

Abstract
Background
It has been reported in the quantitative trait locus (QTL) literature that when testing for QTL location and effect, the statistical power supporting methodologies based on two markers and their estimated genetic map is higher than for the genetic map independent methodologies known as single marker analyses. Close examination of these reports reveals that the two marker approaches are more powerful than single marker analyses only in certain cases.
Simulation studies are a commonly used tool to determine the behavior of test statistics under known conditions. We conducted a simulation study to assess the general behavior of an intersection test and a two marker test under a variety of conditions. The study was designed to reveal whether two marker tests are always more powerful than intersection tests, or whether there are cases when an intersection test may outperform the two marker approach.
We present a reanalysis of a data set from a QTL study of ovariole number in Drosophila melanogaster.
Results
Our simulation study results show that there are situations where the single marker intersection test equals or outperforms the two marker test. The intersection test and the two marker test identify overlapping regions in the reanalysis of the Drosophila melanogaster data. The region identified is consistent with a regression based interval mapping analysis.
Conclusion
We find that the intersection test is appropriate for analysis of QTL data. This approach has the advantage of simplicity and for certain situations supplies equivalent or more powerful results than a comparable two marker test.
Background
Many authors [1-4] have compared the statistical power of different methods for quantitative trait locus (QTL) mapping. These comparisons have shown that the additional information supplied by the genetic distance between identifiable DNA markers unconfounds the QTL effect from the QTL location, thus making two marker models more powerful than single marker models of detection (for review see Doerge et al. [5]).
With the goal of detecting and/or locating QTL, there are two common statistical approaches that can be taken. The first approach is based on ANOVA, or simple linear regression, and performs statistical tests based solely on single DNA marker information. No genetic map is required for single marker analysis, and the calculations are based on phenotypic means and variances within each of the genotypic classes. The second, and more involved approach, is based on two DNA markers, the estimated recombination fraction between them (or the estimated genetic distance), and either a maximum likelihood based calculation or a regression model including multiple (two or more) markers as independent variables. The linear ordering of multiple DNA markers based upon their estimated relationships (i.e., recombination fraction (or genetic distance)) supplies the framework or genetic map for (composite) interval mapping [6,7] and as such unconfounds the QTL effect and the QTL location, thus providing a more precise means for detecting and locating QTL with respect to the estimated genetic map for the organism under investigation.
Rebaï et al. [4] present a comprehensive comparison of the statistical power for many of the commonly used flanking marker or two marker methods employed, and conclude that two marker mapping provides a relatively small gain (5%) in power over single marker methods when the two markers define an interval of width less than 20 cM, but a substantial increase (greater than 30%) in power for intervals upwards of 70 cM, indicating that the gain in power may come from the addition of the second marker to the analysis, or the addition of information from that marker, rather than the map.
Using the findings of Rebaï et al. [4], and others as our motivation, we hypothesize that the power increase between single marker and two marker regression methods is due to additional genotypic information in the second marker. In order to assess this, similar test statistics should be compared. A comparison of maximum likelihood interval mapping to single marker ANOVA is complicated because of the differences that may be observed due to differences between regression and maximum likelihood [3], as well as differences in marker information. In order to avoid this complication, we consider two regression based approaches that differ only in the number of markers included in the initial model (i.e. the statistical methodology is the same, the models are different). First, a set of compound hypotheses are defined for use with a single marker analysis and used to define an intersection test. We then state the equivalent hypotheses for a regression based two marker model for a test of the interval. Last we compare, via simulation, the power of the intersection and the two marker test under the stated hypotheses for each model. We do not consider the case of multiple QTL in a single interval, as this case was not considered by Rebaï et al. [4]. These two approaches are then applied to a 'backcross' population of Drosophila with 76 informative markers [8] to detect QTL associated with ovariole number.
Results
Simulations
An overview of the simulations performed is given in Table 1 and 2. For the purpose of evaluating the relative difference in statistical power between the intersection test and the two marker test, the estimated statistical power of the two marker test was subtracted from that of the intersection test for each of the parameter combinations investigated, and a t-test [9] was performed to test the null hypothesis that the mean difference in power was zero.
Table 1.
Simulation conditions for a binary phenotype, two marker loci M1 and M2, a single locus (Q), sample sizes n = 100, 500, recombination fraction ![]() and
and ![]() , respectively, and effect size. *
, respectively, and effect size. *![]() =
= ![]() = 0.0 not simulated
= 0.0 not simulated
| Population | n | Effect Size | Number of combinations | ||
| Backcross | 100, 500 | 0.0*, 0.10, 0.20, 0.30, 0.40, 0.50 | 0.0*, 0.10, 0.20, 0.30, 0.40, 0.50 | 0.40, 0.60, 0.80, 1.00 | 239* |
| F2 | 500 | 0.0*, 0.10, 0.20, 0.30, 0.40, 0.50 | 0.0*, 0.10, 0.20, 0.30, 0.40, 0.50 | 1.00 | 25 |
Table 2.
Simulation conditions for a normally distributed trait (Q), sample size n = 500, recombination fraction ![]() and
and ![]() , respectively, and effect size. *
, respectively, and effect size. *![]() =
= ![]() = 0.0 not simulated
= 0.0 not simulated
| Population | n | Effect size | Number of combinations | ||
| Backcross | 500 | 0.0*, 0.10, 0.20, 0.30, 0.40, 0.50 | 0.0*,0.10, 0.20, 0.30, 0.40, 0.50 | 0.20, 0.50, 0.80 | 75 |
For the binomial phenotype with a backcross design, sample size of 100, the 100 parameter combinations examined resulted in 34 showing no difference in statistical power between the intersection test and the two marker test (i.e, the value of the difference was exactly zero), 39 favoring the intersection test, and 27 favoring the two marker test. The intersection test was more powerful with a mean difference in power of 0.010, and the t-test of the null hypothesis that the difference in power was zero was rejected (p = 0.020). When n was equal to 500, the 139 parameter combinations examined yielded 116 showing there was no difference in statistical power, 18 parameter combinations indicated the intersection test as more powerful, and 5 indicated the two marker test as more powerful. The mean difference in power was 0.020, and the t-test of the null hypothesis that the difference in power was zero was rejected (p = 0.010). The test of the null hypothesis that the mean difference between the intersection and two marker was zero was rejected for both sets of simulations. The estimated difference between the two approaches was positive, indicating that the intersection test has slightly higher power than the two marker test in these cases.
We also investigated F2 experimental populations for a binomial phenotype, using a sample size of 500. From the 25 parameter combinations investigated, 5 failed to converge consistently for the two marker model due to singularity in the design matrix. The remaining 20 parameter combinations showed 10 as having no difference in statistical power, while the remaining 10 favored the intersection test. Results similar to those found in the initial simulations indicate that the intersection test performs as well as, or better than the two marker test. Comparisons with smaller sample sizes (n = 100) were not conducted because of convergence problems using the two marker model.
For a normally distributed phenotype, 75 parameter combinations for the backcross were examined. From these, 12 showed no difference in statistical power, 34 scenarios favored the intersection test, while 29 indicated the two marker test as more powerful. The mean difference in power was 0.015, and we failed to reject the test of the null hypothesis that the difference was zero, p = 0.064.
Upon investigation of the parameter combinations that showed some difference in power, specifically, for the scenario highlighted by Rebaï et al. [4] (QTL in the middle of the interval with a large distance between markers), we also find the two marker approach to be slightly more powerful than the intersection test. However, when the distance between the QTL and one marker is much smaller than the distance between the QTL and the second marker, the intersection test is more powerful. Although we can point to these cases, it is important to realize that for most of the scenarios no difference in power was observed (see Figure 1).
Figure 1.

Histogram and density plot of difference in power between intersection test and two marker test for 334 simulations.
Drosophila Analysis
Ovariole number is related to reproductive success in Drosophila melanogaster and positively correlated with maximum daily female fecundity [10,11]. The 98 RILs (recombinant inbred lines) for this study were scored for the trait ovariole number and genotyped as described in Wayne et al. [8].
For the 71 marker pairs considered, 36 markers were found to be significant using both the intersection test and the two marker test, and 26 were found to be non-significant with both tests. The intersection and two marker tests were concordant in 62 of the 71 pairs of markers (Table 3). The estimated chance corrected agreement (Kappa coefficient) was 0.75 with a 95% confidence interval of (0.58,0.90). McNemar's test showed no systematic difference in the two approaches (S = 1.00, p = 0.32).
Table 3.
Concordance of significant test results from analysis of 71 unique pairs of adjacent makers using the intersection test and two marker regression test. Yes, indicates that test was significant, No, indicates test was not significant at α = 0.05
| Two marker regression test | Intersection test | |
| Yes | No | |
| Yes | 26 | 6 |
| No | 3 | 36 |
Overlapping regions on chromosome 3 were identified by the intersection test, the two marker test, and the interval mapping test (Table 4). On chromosome 1, the two marker test identified a region between 3E and 7D while the comparable intersection test showed borderline significance for one marker in that region (4F, p = 0.0256). On chromosome 2, the two marker test identified a region from 35B-46C while intersection tests identified a smaller region from 35B-38E and the interval mapping identified a region from 38A-46C. On Chromosome 3 the two marker test identifies two regions 65A-87F and 96F-100A while the intersection test finds the entire region from 63A-100A significant. The interval mapping agrees with the intersection test for interval 63A-65A and finds it significant, while it agrees with the two marker test for the interval 97D-97F and does not find this interval significant. Chromosome 4 was not associated with the trait for any test.
Table 4.
Marker analysis for ovariole number in Drosophila melanogaster from analysis using intersection test, α = 0.025, two marker regression test α = 0.05; and interval mapping; only markers significant in either the two marker test or intersection test listed
| Two marker test | Intersection test | Interval mapping | |||||||||
| Chromosome | Marker | Frequency M2N2 | M2N2 df | M2N2 p-value | Frequency M2 | Frequency N2 | M2 df | N2 df | M2 p-value | N2 p-value | p-value |
| 1 | 3E-4F | 0.67 | 94 | 0.029 | 0.67 | 0.86 | 96 | 95 | 0.14 | 0.0256 | 0.0242 |
| 4F-5D | 0.81 | 93 | 0.032 | 0.86 | 0.84 | 95 | 96 | 0.0256 | 0.056 | 0.0226 | |
| 6E-7D | 0.7 | 93 | 0.0499 | 0.83 | 0.74 | 96 | 95 | 0.069 | 0.1 | 0.0373 | |
| 2 | 35B-38A | 0.69 | 82 | 0.028 | 0.81 | 0.83 | 89 | 84 | 0.33 | 0.017 | 0.3036 |
| 38A-38E | 0.7 | 77 | 0.017 | 0.83 | 0.88 | 84 | 87 | 0.017 | 0.028 | 0.0105 | |
| 38E-43A | 0.71 | 78 | 0.028 | 0.88 | 0.84 | 87 | 84 | 0.028 | 0.038 | 0.0083 | |
| 43A-43E | 0.73 | 79 | 0.03 | 0.84 | 0.9 | 84 | 87 | 0.038 | 0.034 | 0.0177 | |
| 43E-46C | 0.8 | 85 | 0.038 | 0.90 | 0.89 | 87 | 89 | 0.034 | 0.073 | 0.0339 | |
| 3 | 63A-65A | 0.29 | 85 | 0.075 | 0.49 | 0.38 | 94 | 89 | 0.227 | 0.0249 | 0.0187 |
| 65A-65D | 0.3 | 86 | 0.035 | 0.38 | 0.51 | 89 | 93 | 0.0249 | 0.13 | 0.0207 | |
| 65D-67D | 0.42 | 90 | 0.021 | 0.51 | 0.51 | 93 | 94 | 0.13 | 0.004 | 0.0031 | |
| 67D-68B | 0.44 | 91 | 0.00004 | 0.51 | 0.58 | 94 | 94 | 0.004 | 0 | 0 | |
| 68B-68C | 0.46 | 90 | 0.00003 | 0.58 | 0.49 | 94 | 92 | 0 | 0.002 | 0 | |
| 68C-69D | 0.41 | 89 | 0.002 | 0.49 | 0.46 | 92 | 94 | 0.002 | 0.007 | 0.0013 | |
| 69D-70C | 0.34 | 86 | 0.0007 | 0.46 | 0.45 | 94 | 90 | 0.007 | 0.0008 | 0.001 | |
| 70C-71E | 0.35 | 84 | 0.0002 | 0.45 | 0.38 | 90 | 89 | 0.0008 | 0.0006 | 0.0009 | |
| 71E-72A | 0.35 | 84 | 0.0002 | 0.39 | 0.42 | 89 | 90 | 0.0006 | 0.0007 | 0.0016 | |
| 72A-73D | 0.39 | 88 | 0.0001 | 0.42 | 0.46 | 90 | 90 | 0.0007 | 0.0003 | 0.001 | |
| 73D-76A | 0.34 | 83 | 0.0001 | 0.46 | 0.37 | 90 | 87 | 0.0003 | 0.0001 | 0.0007 | |
| 76A-76B | 0.34 | 85 | 0.0001 | 0.37 | 0.45 | 87 | 92 | 0.0001 | 0.0009 | 0.0005 | |
| 76B-77A | 0.41 | 90 | 0.0008 | 0.45 | 0.44 | 92 | 92 | 0.0009 | 0.0009 | 0.0009 | |
| 77A-82D | 0.34 | 83 | 0.0002 | 0.44 | 0.38 | 92 | 87 | 0.0009 | 0.0003 | 0.0011 | |
| 82D-85F | 0.31 | 84 | 0.0006 | 0.38 | 0.38 | 87 | 90 | 0.0003 | 0.0012 | 0.0006 | |
| 85F-87B | 0.27 | 84 | 0.0019 | 0.38 | 0.32 | 90 | 89 | 0.0012 | 0.003 | 0.0012 | |
| 87B-87E | 0.28 | 85 | 0.008 | 0.32 | 0.34 | 89 | 91 | 0.003 | 0.01 | 0.0028 | |
| 87E-87F | 0.27 | 84 | 0.006 | 0.34 | 0.30 | 91 | 89 | 0.011 | 0.004 | 0.0066 | |
| 87F-88E | 0.23 | 85 | 0.13 | 0.3 | 0.32 | 89 | 88 | 0.004 | 0.11 | 0.0061 | |
| 96A-96F | 0.2 | 87 | 0.066 | 0.32 | 0.27 | 94 | 90 | 0.21 | 0.013 | 0.0176 | |
| 96F-97D | 0.23 | 86 | 0.013 | 0.27 | 0.36 | 90 | 92 | 0.013 | 0.048 | 0.0168 | |
| 97D-97E | 0.3 | 88 | 0.024 | 0.36 | 0.32 | 92 | 90 | 0.048 | 0.02 | 0.053 | |
| 97E-98A | 0.26 | 88 | 0.013 | 0.32 | 0.34 | 90 | 94 | 0.02 | 0.018 | 0.016 | |
| 98A-99A | 0.27 | 88 | 0.0001 | 0.34 | 0.4 | 94 | 90 | 0.018 | 0 | 0.0001 | |
| 99A-99B | 0.34 | 88 | 0 | 0.40 | 0.43 | 90 | 90 | 0 | 0.0000 | 0 | |
| 99B-99E | 0.32 | 88 | 0 | 0.43 | 0.33 | 90 | 94 | 0 | 0.0000 | 0 | |
| 99E-100A | 0.33 | 93 | 0 | 0.33 | 0.34 | 94 | 95 | 0 | 0.0000 | 0 | |
The application of the intersection test to these data can be further expanded to include an analysis with all 76 markers. We conducted 76 single marker regression tests at a Bonferroni adjusted α of 6.6 × 10-4. Markers 68B, 71E, 73D, 76A, 82D, 99A, 99B, 99E and 100A were significant using this intersection test (see Figure 2). The regions identified are consistent with a regression-based interval mapping analysis.
Figure 2.

Drosophila melanogaster intersection test. T-test statistics and thresholds for evaluation of significance considering all markers typed (solid lines, T = 3.521, 76 markers; T = 2.29, paired markers). The marker on Chromosome 4 spa was not statistically significant (p = 0.32). Panel a: Chromosome 1 Panel b: Chromosome 2 Panel c: Chromosome 3
Discussion
The findings of Rebaï et al. [4] show differences in the statistical power of the two marker methods (i.e., interval mapping) over single marker tests (e.g., ANOVA, t-test) only when the markers are more than 50cM apart, suggesting that these differences may be due to the addition of information in the second marker. Our simulation study supports this hypothesis.
The application of an intersection test uses information from both markers, and tests the same null hypotheses as the two marker test. The use of the intersection test takes advantage of the additional genotypic information provided in the second marker.
While compound hypotheses are common in statistical theory, and typically seen in the use of union/intersection tests, their use in the quantitative genetics arena and QTL application is relatively novel. Furthermore, the intersection test is simple to implement, the expansion to multiple markers is straightforward, and uses all available marker information. In a framework map, where markers are unlinked, the intersection test is simply the single marker analysis with a Bonferroni correction for the significance level. In cases where markers are correlated, the application of the Bonferroni correction will be overly conservative. This correction guarantees that the nominal α is not exceeded, but is well known to be overly conservative in cases where tests are not independent. In this case, the application of the intersection test will require an alternative correction in order to achieve maximum power.
We demonstrate situations for a pair of adjacent markers where the power of the intersection test is equal to or greater than the power of the two marker test. In the case highlighted by Rebaï et al. [4], markers more than 50 cM apart and large effect size, we also find that the two marker test has higher power than the intersection test. A counter example is when one marker is much closer to the QTL than the other marker, in this case the intersection test is more powerful. Overall, the power of the two approaches is nearly identical and differences between them small.
In the Drosophila reanalysis both methods identify the same general regions. However, six marker pairs were found to be significant using the two marker tests that were not identified using the intersection test. Using the map and notation defined by Nuzhdin et al. [12] they were: Chromosome 1 pairs 3E-4F, 4F-5D, 6E-7D; Chromosome 2 pairs 38E-43A, 43A-43E, and 43E-46C. The p-values for the 6 marker pairs from the intersection tests were small but did not exceed the Bonferroni corrected significance level (see Table 4). The above markers that contribute to these marker pairs are linked indicating that the Bonferroni correction may be overly conservative.
In contrast, three marker pairs on Chromosome 3 were significant using the intersection tests, but were not significant using the two marker tests: 63A-65D, 87F-88E, and 96A-96F (Table 4). In these cases the "internal" marker of the pair is giving signal while the "outer" marker does not. This provides an interesting point of discussion. We could say that marker 65D appears to be associated with ovariole number, but we do not know if the QTL lies to the left or right of this marker. Just because marker 63A does not appear to be significantly associated with ovariole number, we can not infer that the region to the "left" of 65D does not contain the gene of interest.
In some cases, the two marker test results in a larger region than the intersection test, while in others the reverse is true. QTL mapping is usually a first attempt to locate genes, which the biologist uses to identify all possible regions of interest, e.g. is willing to accept type I error. We have discussed different ways to detect underlying QTL and an approach for maximizing or minimizing the potential region containing the QTL. It is also possible to estimate the QTL position directly. Estimates can be obtained using a variety of techniques and the different possible approaches to estimation are reviewed in Doerge et al. [5] and Kao [3]. However, even when the position is estimated, a confidence interval will exist defining the size of the region to be included for further study. Different approaches will result in regions of different sizes with more, or fewer markers included. The differences in the size of the regions are potentially important to a biologist, who relies on QTL mapping analyses to determine regions for further study. Most biologists accept that current QTL mapping methods are best used for identifying broad regions, which subsequently can be dissected with more precise genetical techniques. The question is then: what region should be advanced to fine mapping experiments? The investigator may choose to take only the regions which are significant in both intersection and multiple marker approaches, or s/he may choose to carry forward any marker that shows a positive result according to at least one analysis. We recommend that experimentalists perform both a single marker analysis with an intersection test and a multiple marker analysis and use the information available in both analyses to guide their decisions about what regions to carry forward for further study.
Conclusion
We find that the intersection test has equal or greater power compared to the two marker equivalent. Our analyses were conducted using the Bonferroni correction for the intersection test. When markers are linked, as in many of our simulations, this correction is overly conservative. If the intersection test is used in conjunction with a more appropriate correction, the performance of the intersection test would improve perhaps even surpassing the two marker equivalent in more cases. Thus, our motivation and hope in presenting this investigation of the statistical power of intersection tests versus two marker tests is to make clear the compound framework and resulting evidence under which intersection tests are indeed equal to and/or more powerful than the complicated procedures based on two marker models.
Materials
Statistical Framework
As the framework for our comparison, and in conjunction with the previous simulations and conclusions provided by the work of Rebaï et al. [4], we consider a backcross experimental design originating from a cross of two homozygous inbred lines, differing in the trait of interest, and producing heterozygous lines that are backcrossed to one of the initial homozygous parental lines. We examine both normal and binomial phenotypic distributions. In general, we denote each marker as M1...Mk, where k is the number of markers being examined and allow each marker to have two alleles, M11, M12...Mk1, Mk2. The 2k phenotypic means are differentiated via subscripts (e.g., μM11...Mk1/M11...Mk1 or μM11...Mk1/M12...Mk2) and the frequencies of these classes are denoted as p11, p21...pk1 under the binomial scenario (i.e., 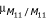 = np11).
= np11).
Single Marker Model and Hypotheses
A simple linear regression backcross model is employed for single marker QTL detection
Yj = β0 + β*X*j + εj; j = 1,...,n (1)
where Yj is the quantitative trait value, X*j is an indicator variable that denotes the state of a particular marker, β0 is the overall mean, and β* is the effect of an allelic substitution at the marker. Ideally, if the marker and QTL are completely linked, the effect of an allelic substitution is the effect of the QTL. If k markers are considered independently, k linear regression models can be considered (i.e., one for each marker, M1, M2,..., Mk) by denoting the allelic substitution associated with marker Mi as β* = βi, for i = 1... k. For k = 2 markers, we denote the allelic substitution associated with marker M1 as β* = β1, where β0 = μM11/M11 and β1 = μM11/M12 - μM11/M11; and the allelic substitution associated with marker M2 as β* = β2, where β0 = μM21/M21 and β2 = μM21/M22 - μM21/M21.
A compound hypothesis testing the effect of an allelic substitution at either or both of these two independent markers is,
![]()
Rejection of this compound null hypothesis indicates an association between a QTL and either or both of the markers, M1 and M2, hence the term intersection test. From a statistical perspective the relative position of the two markers is irrelevant. However, to compare this to a two marker model there is an implicit assumption that the markers considered form an interval, or are adjacent to one another. This marks a departure from the traditional single marker analysis where no consideration to marker order is given. To define an overall level α test, the significance level α must be adjusted for the individual tests to account for multiple testing. There are many ways to account for multiple testing. Assuming the markers are independent, the Bonferroni correction can be applied [9]. The Bonferroni correction is conservative for the intersection test and the lack of independence between markers would tend to make it more difficult for the intersection test to reject.
More generally, for k markers, the compound hypothesis testing the effect of an allelic substitution at any of the independent markers, M1...Mk is
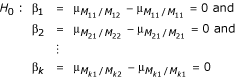
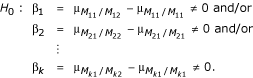
Rejection of this compound null hypothesis indicates an association between a QTL and at least one of the markers, M1...Mk. To define an overall level α test, using a Bonferroni correction [9], each β* is tested at an adjusted significance level of 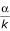 . An association between a QTL and a marker is then indicated when the individual single marker test rejects the null at the adjusted α level.
. An association between a QTL and a marker is then indicated when the individual single marker test rejects the null at the adjusted α level.
The practical result of the application of an intersection test, is the simplicity of calculation of the single marker test statistic, with a correction for multiple testing.
Two Marker Regression Model and Test of the Corresponding Interval
Extending the (backcross) notation defined previously, a multiple linear regression model (based on two markers) can be employed for QTL detection purposes. The model is defined as
Yj = β0 + β1X1j + β2X2j + β3X3j + εj; j = 1,...,n
where X1j and X2j are the genotypic states of the respective markers M1 and M2, along with their respective allelic substitution effects (β1, β2), and X3j is the combined genotypic states of markers M1 and M2 with allelic substitutions at both markers M1 and M2 having effect β3. Interestingly to note, when one is selectively genotyping, the information in β3 is maximized.
In other words,
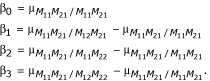
Based upon this two marker model with four parameters, the hypothesis employed to perform a level α test for association between a trait and the marker loci M1 and M2 is the test of β3 where,
![]()
The null hypothesis for this test is that there is no association between either marker (M1 or M2) and the trait. A similar set of hypotheses follow for an F2 experimental design.
This model parameterization differs from the least squares interval mapping approach first introduced by Knott and Haley [2]. In the parameterization proposed here, only one test is performed for the pair of markers. In contrast, the regression based interval mapping approach [2], recalculates the value of the independent variables for each putative position in the interval. Our two marker regression has a different parameterization from Knott and Haley [2]. We chose the alternate parameterization in order to directly compare the two marker model and the single marker model. In the Knott and Haley [2] parameterization, flanking markers are used to define the coefficients of the regression as mean, additive or dominance effects. For s steps along the interval between two markers M1 and M2 values of X are calculated according to the conditional probability of a QTL in that location.
The regression based interval mapping parameterization thus provides a mechanism to test for additive and dominance effects using tests of the regression parameters. In our parameterization, the regression coefficients are tests for detection. Thus, the two parameterizations have different null hypotheses for the tests of the regression coefficients and are not directly comparable in terms of power. We use the alternative parameterization so that the interpretation of the tests is comparable in the single marker and two marker regression models and we can directly compare the power of the two tests.
Simulations
Data were simulated for two marker backcross and F2 populations with binomial trait distributions and two marker backcross populations with normal trait distributions. A total of 339 parameter combinations were examined (Table 1). For each combination of parameters, 1000 data sets were simulated. Traits were simulated from a binomial distribution Bin(n,p) where sample sizes n = 100 and n = 500 were utilized, and from a normal distribution N(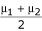 , 1.0) with n = 500. The effect of the binary trait [13] varied based on μ = npi (Table 1). The binomial probabilities p1, p2, and p3 represent the probability that a binary trait is present given a specific BTL genotype (GT), or the penetrance of the trait for the specific genotypes Q1/Q1, Q1/Q2, and Q2/Q2, respectively. The location of the locus relative to marker loci M1 and M2 also varied. Similarly, the effect under the normally distributed phenotype was allowed to vary (Table 2) under seventy five parameter combinations. The effect size is the difference in the penetrances (for binary traits) and between the means (for normally distributed traits). For each phenotypic trait distribution and each parameter combination (Table 1 and 2) we analyzed, via least squares, 1000 simulated data sets using both the single marker regression model and the two marker regression model.
, 1.0) with n = 500. The effect of the binary trait [13] varied based on μ = npi (Table 1). The binomial probabilities p1, p2, and p3 represent the probability that a binary trait is present given a specific BTL genotype (GT), or the penetrance of the trait for the specific genotypes Q1/Q1, Q1/Q2, and Q2/Q2, respectively. The location of the locus relative to marker loci M1 and M2 also varied. Similarly, the effect under the normally distributed phenotype was allowed to vary (Table 2) under seventy five parameter combinations. The effect size is the difference in the penetrances (for binary traits) and between the means (for normally distributed traits). For each phenotypic trait distribution and each parameter combination (Table 1 and 2) we analyzed, via least squares, 1000 simulated data sets using both the single marker regression model and the two marker regression model.
For the intersection test, the null hypothesis was rejected when the empirical p-value for either single marker regression test statistic was less than = 0.025 (Bonferroni adjustment). For the comparable two marker test (i.e., β3 = 0), the null hypothesis was rejected when the empirical p-value was less than α = 0.05. Under each parameter combination, the cumulative assessment of statistical power was evaluated from the 1000 simulated data sets as the proportion of times the empirical (permutation) p-values were less than the specified α level.
Drosophila Analysis
The population of Drosophila melanogaster used in our analysis was a set of 98 RILs (recombinant in lines) derived from a cross of two isogenic lines as described in Wayne et al. [8], for the trait ovariole number. There were 76 informative markers on 4 chromosomes. Markers used were the cytological map positions of the insertion sites of roo transposable element markers, with the exception of the fourth chromosome, where a visible mutation was used as a marker (spa) [12]. A complete linkage map was obtained for chromosome 1 (the X) and chromosome 3, with 15 adjacent marker pairs (16 markers) on 1 and 36 adjacent marker pairs (37 markers) on 3. There was a centromeric break in the genetic map for chromosome 2, such that there were 18 adjacent pairs (19 markers) on the left arm and 2 adjacent pairs (3 markers) on the right arm.
To compare the intersection test to the two marker test, the 71 pairs of markers identified above were examined. For each pair, the two marker regression with the test of the β3 parameter was conducted at α = 0.05. The two individual markers were then separately modeled in a linear regression model (see Equation 1), and the intersection test was conducted. For the 71 unique pairs of markers, concordance between the intersection test and two marker test was estimated using the Kappa coefficient, and McNemar's test [14] was conducted to determine whether systematic differences existed between the two methods. Regression based interval mapping was performed according to the Haley and Knott parameterization [1,2]. Analysis was conducted using S-PLUS 2000 (Insightful Corp.).
Authors' Contributions
Cynthia Coffman is the post-doctoral associate who programmed all simulations and analyzed the Drosophila data. Rebecca Doerge and Lauren McIntyre designed the simulation study and assisted with the interpretation of the results. Marta Wayne provided the Drosophila data, and assisted in the interpretation of the results. All authors contributed to the writing of this manuscript.
Acknowledgments
Acknowledgments
This work is supported by the Purdue University Experimental Research Station; a National Science Foundation Grant (DBI 98-08026/00-96044) to LMM, CJC, and RWD; a National Institute of Health grant (NIA-AG16996) to LMM; a United States Department of Agriculture Grant (98-35300-6173) to RWD; a National Institutes of Health grant (GM59884-02) to MLW, and a Veterans Affairs Health Services Research Postdoctoral Fellowship to CJC.
Contributor Information
Cynthia J Coffman, Email: cynthia.coffman@duke.edu.
RW Doerge, Email: doerge@stat.purdue.edu.
Marta L Wayne, Email: mlwayne@zoo.ufl.edu.
Lauren M McIntyre, Email: lmcintyre@purdue.edu.
References
- Haley C, Knott S. A simple regression method for mapping quantitative trait loci in line crosses using flanking markers. Heredity. 1992;69:315–324. doi: 10.1038/hdy.1992.131. [DOI] [PubMed] [Google Scholar]
- Knott S, Haley C. Aspects of maximum likelihood methods for mapping of quantitative trait loci in line crosses. Genetical Research. 1992;60:139–152. [Google Scholar]
- Kao CH. On the differences between maximum likelihood and regression interval mapping in the analysis of quantitative trait loci. Genetics. 2000;156:855–865. doi: 10.1093/genetics/156.2.855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebaï A, Goffinet B, Mangin B. Comparing powers of different methods for QTL detection. Biometrics. 1995;51:87–99. [PubMed] [Google Scholar]
- Doerge RW, Zeng Z-B, Weir BS. Statistical issues in the search for genes affecting quantitative traits in experimental populations. Statistical Science. 1997;13:195–219. [Google Scholar]
- Lander ES, Botstein D. Mapping mendelian factors underlying quantitative traits using rflp linkage maps. Genetics. 1989;121:185–199. doi: 10.1093/genetics/121.1.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Z-B. Precision mapping of quantitative trait loci. Genetics. 1994;136:1457–1468. doi: 10.1093/genetics/136.4.1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wayne ML, Hackett JB, Dilda CL, Nuzhdin SV, Pasyukova EG. Quantitative trait locus mapping of fitness-related traits in Drosophila melanogaster. Genetical Research. 2001;77:107–116. doi: 10.1017/S0016672300004894. [DOI] [PubMed] [Google Scholar]
- Steele R, Torrie J. Principles and procedures of statistics:a biometrical approach. 3. McGraw-Hill; 1997. [Google Scholar]
- Boulètreau-Merle J, Allemand JR, Cohet Y, David JR. Reproductive strategy in Drosophila melanogaster: Significance of a genetic divergence between temperate and tropical populations. Oecologia. 2001;53:323–329. doi: 10.1007/BF00389008. [DOI] [PubMed] [Google Scholar]
- Cohet Y, David J. Control of the adult reproductive potential by preimaginal thermal conditions. Oecologia. 1978;36:295–306. doi: 10.1007/BF00348055. [DOI] [PubMed] [Google Scholar]
- Nuzhdin SV, Pasyukova EG, Dilda CL, Zeng ZB, Mackay TFC. Sex-specific quantitative trait loci affecting longevity in Drosophila melanogaster. Proceedings from the National Academy of Science. 1997;94:9734–9739. doi: 10.1073/pnas.94.18.9734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntyre LM, Coffman CJ, Doerge RW. Detection and localization of a single binary trait locus in experimental populations. Genetical Research. 2001;78:79–92. doi: 10.1017/S0016672301005092. [DOI] [PubMed] [Google Scholar]
- Agresti A. Categorical Data Analysis. John Wiley and Sons, New York, NY; 1990. [Google Scholar]