

Abstract
The orientation of the substrate camphor in the active site of reduced CO-bound cytochrome P450cam (CYP101) as a function of reduced putidaredoxin (Pdxr) addition has been examined by NMR using perdeuterated CYP101 and peredeuterated Pdx as well as isotopically labeled d-camphor. This permits the 1H resonances of CYP101-bound camphor to be observed without interference from the signals of CYP101 or Pdx, and confirms assignments of the methyl signals of camphor in the bound form. The Cys4Fe2S2 ferredoxin Pdx is the physiological redox partner and effector of CYP101. The addition of Pdx to the reduced CYP101-camphor-CO complex results in a conformational selection that is slow on the chemical shift time scale with spectral effects observed primarily at the 8-CH3 group of the camphor. The camphor signals are ring-current shifted by the heme, and for the 9- and 10-CH3 resonances, these shifts are reasonably well predicted by ring current calculations from the crystal structure of CO-bound CYP101. However, in the absence of Pdx the 8-CH3 resonance of CYP101-bound camphor is observed at considerably higher field than predicted. Dynamic simulations using ring current shift restraints generated a structure with low chemical shift violations in which the hydrogen bond between the camphor carbonyl oxygen and the OH of Tyr 96 is lost, and an expansion of the active site takes place that permits reorientation of the camphor within the active site.
Cytochrome P450cam (CYP101) catalyzes the 5-exo-hydroxylation of camphor 1, the first step of catabolism of 1 by the soil bacterium Pseudomonas putida.1 Two electrons are required for turnover, and the second reduction, of the Fe(II) P450cam-O2-1 ternary complex (CYP-1-O2), is the rate limiting step under physiological conditions.2 The presence of an effector is required for the formation of hydroxycamphor product.3 The Fe2S2 ferredoxin putidaredoxin (Pdx) is the biological effector and reductant of CYP101.3 Structural perturbations have been observed spectroscopically in the CYP101 active site upon binding of Pdx, and it has been proposed that these perturbations are involved in the effector and/or electron transfer activity of Pdx.4–9
The complex between reduced Pdx (Pdxr) and reduced 1- and CO-bound P450cam (CYP-1-CO) is often used a model for the catalytically competent Pdxr-CYP-1 -O2 complex, and is convenient for NMR spectroscopy in that the heme is diamagnetic. Recently, we reported perturbations in the conformational equilibrium of CYP-1-CO as a function of addition of Pdxr.7 These perturbations were monitored by 1H,15N HSQC experiments using sequence-specific resonance assignments in CYP101. In addition to changes that take place in the proposed interface between the two proteins, we identified perturbations in regions of the P450cam molecule that have been shown to be involved in substrate access and orientation, including the B’, F and G helices and portions of the β3 sheet. Based on this, we proposed a dynamic model for the effector activity of Pdx, in which binding of Pdx forces selection of the active conformation of CYP101 from a manifold of conformations and we suggested that the conformational changes observed in the substrate access region are critical to this selection. The conformation thus selected prevents loss of substrate and/or intermediates, and enforces the correct orientation of the substrate with respect to the activated oxygen. In other work, Tosha et al. reported perturbations in the 1HNMR spectrum of CYP-1-CO as a function of Pdxr addition that indicate changes in the orientations of 1, the active site Thr 252 and the distal Fe ligand cysteine thiolate CH2 group in the presence of Pdxr.6 Here we present evidence for a high-barrier conformational shift in CYP-1-CO upon binding of Pdxr that results in changes in the environment of camphor in the active site.
Highly perdeuterated C334A CYP101 was expressed, purified, reduced and prepared for NMR spectroscopy in D2O buffer as described previously.7 The C334A mutant of CYP101 has been shown to be spectroscopically and enzymatically identical to wild-type enzyme, but does not form dimers in solution.10 Perdeuterated Pdx was expressed in a manner similar to CYP101, reconstituted and purified according to the method of Jain,11 and then reduced with sodium dithionite for titration as described previously.7 8- d1-1 was synthesized by the method of Dadson et al.12, 13 NMR spectroscopy was performed on a Varian INOVA 600 MHz NMR spectrometer. All 1H and 13C chemical shifts are reported in ppm relative to TSP (trimethylsilylpropionic acid sodium salt).
Three methyl signals are observed in the upfield region of the 1H spectrum of perdeuterated CYP-1-CO that were assigned from 1H, 13C HSQC data to the 10-CH3, 8-CH3 and 9-CH3 resonances of CYP101-bound 1. The assignment of the 10-CH3 is based on the distinctive upfield 13C shift of that resonance.14 The assignment of the 8-CH3 was confirmed by the attenuation and isotope shift of that signal when 8-d1-1 is used to prepare the CYP101 sample (Figure 1). The 9-CH3 1H assignment is by elimination and agrees with that of Tosha et al.6 Using the published structure of CO-bound CYP101 (PDB entry 3CPP),15 ring current shifts expected to bound 1 due to the heme porphyrin ring and nearby aromatic residues were calculated using SHIFTS (D. Case, Scripps Res. Inst., La Jolla).16 The agreement between observed and calculated 1H shifts is within error limits (Δδ = +/−0.3) for the 10-CH3 (ΔδH = −0.20) and the 9-CH3 (ΔδH = −0.16). However, the 1H signal of the 8-CH3 group of 1 is shifted well upfield of predicted values (ΔδH = −0.80), indicating that this methyl group is experiencing considerably more ring current shift than is predicted from the 3CPP structure (Fig. 1, bottom and Table I.).
Figure 1.
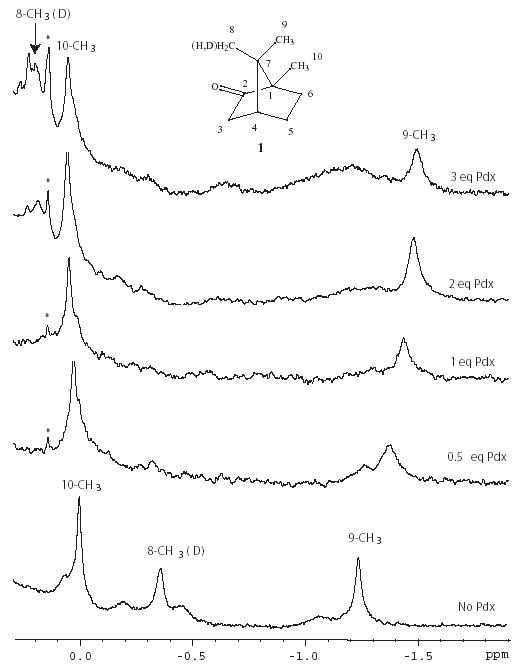
Series of 1H NMR spectra showing titration of perdeuterated CYP101-1-CO at 14 T (600 MHz 1H) with perdeuterated Pdxr. Starting concentration of CYP101 is 0.3 mM. Both proteins were dissolved in 2 mM 8-d1-camphor 1, 100% D2O, pH 7.4 phosphate buffer (uncorrected for isotope effects), 298 K, with perdeuterated Pdxr. The signals observed are primarily those of CYP-bound 8-d1-1. Note that the integration of the 8-CH3 signal is less than that of the other two methyl groups due to the presence of a single deuterium at that position, and the peak is also shifted slightly upfield from the position observed with all protio-1 (data not shown) due to an isotope shift. Peak marked with a * is an exchangable proton observed in CYP101 when H2O is present. The inset shows the structure of camphor 1 with carbon atom numbering.
Table 1.
1H chemical shifts of CYP-1-CO bound 1 methyls as a function of Pdxr binding (298 K). Calculated shifts are obtained using the SHIFTS program14 with PDB entry 3CPP. The difference between observed and calculated shifts (δcalc−δobs) are shown in parentheses next to δobs.
| methyl | 13C δobs | 1Hδcalc | 1Hδobs, no Pdx | 1Hδobs + 3 eq. Pdxr |
|---|---|---|---|---|
| 8-CH3 | 21.1 | 0.51 | −0.33 (−0.84) | 0.20 (−0.31) |
| 9-CH3 | 18.8 | −1.38 | −1.22 (+0.16) | −1.48 (−0.08) |
| 10-CH3 | 10.4 | 0.22 | 0.02 (−0.20) | 0.06 (−0.04) |
Using the experimental ring current shifts for the methyl groups of 1 in the absence of Pdx as an energetic constraint, we performed a 170 ps vacuum molecular dynamics simulation at 300K in order to sample conformations of the CYP101 active site and orientations of 1 that could give rise to the chemical shifts observed in the absence of Pdx. The 3CPP crystal structure with hydrogens added but without external waters was used as a starting point for the simulation. Initially, the structure relaxed to a manifold of states (average RMS deviation = 2 Å) where it remained until 80 ps had elapsed. In this manifold, the ring current shifts constraints showed an average total violation for the three methyls of 1 |Δδ| = 1.41. After 80 ps, a transition takes place over 30 ps into a manifold of states that persist until the end of the simulation. This manifold has a higher overall RMSD (average RMSD = 2.5 Å) than earlier in the simulation and gives a lower average ring current shift violation (|Δδ| =1.02). The transition between 80 and 110 ps is correlated with (and apparently driven by) the lowering of the total ring current shift violation. The structure with the lowest total ring current shift violations (at 151 ps, |Δδ| = 0.56) was selected for examination. The most obvious difference between that structure and 3CPP is a ~20° rotation of 1 around a vector between atoms C1 and C5 (Fig. 2). The rotation does not displace the 10-CH3 significantly relative to the heme, but moves the 8-CH3 into a position more directly over the heme ring. The 9-CH3 remains at about the same distance from the heme as in 3CPP, but is slightly closer to the heme normal. The rotation increases the distance between the carbonyl oxygen of 1 and the hydroxyl group of Tyr 96 out of hydrogen bonding range. There are fewer van der Waals contacts between distal residues and the 8-CH3 in the ring-current driven structure, and the side chains of Phe 87 and Asp 297 are both visibly displaced to accommodate the new orientation of 1. These changes represent an expansion of the distal binding pocket in order to accommodate the ring-current shift driven orientation (Figure 2).
Figure 2.
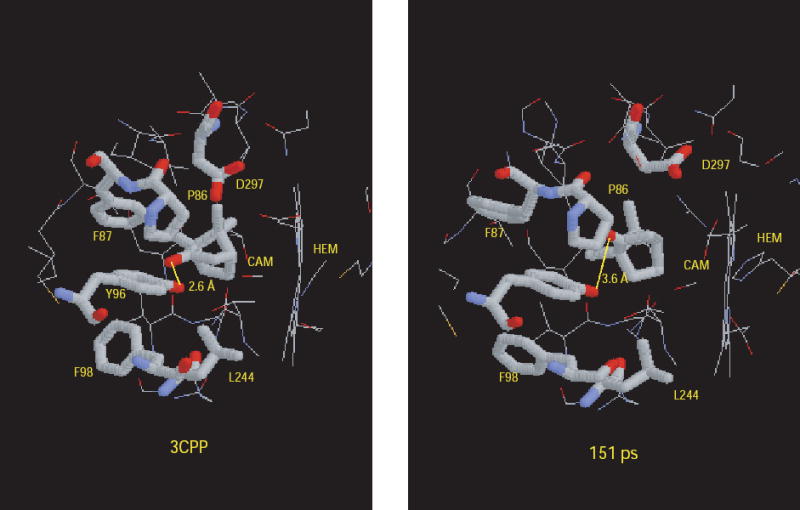
Comparison of CYP101 active site from PDB structure 3CPP (ref. 15), left, and structure identified after 151 ps of restrained molecular dynamics simulation that minimizes ring current shift violations (right). See text for details.
Titration of samples of perdeuterated CYP-S-CO in D2O with perdeuterated Pdxr allowed us to observe perturbations to camphor 1H signals resulting from the formation of the Pdxr-CYP-S-CO complex without interference from protein 1H signals. Titration curves derived from chemical shift perturbations were consistent with the Kd for the Pdxr-CYP-S-CO complex measured previously by our group7 and Tosha et al.6 indicating that the same event is being monitored. Of the three camphor methyl signals, the most dramatic response to the presence of Pdxr is observed at the 8-CH3 resonance, which undergoes an exchange phenomenon that is slow on the chemical shift time scale at 298 K and 14 T magnetic field. The 8-CH3 resonance does not shift, but broadens drastically, and is essentially undetectable after 0.5 eq. of Pdxr has been added (Figure 1). At saturating Pdx concentrations, a new resonance at δ = 0.20 ppm appears that gives rise to an NOE cross peak to the 9-CH3 resonance (Figure 3). The identity of this resonance as 8-CH3 was confirmed using 8-d1-camphor. From the difference in chemical shift between the two resonances assigned to the 8-CH3, the exchange rate must be no greater than 300 s−1 at 298 K. The shift change of the 9-CH3 signal (which is at fast exchange) gives a lower limit of 150 s−1.
Figure 3.
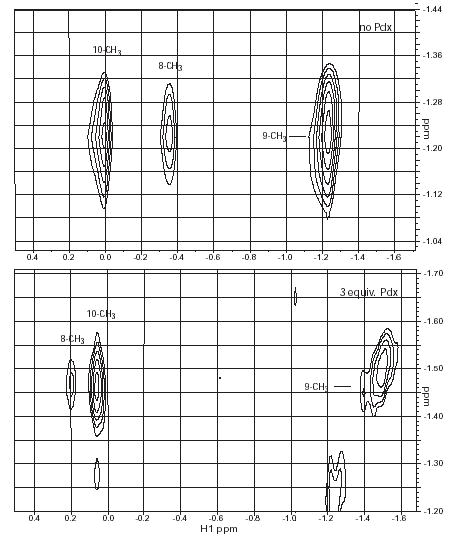
Region of 600 MHz 1H NOESY spectra (tmix=500 ms) showing cross peaks between 9-CH3 of 1 (vertical axes) and the 8- and 10-CH3 groups of 1 bound to perdeuterated CYP101-CO. Top spectrum, no Pdxr, bottom spectrum, 3 eq. of perdeuterated Pdxr. Sample conditions are the same as in Fig. 1. The signal intensity at the 8-CH3 is attenuated by deuteration.
For the 10-CH3 1H resonance, only a small difference is seen between the chemical shift in the absence of Pdx and when Pdx is saturating (Δδmax = +0.05). The line width of the 10-CH3 remains essentially constant throughout the titration. As reported by Tosha et al.,6 the 9-CH3 1H resonance showed a somewhat larger chemical shift change (Δδmax = −0.24) from 0 to 3 eq. of Pdx with noticeable broadening near the midpoint of the titration. It is interesting that the chemical shifts observed for the methyl groups of 1 at saturating Pdx concentrations are in better agreement with those predicted from the 3CPP structure than in the absence of Pdx (Table I). This suggests to us that the crystallographic structure may better represent the active site conformation selected upon Pdx binding (that is, the enzymatically competent conformation) than the manifold of conformations present in solution in the absence of Pdx. This would also explain why X-ray generated photoelectrons are capable of supporting hydroxylation of 1 in a CYP101 crystal in the absence of effector.17
The observation of a process at slow exchange on the chemical shift time scale in protein structure is often associated with cis-trans isomerization of a X-Pro peptide linkage.18 We note that Pro 86 is in close proximity to bound 1 in the active site, and is structurally and spatially adjacent to many of the residues that we observed previously to be affected by the addition of Pdx, including residues in the B' helix and the β3 sheet. Although the Cys 85-Pro 86 peptide bond is in the all-trans conformation in 3CPP, the possibility that it or other proline residues might be capable of cis-trans isomerization in solution should not be discounted. We are currently testing this hypothesis experimentally.
Supplementary Material
Acknowledgments
This work was supported in part by grants from the U.S. National Institutes of Health R01-GM44191 and R01-GM067786 (T.C.P.)
Footnotes
Supporting Information Available:. Input files for AMBER, selected dynamics traces, and supporting NMR information are available as Supplementary Material.
References
- 1.Mueller EJ, Loida PJ, Sligar SG. In: In Cytochrome P450: Structure, Function and Biochemistry. 2nd ed. Ortiz de Montellano P, editor. Plenum Press; New York: 1995. pp. 83–124. [Google Scholar]
- 2.Brewer CB, Peterson JA. J Biol Chem. 1988;263:791–798. [PubMed] [Google Scholar]
- 3.Lipscomb JD, Sligar SG, Namtvedt MJ, Gunsalus IC. J Biol Chem. 1976;251:1116–1124. [PubMed] [Google Scholar]
- 4.Unno M, Christian JF, Sjodin T, Benson DE, Macdonald IDG, Sligar SG, Champion PM. J Biol Chem. 2002;277:2547–2553. doi: 10.1074/jbc.M108917200. [DOI] [PubMed] [Google Scholar]
- 5.Sjodin T, Christian JF, Macdonald IDG, Davydov R, Unno M, Sligar SC, Hoffman BM, Champion PM. Biochemistry. 2001;40:6852–6859. doi: 10.1021/bi002510b. [DOI] [PubMed] [Google Scholar]
- 6.Tosha T, Yoshioka S, Takahashi S, Ishimori K, Shimada H, Morishima I. J Biol Chem. 2003;278:39809–39821. doi: 10.1074/jbc.M304265200. [DOI] [PubMed] [Google Scholar]
- 7.Pochapsky SS, Pochapsky TC, Wei JW. Biochemistry. 2003;42:5649–5656. doi: 10.1021/bi034263s. [DOI] [PubMed] [Google Scholar]
- 8.Nagano S, Tosha T, Ishimori K, Morishima I, Poulos TL. J Biol Chem. 2004;279:42844–42849. doi: 10.1074/jbc.M404217200. [DOI] [PubMed] [Google Scholar]
- 9.Tosha T, Yoshioka S, Ishimori K, Morishima I. J Biol Chem. 2004;279:42836–42843. doi: 10.1074/jbc.M404216200. [DOI] [PubMed] [Google Scholar]
- 10.Nickerson DP, Wong LL. Prot Eng. 1997;10:1357–1361. doi: 10.1093/protein/10.12.1357. [DOI] [PubMed] [Google Scholar]
- 11.Jain NU, Pochapsky TC. Biochem Biophys Res Commun. 1999;258:54–59. doi: 10.1006/bbrc.1999.0583. [DOI] [PubMed] [Google Scholar]
- 12.Dadson WM, Money T. J Chem Soc Chem Commun. 1982:112–113. [Google Scholar]
- 13.Dadson WM, Hutchinson JH, Money T. Can J Chem. 1990;68:1821–1828. [Google Scholar]
- 14.Crull GB, Garber AR, Kennington JW, Prosser CM, Stone PW, Fant JW, Dawson JH. Magn Reson Chem. 1986;24:737–739. [Google Scholar]
- 15.Raag R, Poulos TL. Biochemistry. 1989;28:7586–7592. doi: 10.1021/bi00445a013. [DOI] [PubMed] [Google Scholar]
- 16.Case DA. J Biomol NMR. 1995;6:341–346. doi: 10.1007/BF00197633. [DOI] [PubMed] [Google Scholar]
- 17.Schlichting I, Berendzen J, Chu K, Stock AM, Maves SA, Benson DE, Sweet BM, Ringe D, Petsko GA, Sligar SG. Science. 2000;287:1615–1622. doi: 10.1126/science.287.5458.1615. [DOI] [PubMed] [Google Scholar]
- 18.Bosco DA, Eisenmesser EZ, Pochapsky S, Sundquist WI, Kern D. Proc Natl Acad USA. 2002;99:5247–5252. doi: 10.1073/pnas.082100499. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.