

Abstract
Rheumatoid arthritis (RA) is a chronic systemic inflammatory disorder which causes deforming joint disease and a spectrum of extraarticular manifestations. Poor disease control may lead to functional impairment and loss of independence. In recent times a prominent role for B cells in the pathogenesis of RA has been suggested. Two major theories have been postulated to explain the role of rheumatoid factor (RF) in the RA inflammatory process and the reason for RF overproduction; the loss of tolerance model and the autonomous mutated B cell model. With this in mind, strategies have been adopted to deplete B cells including the use of the anti-CD20 antibody rituximab. Rituximab leads to complement mediated lysis of B cells as well as antibody-dependant cellular cytotoxicity. It has been hypothesized that rituximab may also initiate apoptosis in RA and alter the ability of B cells to respond to antigen and other stimuli. Several recent studies using rituximab have demonstrated significant declines in RA activity providing evidence for the role of B cells in RA. Rituximab would appear to be a major addition to the increasing therapeutic options for sufferers of RA.
Keywords: rheumatoid arthritis, rituximab, autoimmune diseases
Introduction
Rheumatoid arthritis (RA) is a chronic systemic inflammatory disorder with consequences extending well beyond the recognizable joint disease. Poor disease control leads to pain and deformity with consequent functional impairment and loss of independence. The RA sufferer may also experience significant systemic illness due to extraarticular disease which includes nodules, vasculitis, pulmonary fibrosis, nerve entrapment, and myelopathy due cervical disease (Lee and Weinblatt 2001).
The RA syndrome and, in particular, extraarticular disease is associated with the production of the autoantibody known as rheumatoid factor (RF). This is an antibody directed against the Fc portion of immunoglobin G (IgG) constant regions (Edwards and Cambridge 1998). RF is found in 80% of patients with RA, but also may be found in normal individuals.
Several biological therapeutic approaches have been introduced for use typically in patients refractory to conventional agents such as methotrexate. These include tumor necrosis factor-α (TNF-α) and interleukin (IL)-1 receptor antagonists in an attempt to block the well described inflammatory cytokine pathways present in the disease (Choy and Panayi 2001). A more prominent role for B cells in the pathogenesis of RA has been suggested in recent times (Edwards and Cambridge 1995). With this in mind, strategies have been adopted to deplete B cells including the use of the anti-CD20 antibody, rituximab. Several recent studies using rituximab have demonstrated significant declines in RA activity providing evidence for the role of B cells in RA. In this review we provide an overview of the rationale for and experience with rituximab in RA.
Pathogenesis of RA
Current theories of the aetiopathogenesis of RA suggest that the disease is due to a combination of synovial hyperplasia and an inflammatory cell infiltrate secreting cytokines leading to joint destruction and deformity (Lee and Weinblatt 2001). Cytokines are believed to play a major role in the pathogenesis of the disease (Choy and Panayi 2001) and interact at various levels as outlined in schematic form in Figure 1. Tumor necrosis factor-α has often been considered to be the central cytokine responsible for the inflammatory infiltrate, particularly as one of the major cell populations in the rheumatoid joint is the macrophage. Feldmann et al (1996) and Firestein et al (1990) demonstrated in synovial cultures that TNF-α protein and mRNA expression were high in the macrophage population of RA patients compared with normal. In addition, inhibition of synovial TNF-α expression with TNF-α antibodies resulted in suppression of not only TNF-α but other inflammatory cytokines such as IL-1, IL-6, IL-8 and granulocyte-macrophage colony-stimulating factor (GMCSF) (Brennan et al 1989).
Figure 1.
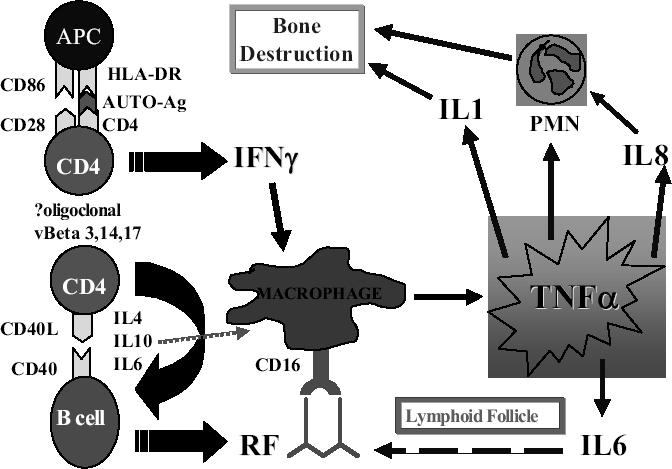
Proposed pathways in the inflammatory cascade of rheumatoid arthritis. Rituximab would be expected to act on B cells and RF production, thus decreasing TNF-α production by macrophages via Fc receptor binding.
Abbreviations:APC, antigen-presenting cell; IL, interleukin; INFγ, interferonγ; HLA-DR, human leukocyte antigen DR; PMN, polymorphonuclear cell; RF, rheumatoid factor; TNFα, tumor necrosis factor-α.
A natural progression of this work was to assess the role of anti-TNF-α−antibodies in clinical trials involving patients with RA. Recent trials using synthetic TNF-α antibodies have resulted in substantial remissions in RA patients compared with methotrexate or placebo reinforcing the invitro and animal data (Maini et al 1999; Weinblatt et al 1999). Where available, these anti-TNF-α−antibodies are now considered standard of care in RA for patients that have failed methotrexate or other disease-modifying anti-rheumatic drugs (DMARDs).
Some proponents of a role for T cells in RA point to the evidence for a genetic component in the disease. Approximately 80% of RA patients express human leukocyte antigen (HLA) DRB1 0404, 0401 subtypes compared with 28% in the general population (Lanchbury 1992). Given that the main function of HLA Class II molecules is to present antigen to CD4, it is a proposed that an inflammatory cascade is initiated through co-stimulatory molecules (Fox 1997). It is possible that secretion of interferon gamma from these activated T cells may activate macrophages to produce TNF-α. However, anti-CD4 antibody therapy was not beneficial in a randomised trial suggesting that T cells are not central to the pathogenesis of RA (van der Lubbe et al 1997). In contrast inhibition of costimulatory molecules such as CTLA-4Ig was recently successful (Kremer et al 2005) in ameliorating disease pointing to activation of T cells as a target for therapy in RA.
Rationale for treatment with rituximab (the role of B cells in RA)
Although T cell and macrophage populations dominate the current theories of pathogenesis of RA, there is emerging evidence that B cells and their production of RF may also be intrinsic to RA (Mitchison et al 2000). The possibility that RF is implicated in disease pathogenesis is raised by the consistent finding that 80% of patients with RA have elevated serum levels of the autoantibody which correlates with disease severity and extra-articular manifestations (Edwards et al 1999).
Rheumatoid factor is an antibody directed against the Fc fragment of the IgG molecule (Sutton et al 2000). In RA, RF can be of all subclasses, but is chiefly an IgG antibody that has a high affinity to receptors on macrophages and neutrophils, fixes complement, and can migrate into the extravascular space. RF in RA patients also shows a high degree of substitution mutations indicating affinity maturation (Randen et al 1993). This suggests that RF in RA patients is produced in response to an antigen/T cell-driven process of somatic mutation of the immunoglobulin gene sequence. In contrast, the RF produced by normal individuals in response to infection or immunization is usually IgM, transient, with low affinity, and of germ line origin suggesting it is part of the innate immune response (Thompson et al 1995). Given that RF is different in RA patients, it has been important to discover how it would contribute to inflammation and persist in patients with RA.
There are two major theories that have been postulated to explain the role of RF in the RA inflammatory process and the reason for RF overproduction: i) The loss of tolerance model due to CD40–CD40L activation signals (Kyburz et al 1999); ii) The autonomous mutated B cell model (Edwards et al 1999).
The loss of tolerance model due to CD40–CD40L activation signals
B cells can act as antigen-presenting cells. After engulfing foreign material they present antigen fragments to T cells in association with Major Histocompatibility Complex (MHC) class II. CD40 and CD40L are co-stimulatory molecules essential to the antigen-presenting role of B cells, particularly in RA (Sekine et al 1998). On engagement of T and B cells, the T cell becomes activated and expresses CD40L, which in turn binds CD40 on the B cell, initiating the process of somatic hypermutation of the immunoglobulin gene and class switching. In follicular centres of lymph nodes, antigen complexed with the C3d complement fragment can also signal to the B cell to proliferate without the assistance of T cells. In the case of a self-antigen, tolerance is generally initiated. This may be either by a lack of co-stimulation by the CD40–CD40L system or changed specificity of the B cell. Provision of either of these signals leads to apoptosis of the B cell.
Kyburz et al (1999) conducted experiments in a transgenic animal model suggesting overactivity of the CD40–CD40L system may occur in RA (Kyburz et al 1999). This overactivity results in excess IL-4 production, leading to the inhibition of B cell apoptosis. There is also differentiation of B cells into plasma cells, thus maintaining them in germinal centres in a state of continuous RF production. CD40 is expressed on synovial monocytes, which potentially leads to TNF-α production. CD40 is also found on synovial fibroblasts which may provide a supportive stroma for B cell differentiation within the joint (van Kooten and Banchereau 2000). CD40L is expressed on activated T cells within the lymphoid aggregates of joints in RA patients (Sekine et al 1998). This theory does not explain the actual role of RF, but can account for the activation signals that potentiate its production.
The autonomous mutated B cell model
Edwards and Cambridge (1998) have promoted a hypothesis that RF itself is pathogenic due to a random immunoglobulin gene mutation leading to dysfunctional IgG which initiates inflammation by Fc receptor attachment, TNF-α production and complement activation. This hypothesis predicts that B cell production of IgG RF arises due to a random mutation when immunoglobulin gene rearrangement occurs in the germinal centre upon antigen stimulation (Pulendran et al 1997). If the antigen is a protein involved in the immune system (such as IgG) it is possible that RF-producing B cells will undergo apoptosis as part of tolerance or may escape tolerance leading to autonomous production such as in RA (Edwards and Cambridge 1995). Once RF is produced by a clone of B cells which have escaped tolerance, it is likely that because it is an IgG immunoglobulin, it has the ability to pass out of the circulation and attach to the IgG Fc receptor on macrophages in the synovium. The FcγRIIIa receptor is abundantly expressed on synovial macrophages (Edwards, Blades, et al 1997) and when activated by dimers of IgG, such as RF, it facilitates the release of TNF-α (Abrahams et al 2000). TNF-α in turn upregulates the expression of vascular cell adhesion molecule (VCAM) and other adhesion molecules (Edwards, Leigh, et al 1997), promoting survival of B cells in intimate contact with the macrophages. ‘Nurse-like’ cells in bone marrow and synovium in RA patients are also likely to contribute to prolonged survival and IgG production via upregulation of VCAM and intercellular adhesion molecule (ICAM) (Shimaoka et al 1998). The dimerization of RF IgG molecules can also activate complement which will contribute to the inflammatory cell infiltrate in the pannus (Edwards and Cambridge 1998b).
Thus it is clear that B cells can be pathogenic in RA at numerous levels. B cells are the source of both RF and IL-6 thus potentiating their own secretion of autoantibody. These pathways can interact with the production of TNF-α via the Fc portion of macrophages (Abrahams et al 2000). In addition, the B cell’s ability to act as an antigen-presenting cell initiates the CD40–CD40L cascade and the T cell cytokine cascade. The availability of rituximab provides an attractive therapeutic option to deplete B cells in RA and thus disrupt these pro-inflammatory pathways which are central to the pathogenesis of RA.
Clinical trials using rituximab in RA
Rituximab is a mouse–human chimeric monoclonal antibody that has a murine variable region that is specific for CD20, an antigen found on the surface of B cells in their earlier stages of development prior to the plasma cell stage. The IgG1 heavy and light chain constant regions are human.
CD20 is also found on most malignant B cell lymphoma cells and is postulated to be involved in regulation of the cell cycle and cell differentiation. Rituximab leads to complement-mediated lysis of B cells as well as antibody-dependant cellular cytotoxicity. It has been hypothesized that rituximab may also initiate apoptosis in RA and alter the ability of B cells to respond to antigen and other stimuli (Tsokos 2004). This agent was originally introduced in 1997 for the treatment of low grade lymphoma alongside combined chemotherapy. There has since been a proliferation of potential indications for its use including an extended spectrum of hematological malignancies, nonmalignant hematological disease such as immune thrombocytopenic purpura (ITP), autoimmune hemolytic anemia, and other autoimmune diseases. There is now considerable experience in rituximab use, with the agent being generally well tolerated. The main problems with its use include infusional reactions, cytopenias, and immunosuppression. It is probable that the lack of depletion of plasma cells with rituximab has avoided significant immunoglobulin deficiency in most clinical trials including rheumatoid patients (Edwards et al 2004; Moore et al 2004).
CD20 is not expressed on hematopoietic stem cells and for this reason B cells may be depleted by rituximab without preventing their regeneration whilst hopefully eliminating the RF-producing clone. Rituximab has been used to date in patients that have been heavily pre-treated with more conventional agents. It has generally been combined in use with other agents, most commonly cyclophosphamide and methotrexate.
In the first published data by Edwards and Cambridge (2001), rituximab was used in combination with cyclophosphamide in 5 patients leading to an American College of Rheumatology (ACR)50 in 5/5 and ACR70 in 3/ 5 at 26 weeks. This was followed by other studies by Leandro et al (2002), De Vita et al (2002), Kneitz et al (2004), and Higashida et al (2005) demonstrating efficacy of rituximab (see Table 1). Leandro et al (2002) studied the use of rituximab alone compared with rituximab and differing combinations of cyclophosphamide and methotrexate in five different cohorts. The results of this study suggested that rituximab alone may not be as effective and that cyclophosphamide or methotrexate would be required to provide activity on non-B cell populations.
Table 1.
Major clinical trials reporting rituximab use in rheumatoid arthritis
| Trial | Number of patients | ACR50 at 6 months (%) |
|---|---|---|
| Edwards and Cambridge 2001 | 5 | 100 |
| De Vita et al 2002 | 5 | 40 |
| Leandro et al 2002 | 291 | 59 |
| Moore et al 2004 | 10 | 80 |
| Edwards et al 2004 | 161 | 422 |
| Higashida et al 2005 | 17 | 33 |
Note: Kneitz et al 2004 reported a significant improvement in disease activity score at 28 weeks (>1.2) in 4/5 patients (ACR50 not reported); 122 patients underwent 29 courses; 242% of 81 patients receiving rituximab combined with cyclophosphamide or methotrexate
Abbreviations: ACR, American College of Rheumatology.
Cyclophosphamide may confer a currently unexplained synergistic action with rituximab that may be important in the mechanism of the response. In addition to its B cell depleting properties, cyclophosphamide may have other immunosuppressive properties, such as an effect on T cell co-stimulation as has been demonstrated in systemic lupus erythematosus (SLE) patients (Amano et al 2000). We tested the hypothesis that cyclophosphamide was a key contributor to the effect of rituximab in a trial of patients who had previously received cyclophosphamide a median of 25 months earlier as part of hemopoietic stem cell transplant (HSCT) treatment (Moore et al 2002). During the HSCT trial, it was demonstrated that the major responses to HSCT was during the period of B cell lymphopenia (months 3–9). Similarly, the use of rituximab in the same cohort of patients produced the same pattern of B cell depletion and response during the 3–9 month period (Moore et al 2004). These heavily pre-treated patients did not demonstrate major toxicity from rituximab and importantly 9/10 did not experience hypogammaglobulinemia.
Edwards followed up his pioneering work with a much larger series of 161 patients in the only published randomized study using rituximab in RA (Edwards et al 2004). Patients all had active RA despite treatment with methotrexate and were treated in 4 groups; methotrexate alone, rituximab, rituximab plus cyclophosphamide (750 mg on days 3 and 17), or rituximab plus methotrexate. In the three rituximab groups the dose was a 1000 mg intravenous infusion on days 1 and 15. At 24 weeks ACR50 was achieved in 43% with the rituximab–methotrexate combination and 41% in the rituximab–cyclophosphamide combination. This was significantly better than the 13% in methotrexate alone (p=0.005). Responses were also noted in the rituximab alone arm but were inferior to that achieved in the combinations arms.
Concluding Remarks
Thus the trials outlined in Table 1 clearly signify the importance of the B cell in the immunopathogenesis of RA. Edwards et al (2004) and our own data point more to the importance of RF eradication which is reliant on and naturally follows B cell depletion. Future work will need to assess the optimal dosing schedule to obtain RF eradication and studies are currently underway and only reported in abstract form.
The data with rituximab in RA is equivalent to and perhaps even superior to some of the published TNF-α antagonist trials (Maini et al 1999; Weinblatt et al 1999) where ACR50 rates at 6 months ranged between 26%–39%. In addition, Abatacept, a newer constimulator modulator has recently been reported to have a similar response rate (Kremer et al 2005). The major advantages of rituximab over these agents would appear to be the shorter dosage schedule, which avoids the need for regular hospital visits. Another potential advantage is less infection due to the sparing of immunoglobulin deficiency, although a longer follow up will be required to confirm this observation. Further studies are required at this stage to assess infection rate and optimal dosage scheduling, but rituximab would appear to be a major addition to the increasing therapeutic options for sufferers of this debilitating disease.
References
- Abrahams VM, Cambridge G, Lydyard PM, et al. Induction of tumor necrosis factor alpha production by adhered human monocytes: a key role for Fc gamma receptor type IIIa in rheumatoid arthritis. Arthritis Rheum. 2000;43:608–16. doi: 10.1002/1529-0131(200003)43:3<608::AID-ANR18>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Amano H, Morimoto S, Kaneko H, et al. Effect of intravenous cyclophosphamide in systemic lupus erythematosus: Relation to lymphocyte subsets and activation markers. Lupus. 2000;9:26–32. doi: 10.1177/096120330000900106. [DOI] [PubMed] [Google Scholar]
- Brennan FM, Chantry D, Jackson A, et al. Inhibitory effect of TNF alpha antibodies on synovial cell interleukin-1 production in rheumatoid arthritis. Lancet. 1989;2:244–7. doi: 10.1016/s0140-6736(89)90430-3. [DOI] [PubMed] [Google Scholar]
- Choy EH, Panayi GS. Cytokine pathways and joint inflammation in rheumatoid arthritis. N Eng J Med. 2001;344:907–16. doi: 10.1056/NEJM200103223441207. [DOI] [PubMed] [Google Scholar]
- De Vita S, Zaja F, Sacco S, et al. Efficacy of selective B cell blockade in the treatment of rheumatoid arthritis: evidence for a pathogenetic role of B cells. Arthritis Rheum. 2002;46:2029–33. doi: 10.1002/art.10467. [DOI] [PubMed] [Google Scholar]
- Edwards JC, Blades S, Cambridge G. Restricted expression of Fc gammaRIII (CD16) in synovium and dermis: implications for tissue targeting in rheumatoid arthritis (RA) Clin Exp Immunol. 1997;108:401–6. doi: 10.1046/j.1365-2249.1997.3941286.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards JC, Cambridge G. Is rheumatoid arthritis a failure of B cell death in synovium? Ann Rheum Dis. 1995;54:696–700. doi: 10.1136/ard.54.9.696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards JC, Cambridge G. Rheumatoid arthritis – the predictable effect of small immune complexes in which antibody is also antigen. Br J Rheumatol. 1998;37:126–30. doi: 10.1093/rheumatology/37.2.126. [DOI] [PubMed] [Google Scholar]
- Edwards J, Cambridge G. Sustained improvement in rheumatoid arthritis following a protocol designed to deplete B lymphocytes. Rheumatology. 2001;40:205–11. doi: 10.1093/rheumatology/40.2.205. [DOI] [PubMed] [Google Scholar]
- Edwards JC, Cambridge G, Abrahams VM. Do self-perpetuating B lymphocytes drive human autoimmune disease? Immunology. 1999;97:188–96. doi: 10.1046/j.1365-2567.1999.00772.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards JC, Leigh RD, Cambridge G. Expression of molecules involved in B lymphocyte survival and differentiation by synovial fibroblasts. Clin Exp Immunol. 1997b;108:407–14. doi: 10.1046/j.1365-2249.1997.4061306.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards JC, Szczepanski L, Szechinski J, et al. Efficacy of B-cell-targeted therapy with rituximab in patients with rheumatoid arthritis. N Eng J Med. 2004;350:2572–81. doi: 10.1056/NEJMoa032534. [DOI] [PubMed] [Google Scholar]
- Feldmann M, Brennan FM, Williams RO, et al. Cytokine expression and networks in rheumatoid arthritis – rationale for anti-Tnf-alpha antibody therapy and its mechanism of action. J Inflamm. 1996;47:90–6. [PubMed] [Google Scholar]
- Firestein GS, Alvaro-Garcia JM, Maki R. Quantitative analysis of cytokine gene expression in rheumatoid arthritis. J Immunol. 1990;144:3347–53. [PubMed] [Google Scholar]
- Fox DA. The role of T cells in the immunopathogenesis of rheumatoid arthritis - new perspectives. Arthritis Rheum. 1997;40:598–609. doi: 10.1002/art.1780400403. [DOI] [PubMed] [Google Scholar]
- Higashida J, Wun T, Schmidt S, et al. Safety and efficacy of rituximab in patients with rheumatoid arthritis refractory to disease modifying antirheumatic drugs and anti-tumor necrosis factor-alpha treatment. J Rheum. 2005;32:2109–15. [PubMed] [Google Scholar]
- Kneitz C, Wilhelm M, Tony HP. Improvement of refractory rheumatoid arthritis after depletion of B cells. Scand J Rheum. 2004;33:82–6. doi: 10.1080/03009740310004379. [DOI] [PubMed] [Google Scholar]
- Kremer JM, Dougados M, Emery P, et al. Treatment of rheumatoid arthritis with the selective costimulation modulator abatacept: twelve-month results of a phase iib, double-blind, randomized, placebo-controlled trial. Arthritis Rheum. 2005;52:2263–71. doi: 10.1002/art.21201. Erratum, p 3321. [DOI] [PubMed] [Google Scholar]
- Kyburz D, Corr M, Brinson DC, et al. Human rheumatoid factor production is dependent on CD40 signalling and autoantigen. J Immunol. 1999;163:3116–22. [PubMed] [Google Scholar]
- Lanchbury JS. The HLA association with rheumatoid arthritis. Clin Exp Rheum. 1992;10:301–4. [PubMed] [Google Scholar]
- Leandro MJ, Edwards JC, Cambridge G. Clinical outcome in 22 patients with rheumatoid arthritis treated with B lymphocyte depletion. Ann Rheum Dis. 2002;61:883–8. doi: 10.1136/ard.61.10.883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DM, Weinblatt ME. Rheumatoid arthritis. Lancet. 2001;358:903–11. doi: 10.1016/S0140-6736(01)06075-5. [DOI] [PubMed] [Google Scholar]
- Maini R, St Clair EW, Breedveld F, et al. Infliximab (chimeric anti-tumour necrosis factor alpha monoclonal antibody) versus placebo in rheumatoid arthritis patients receiving concomitant methotrexate: a randomised phase III trial. Lancet. 1999;354:1932–9. doi: 10.1016/s0140-6736(99)05246-0. [DOI] [PubMed] [Google Scholar]
- Mitchison NA, Wedderburn LR. B cells in autoimmunity. Proc Natl Acad Sci U S A. 2000;97:8750–1. doi: 10.1073/pnas.97.16.8750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore J, Brooks P, Milliken S, et al. A pilot randomized trial comparing CD34-selected versus unmanipulated hemopoietic stem cell transplantation for severe, refractory rheumatoid arthritis. Arthritis Rheum. 2002;46:2301–9. doi: 10.1002/art.10495. [DOI] [PubMed] [Google Scholar]
- Moore J, Ma D, Will R, et al. A phase II study of Rituximab in rheumatoid arthritis patients with recurrent disease following haematopoietic stem cell transplantation. Bone Marrow Transplant. 2004;34:241–7. doi: 10.1038/sj.bmt.1704570. [DOI] [PubMed] [Google Scholar]
- Pulendran B, van Driel R, Nossal GJ. Immunological tolerance in germinal centres. Immunol Today. 1997;18:27–32. doi: 10.1016/s0167-5699(97)80011-4. [DOI] [PubMed] [Google Scholar]
- Randen I, Pascual V, Victor K, et al. Synovial IgG rheumatoid factors show evidence of an antigen-driven immune response and a shift in the V gene repertoire compared to IgM rheumatoid factors. Eur J Immunol. 1993;23:1220–5. doi: 10.1002/eji.1830230604. [DOI] [PubMed] [Google Scholar]
- Sekine C, Yagita H, Miyasaka N, et al. Expression and function of CD40 in rheumatoid arthritis synovium. J Rheum. 1998;25:1048–53. [PubMed] [Google Scholar]
- Shimaoka Y, Attrep JF, Hirano T, et al. Nurse-like cells from bone marrow and synovium of patients with rheumatoid arthritis promote survival and enhance function of human B cells. J Clin Invest. 1998;102:606–18. doi: 10.1172/JCI3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton B, Corper A, Bonagura V, et al. The structure and origin of rheumatoid factors. Immunol Today. 2000;21:177–83. doi: 10.1016/s0167-5699(00)01589-9. [DOI] [PubMed] [Google Scholar]
- Thompson KM, Borretzen M, Randen I, et al. V-gene repertoire and hypermutation of rheumatoid factors produced in rheumatoid synovial inflammation and immunized healthy donors. Ann N Y Acad Sci. 1995;764:440–9. doi: 10.1111/j.1749-6632.1995.tb55861.x. [DOI] [PubMed] [Google Scholar]
- Tsokos GC. B cells, be gone – B-cell depletion in the treatment of rheumatoid arthritis. N Eng J Med. 2004;350:2546–8. doi: 10.1056/NEJMp048114. [DOI] [PubMed] [Google Scholar]
- van der Lubbe PA, Breedveld FC, Tak PP, et al. Treatment with a chimeric CD4 monoclonal antibody is associated with a relative loss of CD4+/CD45RA+ cells in patients with rheumatoid arthritis. J Autoimmunity. 1997;10:87–97. doi: 10.1006/jaut.1996.0113. [DOI] [PubMed] [Google Scholar]
- van Kooten C, Banchereau J. CD40-CD40 ligand. J Leukocyte Biol. 2000;67:2–17. doi: 10.1002/jlb.67.1.2. [DOI] [PubMed] [Google Scholar]
- Weinblatt ME, Kremer JM, Bankhurst AD, et al. A trial of etanercept, a recombinant tumor necrosis factor receptor: Fc fusion protein, in patients with rheumatoid arthritis receiving methotrexate. N Eng J Med. 1999;340:253–9. doi: 10.1056/NEJM199901283400401. [DOI] [PubMed] [Google Scholar]