overall structure. Also, after incorporation of RDC and improved TALOS restraints, several very weak NOEs obtained from a high signal-to-noise 1H, 15N-edited NOESY used in the original structure calculations consistently gave violations. These were presumed to be due to spin diffusion and removed. As a point of interest, current evidence from work with FeARD and non-metal containing mutants suggest the interactions between the E′ helix, strands F and N and turn-loop O are involved in determining the structural differences between FeARD and NiARD, and that rearrangement of the position of helix E′ may be critical in those structural differences (T. Ju, unpublished results).
Figure 3.

Superposition of mean backbone atom positions of the NiARD structure from 1M4O onto average structure from the current refinement. Superposition was performed for backbone atoms in the β-sandwich and helices C and G (residues 2-36 and 95-154 inclusive) to emphasize changes in positions of helices E and E′ (blue neon). RMSD for this superposition is 1.84 Å. If helix P is included in the fit (residues 170–175), RMSD is 1.93 Å. Modeled residues Trp 162- Phe 166 are shown in red neon, with the large shift in position due to the incorporation of hydrogen bonds between residues Ile 163 and Thr 97 based on the 1VR3 crystal structure (see text). Residues 155–160 are shown in green neon. Figure generated using MOLMOL.
The other important contribution to the large displacement of turn-loop O in the current structures relative to 1M4O is the result of modeling of the paramagnetically broadened Trp 162-Phe 166. In the 1M4O calculations, we constrained backbone atoms in those residues to be within 10 Å of the Ni2+ ion, but did not place any further restraints on them. As a result, these residues fell behind the two strands forming the metal binding site, but no further structural information could be obtained to confirm this placement. Inspection of the MmARD structure indicates a short antiparallel hydrogen bonding arrangement for residues homologous to Thr 97 in the active site and a residue on in the turn-loop region O. We tested both residues Ile 163 and Gln 165 as the hydrogen bonding partner for Thr 97. The Ile 163/Thr 97 pairing consistently gave the lowest energy structures. Inclusion of the Thr 97-Ile 163 hydrogen bonds places residues 162–166 close enough to the metal center to satisfy paramagnetic broadening restraints, and furthermore places the side chain of Trp 162 in a position homologous with that of Trp 155 in the MmARD structure.
The new structures reveal symmetry not apparent in the 1M4O structures. Helices C, E′ and P are now clearly parallel. An axis of pseudosymmetry is generated by the intersection of planes bisecting the β-sandwich along the long and short axes perpendicular to the plane of the page as viewed in Figure 1. C2 rotation around this axis interchanges antiparallel strands A, B with F, N, as well as helices E′ and P.
Ni2+ ligation and active site environment in NiARD
Based on XAS and NMR results (Al-Mjeni, Ju et al. 2002; (Szajna et al., 2004), the Ni2+ is expected to be ligated by six O/N ligands, at least three of which are histidine imidazole nitrogens. We have previously proposed that the side chain imidazoles of His 96, His 98 and His 140 as well as the carboxylate of Glu 102 provide four of the six ligands required for pseudooctahedral ligation of Ni2+ in NiARD (Pochapsky et al., 2002). His 96 and Glu 102 provide the axial ligands, while His 98 and His 140 provide two cis equatorial ligands. The two missing cis equatorial ligands are expected to be provided by water or substrate. In the absence of information regarding these ligands, we have left them as water in the current refinement.
The recently reported crystal structure of MmARD supports the proposed metal ligation scheme in NiARD. In the Mm structure, His 88, His 90, Glu 94 and His 133 provide the protein-based ligands for the metal (presumed but not proven to be Ni2+). All of these residues are directly homologous to the proposed metal ligands in NiARD. Despite this independent support, our original caution regarding ligand identity in NiARD remains (Pochapsky et al., 2002). The fact that binding of different metals changes both the enzymatic activity and chromatographic behavior of ARD suggests that metal ligation is plastic in this enzyme, and could depend on the type of metal bound or even the presence or absence of substrate (Al-Mjeni et al., 2002). We are currently attempting to obtain a crystallographic structure with a defined metal present in order to confirm the proposed ligation pattern.
Comparison of the NiARD structure with the MmARD structure
The structure of the ARD homologue from Mus musculus (MmARD) has been determined crystallographically and deposited as PDB entry 1VR3. Figure 5 shows that structure after minimization to regularize bond lengths while keeping α-carbon positions fixed. Sequence homology between the Mm and Klebsiella ARD sequences is shown in Figure 6. Although sequence identity is low (23% identity, 41% similarity), the highest degree of similarity is in the conserved β-helix, particularly in the vicinity of the active site. The Mm structure shows the same folding topology as NiARD from Klebsiella (hereafter called the Ko structure for comparison purposes), and as noted above, shows the same metal ligation scheme as that proposed for Ko, using homologous residues. The β-sandwich is formed with homologous interstrand registry in both proteins. Still, several differences are immediately apparent. Helix E, which is well-formed in KoARD, appears less well organized in MmARD. A large non-homologous insert between strands A and B in the Mm structure occupies space on the top of the β-sandwich in the Mm structure that in the Ko structure is occupied by the C-terminal 3,10 helix P. In the Mm stucture the C-terminal polypeptide (residues 165 to 179) forms a helix that lies parallel to the long axis of the β-sandwich.
Figure 5.
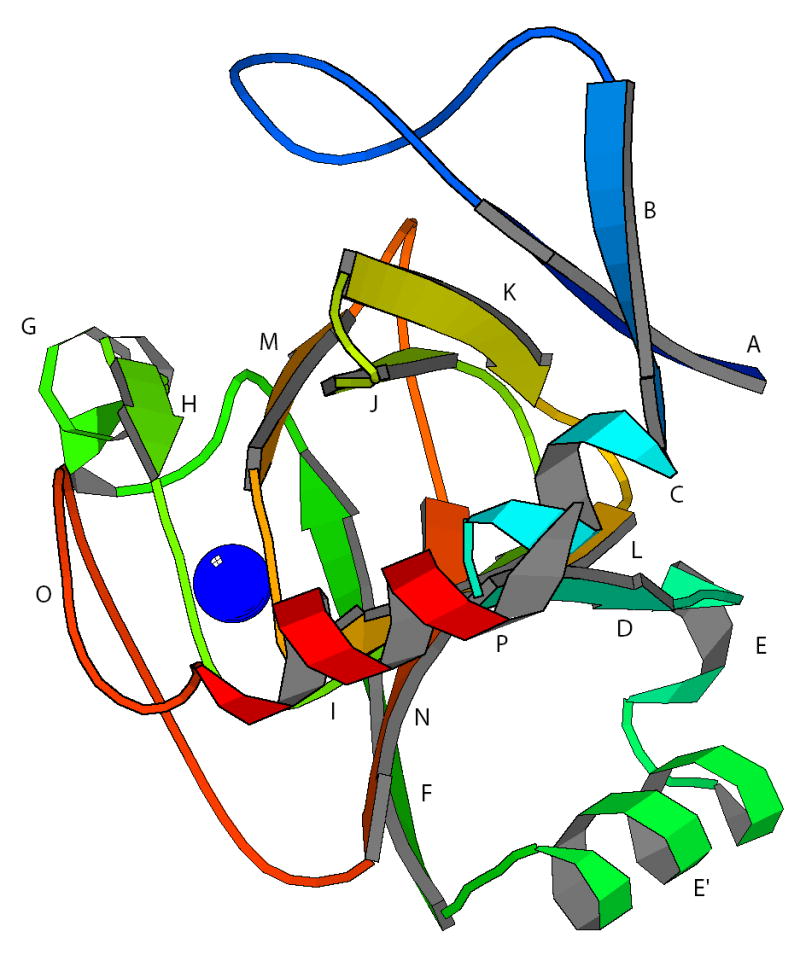
Crystal structure of MmARD (PDB entry 1VR3, JCSG, 2005) with structural features homologous to NiARD indicated by lettering. Structure is viewed in approximately the same orientation as KoARD in Fig. 1. The same letters are used in Figures 1, 5 and 6 to indicate structurally homologous features. A (Val 2-Tyr 6), B (Arg 24-Ser 27), C (Glu 29-Leu 34), D (Val 35-Trp 39), E (Lys 45-Asn 48), E′ (Leu 52-Arg 59), F (Ser 62-Ile 69), G (Asn 76-Lys 82), H (Phe 84-Glu 87), I (Arg 96-Leu 99), J (Gly 103-Asp 109), K (Lys 113-Ser 118), L ( Gly 122-Ile 125), M (His 133-Thr 136), N (Val 143-Phe 149), O (Trp 155- Asn 159) and P (Gln 170-Ala 179). Position of metal (presumed to be Ni2+) is shown as blue sphere.
Figure 6.

Sequence alignment of ARD homologue from Mus musculus (Mm) and from Klebsiella oxytoca (Ko, ATCC strain 8724). BLOSUM62 was used for the alignment. Conserved residues that act as ligands for the metal are boxed vertically and shaded red. Secondary structural features in Figures 1 and 5 are correlated with sequence by letter and color-coding. Note that for C-terminal feature P in either protein, no structural or sequence homology is implied; color coding is for convenience only.
The significance of this difference is as yet unknown. We have found that in a metal-free mutant of KoARD, H98S, the C-terminal polypeptide (residues 160 to 179) is largely unconstrained, and none of the NOEs observed in the NiARD (Ko) structure between the C-terminus and residues on the top face of the β-sandwich are detected in the H98S mutant. We also note that the position of the E′ helix and strand F are strongly perturbed in this mutant, and that the chemical shift pattern of residues in contact with the E′ helix are similar to those observed in FeARD (T. Ju, unpublished results). However, whether these changes represent the primary structural difference between NiARD and FeARD remains to be determined. If this were true, the MmARD structure might in fact more closely resemble FeARD than NiARD. We expect that continuing work on the structures of both FeARD and H98S KoARD in our laboratory will shed more light on this issue.
One surprising observation that may be related to C-terminal plasticity in ARD is the relative instability of NiARD to both orienting media. While the protein proved stable enough to obtain usable RDC data in the 2D experiments described here, after 24 hours the protein had decomposed significantly in both fd and C12E5 oriented phases. Practically, this precluded us from using multidimensional NMR methods for obtaining other RDCs (CN, C′-N, Cα-Cβ). It also raises an interesting question as to the origin of this instability. If, as the Mm and H98S structures suggest, the C-terminal in the configuration observed in NiARD is either metastable or kinetically trapped, interactions with the orienting medium may be sufficient to either lower the conformational barrier to other conformations of the C-terminal peptide or stabilize those orientations after formation.
Structural data deposition
Coordinates and experimental restraints for NiARD have been deposited with the RCSB Protein Data Bank (PDB ID code 1ZRR). NMR resonance assignments for NiARD are available through the BioMagResBank database, accession number 4313.
Figure 4.

Comparison of mean structures calculated as described in text with all constraints identical, but with (neon) and without (thin lines) RDC restraints. Mean structures were calculated each from ensembles of 17 structures. All structures in both ensembles gave no NOE violations > 0.5 Å. The two mean structures show a backbone RMSD of 1.24 Å. Fits of experimental RDC restraints averaged over both ensembles calculated using SSIA (Zweckstetter and Bax, 2000) are as follows: Structures calculated with RDC restraints gave goodness of fit values for C12E5 RDCs, R = 1.00, Q = 0.05 and for fd RDCs, R = 0.99, Q = 0.12. Structures calculated without RDC restraints gave goodness of fit values for C12E5 RDCs, R = 0.57, Q = 0.77 and for fd RDCs, R = 0.75, Q = 0.65.
Acknowledgments
This work was supported by the U. S. Public Health Service (GM067786, TCP). The 600 MHz NMR spectrometer used was purchased via a grant from the National Science Foundation (DBI-9871130). We acknowledge assistance provided by Charles Schweiters and John Kuszewski in using the XPLOR-NIH package, and thank Robert Pejchal for useful discussions and post-facto refinement of the MmARD structure.
References
- Al-Mjeni F, Ju T, Pochapsky TC, Maroney MJ. Biochemistry. 2002;41:6761–6769. doi: 10.1021/bi012209a. [DOI] [PubMed] [Google Scholar]
- Anand R, Dorrestein PC, Kinsland C, Begley TP, Ealick SE. Biochemistry. 2002;41:7659–7669. doi: 10.1021/bi0200965. [DOI] [PubMed] [Google Scholar]
- Bartels C, Xia TH, Billeter M, Guntert P, Wuthrich K. J Biomol NMR. 1995;6:1–10. doi: 10.1007/BF00417486. [DOI] [PubMed] [Google Scholar]
- Cornilescu G, Delaglio F, Bax A. J Biomol NMR. 1999;13:289–302. doi: 10.1023/a:1008392405740. [DOI] [PubMed] [Google Scholar]
- Dai Y, Wensink PC, Abeles RH. J Biol Chem. 1999;274:1193–1195. doi: 10.1074/jbc.274.3.1193. [DOI] [PubMed] [Google Scholar]
- Dai Y, Pochapsky TC, Abeles RH. Biochemistry. 2001;40:6379–6387. doi: 10.1021/bi010110y. [DOI] [PubMed] [Google Scholar]
- Dunwell JM, Culham A, Carter CE, Sosa-Aguirre CR, Goodenough PW. Trends in Biochemical Sciences. 2001;26:740–746. doi: 10.1016/s0968-0004(01)01981-8. [DOI] [PubMed] [Google Scholar]
- Hansen MR, Mueller L, Pardi A. Nature Struct Biol. 1998;5:1065–1074. doi: 10.1038/4176. [DOI] [PubMed] [Google Scholar]
- Hooft RWW, Vriend G, Sander C, Abola EE. Nature. 1996;381:272–272. doi: 10.1038/381272a0. [DOI] [PubMed] [Google Scholar]
- JCSG, the Joint Center for Structural Genomics. Protein Data Base Entry 1VR3. 2005. [Google Scholar]
- Kestell P. Charged sulfur compounds. In: Mitchell S, editor. Biological Interactions of Sulfur Compounds. Taylor & Francis: London; 1996. pp. 180–221. [Google Scholar]
- Ko TP, Day J, Mcpherson A. Acta Crystall Sec D-Biol Crystallogr. 2000;56:411–420. doi: 10.1107/s0907444900002237. [DOI] [PubMed] [Google Scholar]
- Koradi R, Billeter M, Wuthrich K. J Mol Graphics. 1996;14:51. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
- Kostic M, Pochapsky SS, Pochapsky TC. J Am Chem Soc. 2002;124:9054–9055. doi: 10.1021/ja0268480. [DOI] [PubMed] [Google Scholar]
- Kraulis PJ. J Appl Crystallogr. 1991;24:946–950. [Google Scholar]
- Kuszewski J, Gronenborn AM, Clore GM. Prot Sci. 1996;5:1067–1080. doi: 10.1002/pro.5560050609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuszewski J, Gronenborn AM, Clore GM. J Magn Reson. 1997;125:171–177. doi: 10.1006/jmre.1997.1116. [DOI] [PubMed] [Google Scholar]
- Kuszewski J, Clore GM. J Magn Reson. 2000;146:249–254. doi: 10.1006/jmre.2000.2142. [DOI] [PubMed] [Google Scholar]
- Laskowski RA, Rullmann JAC, Macarthur MW, Kaptein R, Thornton JM. J Biomol NMR. 1996;8:477–486. doi: 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
- Mo HP, Dai Y, Pochapsky SS, Pochapsky TC. J Biomol NMR. 1999;14:287–288. doi: 10.1023/a:1008396624784. [DOI] [PubMed] [Google Scholar]
- Muhandiram DR, Kay LE. J Magn Reson Series B. 1994;103:203–216. [Google Scholar]
- Pochapsky TC, Pochapsky SS, Ju TT, Mo HP, Al-Mjeni F, Maroney MJ. Nature Struct Biol. 2002;9:966–972. doi: 10.1038/nsb863. [DOI] [PubMed] [Google Scholar]
- Prestegard JH, Bougault CM, Kishore AI. Chem Rev. 2004;104:3519–3540. doi: 10.1021/cr030419i. [DOI] [PubMed] [Google Scholar]
- Ruckert M, Otting G. J Am Chem Soc. 2000;122:7793–7797. [Google Scholar]
- Szajna E, Dobrowolski P, Fuller AL, Arif AM, Berreau LM. Inorg Chem. 2004;43:3988–3997. doi: 10.1021/ic040002a. [DOI] [PubMed] [Google Scholar]
- Tjandra N, Omichinski JG, Gronenborn AM, Clore GM, Bax A. Nature Struct Biol. 1997;4:732–738. doi: 10.1038/nsb0997-732. [DOI] [PubMed] [Google Scholar]
- Woo EJ, Dunwell JM, Goodenough PW, Marvier AC, Pickersgill RW. Nature Struct Biol. 2000;7:1036–1040. doi: 10.1038/80954. [DOI] [PubMed] [Google Scholar]
- Wray JW, Abeles RH. J Biol Chem. 1993;268:21466–21469. [PubMed] [Google Scholar]
- Zweckstetter M, Bax A. J Am Chem Soc. 2000;122:3791–3792. [Google Scholar]