

Abstract
The Self-Nonself discrimination is germline encoded for defense mechanisms (“innate immune systems”) whereas it is somatically learned for immune systems (“adaptive immune systems”). It is proposed that immune systems evolved from defense mechanisms by adding large recognitive repertoires that, by aggregating with antigens, were able to trigger the already existent effector functions of defense mechanisms. Thus today there are two pathways to triggering each class of effector function (MΦ opsonization, C′ lysis or NK/NC activity). The (Ag-Ab)n complex and the T-cell antigen-receptor interaction trigger the immune pathway; the receptors of the defense mechanism trigger the “alternate” or “innate” pathway.
The evolutionary selection pressure on defense mechanisms was to increase the size of the recognitive repertoire, which in turn, necessitated the emergence of a somatically learned S-NS discrimination. By contrast with defense mechanisms that are triggered eTh-independently by polymers (Signal[3]), immune systems can be activated by monomers, a pathway that requires associative recognition of monomer and the reading of two Signals, Signal[1] resulting from the binding of an epitope to the antigen-receptor complex, Signal[2] delivered by an effector T-helper (eTh).
The evolving immune system hijacked part of this eTh-independent pathway (Signal[3]) into which Signal([1]+[2]) merged. A model of these relationships is proposed.
INTRODUCTION
Over the years Göran Möller and Antonio Coutinho have differed sharply with us concerning the role and interpretation of so-called “thymus independence” of responses to given antigens. We have tended to trivialize the phenomenon by extrapolating from “thymus dependence” whereas they have tended to canonize it by extrapolating from “thymus independence.” However, both phenomena exist and the relationship between them must be faced. This essay is an attempt on our part to bring the general phenomenon of effector T-helper-independent responsiveness into a biological context, maybe even to convince Möller and Coutinho to meet us halfway.
The subject of eTh-independent induction of B-cells has been reviewed recently from the point of view of mechanism (1,2), but to date, the biology of the phenomenon has been ignored.
BACKGROUND
Protective mechanisms fall into two categories
All organisms have protective mechanisms against parasitism. For example, bacteria have restriction enzymes that cleave the DNA of an invading virus but do not cleave the host DNA. Insects produce antibiotic peptides that lyse invading bacteria and fungi but do not lyse host cells. Plants have lectins that agglutinate bacteria to facilitate their ridding but are not destructive and ridding for host cells. All animals possess phagocytes that engulf and digest particles (bacteria, debris), detected by particle sensors or carbohydrate receptors. We will refer to these examples as defense mechanisms.
Defense mechanisms are found in both invertebrates and vertebrates. Invertebrates do not have immune systems; only vertebrates have immune systems.
As protective mechanisms of the kind referred to here are destructive and ridding, they pose a problem for evolutionary selection.
What does it take to destroy and rid a pathogen without debilitating the host?
Historically this problem has been referred to as horror autotoxicus or more recently as the Self-Nonself discrimination, a description that has allowed all kinds of semantic confusionism to cloud discussion of the question. The Self-Nonself (S-NS) discrimination is not a semantic problem; it is a requirement of the proper functioning of a mechanism that is destructive and ridding of invaders. If the destructive and ridding mechanism operated on Self, as well as Nonself, the individual would be eliminated.
Why make a distinction between defense mechanisms and immune systems?
The S-NS discrimination is germline encoded in the case of defense mechanisms, whereas it is somatically learned in the case of immune systems. The need to make a S-NS discrimination operates as an evolutionary selection pressure on defense mechanisms by ridding individuals, whereas it operates on immune systems by ridding cells.
As a digression in nomenclature, immunologists use the terms innate immune system and adaptive immune system. We would have no objection to using those terms, innate versus adaptive, if they were defined as we have above, namely innate means a germline encoded S-NS discrimination, whereas adaptive means a somatically learned S-NS discrimination. As this is not agreed upon, we will remain with our terminology.
In summary then,

THE BIOLOGY OF eTh-INDEPENDENCE
What was it that made defense mechanisms inadequate and set up the selection pressure for an immune system?
When animals were short-lived and occupied defined niches in the ecosystem, a near steady state existed between a small defined pathogenic load and the host. The defense mechanism operated to keep the proportion of hosts sacrificed by the pathogenic load sufficiently low so as not to be the limiting factor to the procreation of the species. As animals became long-lived and wandered over great distances, they were subjected to large, diverse parasitic loads for which defense mechanisms were inadequate because their recognitive repertoires were too small. However, increasing the repertoire required the appearance of a learned S-NS discrimination. Why?
As the size of the repertoire increases, the chance that it will contain at least one anti-Self specificity increases until it is incompatible with a further increase in the size of the repertoire. A germline encoded mechanism for the S-NS discrimination that relies on eliminating individuals puts a cap on repertoire size when it is still too small to effectively protect a long-lived peripatetic population.
What is the present day relationship between defense mechanisms and immune systems in vertebrates?
Evolutionary past determines the present with the result that many of the properties of defense mechanisms are used by immune systems. Once a crucial decision is made early in evolution the subsequent steps are limited. For example, once the primordial horse took the evolutionary pathway to escape predators by running, the physical changes selected upon in order to do this, made it impossible to change the pathway in mid-evolution to escaping predators by flying. This is what gives evolution the illusion of having a goal, the view of teleology. Evolution is a historical process; choices based on past experience limit and shape the available choices in the present.
As a somewhat idealized picture, immune systems superimposed large recognitive repertoires on the already existent effector mechanisms of defense systems. This resulted in two pathways into the triggering of effector function. A general schema would be:

In essence the effector mechanisms used by Defense Systems became responsive to the antigen-antibody complexes, (Ag-Ab)n, of immune systems by adding Fc-receptors (FcR) as sensing devices. Also cell-mediated MHC-restricted cytotoxicity was added to cope with intracellular pathogens (viruses). This permitted two pathways to the panoply of effector functions, one via defense mechanisms, the other via immune systems. As a byproduct of the immune system pathway, the effector functions could be triggered at much lower “concentrations” of pathogen, thus increasing enormously the effectiveness of the protection.
The somatically learned S-NS discrimination: an eTh-dependent decision
When the S-NS discrimination is germline encoded, as is the case for defense mechanisms, the cells are born as effectors (e-cells).

Upon interaction with their targets they are triggered to deliver an effector function.
There would be no way to make a S-NS discrimination that is somatically learned if cells were born as effectors. An intermediate stage (i-cell) is required. So immune systems add an extra step.

The i-state, usually referred to as the “antigen-responsive state”, is the stage of differentiation at which the S-NS discrimination is made. The i-cell differs from the e-cell in two ways upon interaction with antigen.
The i-cells have no effector function. They receive signals; they do not send them. They listen; they do not talk. The e-cells carry out effector functions. They send signals; they do not receive them. They talk; they do not listen.
It is only when in the i-state that a learned S-NS discrimination is made. The decision step resulting in a S-NS discrimination, is a property of i-cells only. Once the cell is activated and regulated by decisions optimizing the class of effector function, a S-NS discrimination is no longer possible; anti-Self and anti-Nonself cells are driven equally to effectors.
Two pathways, inactivation or activation, requires two signals, the origin of the two signal model of the S-NS discrimination.
A summary of the pathway of decision leading to effectors is schematized below:

The anticipatory or a-cell mandates that Signal[1] be common to both pathways. The requirement for Signal[1] by both pathways assures that no i-cell can be activated that, in principle, could not have been inactivated. Clearly, a mutation that knocked out Signal[1] would be lethal due to induction of anti-S, if Signal[2] alone were activating. Only a-cells can read Signal[2]. Signal[2] is meaningless to a cell that is not receiving Signal[1]. In essence, Signals([1]+[2]) = Signal[3], the activating signal. Once the a-cell is activated, it becomes responsive to interleukins (IL) and is put under regulation referred to as the determination of effector class. This is outside of our discussion here.
Several points require comment:
-
How is the S-NS learned?
There is a period during embryogenesis when no eTh are present. All cells are born as i-cells and upon interaction with antigen, are inactivated; that is, they receive Signal[1] in the absence of Signal[2]. Those interacting antigens that are present during this period and that persist throughout life, are defined by the immune system as Self. Self is persistent. After a given time period, often after birth, eTh anti-nonself appear by the antigen-independent pathway and the immune system becomes responsive to nonself, which is transient because an effective effector response rids it. Nonself is transient.
-
Where does the first eTh come from?
It is the presence or absence of Signal[2] that determines which pathway is taken by the i-cell. Signal[2] is delivered by effector T-helpers (eTh). Given that they are born like all other cells, as iTh, we are faced with the question, “Where does the first eTh come from?” This question arises as a consequence of the model and it has taken many years of dead-end suggestions to produce a reasonable solution. The problem referred to as the primer question was first posed in 1968 but it took until 1978 to understand that helpers must be special in that they undergo a unique pathway, i.e., that is antigen-independent. Essentially the iTh anti-self are filtered out and the remainder iTh anti-nonself differentiate Ag-independently to eTh. This is a slow process designed only to provide primer eTh that initiates responsiveness. It is the iTh-APC-eTh interaction that is the rapid process necessary for an effective rate of induction. This assumption is a requirement of the two signal model that has not been analyzed experimentally as yet. Parenthetically, we might add that all models of the mechanism of the S-NS discrimination must answer to the primer problem.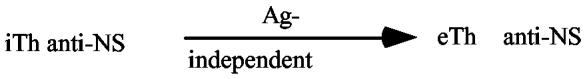
-
eTh-iB interactions cannot be required for non-protein antigens
Given that eTh can only recognize peptide (P)-restricting element (R) complexes specific activation of iB by non-protein antigens must be eTh-independent. The most important naturally occurring non-protein antigens are carbohydrates (CHO). Thus the immune system appears to have borrowed part of the already functional pathways used by defense mechanisms to cope with antigens categorized as eTh-independent.
-
Separating activation from induction of effector function
As the S-NS discrimination is germline encoded in the case of eTh-independent antigens, we need only deal with the activation mechanism. Once activated, the cell becomes responsive to interleukins (IL) that regulate the second set of decisions, the determination of the effector class optimal to destroy and rid the pathogen. This second decision process is the same for all antigens whether eTh-dependent or eTh-independent. Once the i-cell is activated, there is no way for that cell to know if it had been activated by an eTh-dependent or -independent mechanism.
The comprehensive recent reviews by Mond et al. (1,2) show that experimental work has dealt almost exclusively with the steps following activation of the iB-cell. They point out that the eTh-independent antigens mediate two events, activation of the iB-cell and triggering of effector mechanisms (MΦ and NK) to secrete cytokines that drive the activated B-cell to become an Ig-secreting eB-cell (Decision 2). This predicts two categories of eTh-independent antigens, those that can activate i-cells but cannot induce effector function (e.g., Ig secretion) because they cannot, in addition, stimulate cytokine production and those that can induce effector function because they act at both levels. Here we are considering only the first step, the activation of i-cells.
THE LEGACY OF eTh-INDEPENDENCE
Let us carry out a Gedanken evolutionary experiment. Consider a primordial animal with a defense mechanism that was eventually to evolve into an immune system. We recall that defense mechanisms are triggered eTh-independent. The interaction of pathogens with receptors on the effectors of that animal triggered a response. This was an ON signal that we will refer to as Signal[3]. The most likely target of that defense mechanism was a polymer (e.g. a CHO), which possessed a large number of repeating determinants or epitopes appropriately spaced so that the antigen-receptors of the defense mechanism read them as a polymer. Here the language and thinking of Dintzis, et al. (3) are useful. The polymer must be capable of forming an Immunon, which is a minimum triggering unit referred to here as Signal[3]. A sharp threshold number of antigen-receptors must be aggregated per unit of surface area to become signaling. In this primordial animal, Signal[3] triggered e-cells to express their effector function. In this case, the S-NS discrimination had to be germline encoded. The evolution to a somatically learned S-NS discrimination dependent on Signal[2] delivered by eTh had to build upon the already existent Signal[3] as an activating mechanism. The primordial “i-cells” of the emerging immune system probably retained much of the Signal[3] pathway when they added antigen-receptors of immune systems.
However, two factors leading to the same problem come into play. First, the pathogenic universe is under strong selective pressure to space its repeat determinants such that effectively they would be viewed as monomers by the primordial i-cell. This cell would be unable to respond to monomers (i.e., Signal][3] would be inoperative) unless it evolved a Signal[1] pathway. Second, the pathogenicity of many organisms depends on monomeric toxins and enzymes, an effective ridding response to which is critical for protection. The limit case operating as an evolutionary selection pressure is to achieve an effective effector response to nonself monomers.
When the immune system responds in an eTh-dependent way, it is treating the antigen as a monomer, independent of how the experimenter defines it. Signal[1] then must be initiated by a conformational change, as monomers cannot cross-link the antigen-receptors on a monospecific i-cell. This conformational change could either be read directly or result in self-complementation of the antigen-receptor complex to initiate Signal[1] that is read as apoptosis. The role of the eTh delivering Signal[2] is to divert Signal[1] from being read as apoptosis to its being read as activation; that is, Signal([1]+[2]) is postulated to join the pathway initiated by Signal[3]. This is diagrammed as follows:

The antigens that induce iB-cells eTh-independently, are of diverse chemistry: synthetic polymers (PVP, polyacrylamide), D-amino acid polymers, certain carbohydrates (α(1,3) dextran, galactans, levans, etc.) and certain viruses (vaccinia, VSV). These substances can be envisaged to have two properties, a minimum size repeat number of a given determinant and an appropriate spacing between the determinants as defined by the Immunon (3). Interactions with such antigens result in Signal[3]. This leads to the eTh-independence of activation. Most antigens do not possess these properties and interact, in essence, as monomers and deliver Signal[1] (tolerance). These antigens are eTh-dependent for activation. This interpretation has two consequences:
The S-NS discrimination for eTh-independent antigens must be germline encoded as it is for defense mechanisms. No Self antigen can behave eTh-independently as that would be lethal.
As the spacing between determinants on an eTh-independent antigen is increased, it should become tolerigenic. This antigen should become inductive if help is provided. Tolerance should not be found in the case of eTh-independent antigens as they deliver a single ON signal only (Signal[3]). Rather apparent unresponsiveness to them must be due to suppression or treadmilling.
A model comparing activation by eTh-dependent and eTh-independent antigens
The activation step in response to eTh-independent antigens has been described for the past 30 years as requiring cross-linking of the antigen-receptor complex in a multivalent manner (Signal[3]). This mechanism must be distinct from Signal[1] the inactivation mechanism initiated by eTh-dependent antigens. However, Signal ([1]+[2]), the activating mechanism for eTh-dependent antigens, must join the pathway initiated by Signal[3].
One possible scenario might be:
1) The antigen-receptor complex (BCR-Igα2β2 or TCR-αβ-CD3) exists in two conformations, unbound (U) and bound (B). In the presence of ligand (L) there is an allosteric conformational change eventually read as Signal[1].
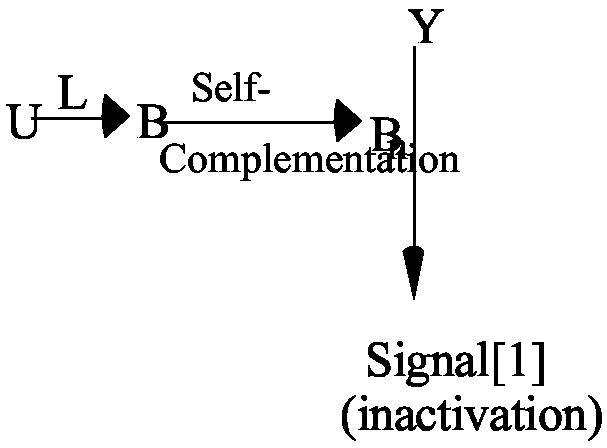
The B conformation itself can be assumed to initiate Signal[1] or it can be self-complementing to Bn, a polymer that catalyzes Y to deliver Signal[1] for inactivation. If n=1, the B conformation itself must be read directly as being at the origin of Signal[1]. If n >1, the dimer or linear polymer resulting from self-complementation, initiates Signal[1].
The activation step driven by eTh might be pictured as follows:

Bn might be supra-polymerized to (Bn)m to form a two dimensional patch, possibly by addition of another protein, which catalyzes X to initiate activation. Multivalent cross-linking eTh-independent Ags that deliver Signal[3] aggregate U to (Bn)m.
This model predicts that iB-cells are not blind to monomers as presently believed (see ref. 4 for further discussion). Rather, the entire activation machinery requiring eTh was evolutionarily selected to respond to monomers. The postulate that the B conform is self-complementing is not driven by a necessity of theory but by the claims that aggregation of the antigen-receptor complex by anti-Ig triggers apoptosis. If this is correct, self-complementation of B to Bn would be indicated for the Ag-driven Signal[1]. Under this model, the effects of anti-Ig would be expected to be weak compared to antigen and a property of unique monoclonals that favor binding to the B conformation.
As an aside, the demonstration of the existence of a U to B conformational change on binding L would open up a totally new area of research into immunosuppressants that act to drive U→B Ag-independently or greatly increase the sensitivity to Ag-specific Signal[1] driven deletion.
Predictions of the model
Consider a set of antigens with increasing spacing between a given unique epitope. This set of antigens would be expected to behave as illustrated below:
| Antigen | Property | Interpretation | Comment |
|---|---|---|---|
| 1-1-1-1-1-1-1 | eTh-independent immunogen, non-tolerigenic | Signal[3] | S-NS is germline encoded |
| 1-0-1-0-1-0-1 | Tolerigenic and eTh-dependent immunogen (behaves as a monomer) | Signal[1] +Signal[2] | S-NS is somatically learned. |
| 1-0-0-1-0-0-1-0-0 | Blocking, not tolerogenic or immunogenic | No signal | If self-complementation of antigen-receptor obtains, it is inhibited. (see text) |
1 = repeat epitope
0 = spacing between repeat epitopes
The antigen, 1-1-1-1-1-1-1, is eTh-independent because it forms (Bn)m, the initiator of Signal[3], on interaction with the antigen-receptor complex, U. The antigen, 1-0-1-0-1-0-1, behaves as a monomer, inducing U→Bn. Here, the value of n modifies the interpretation. If n=1 (i.e. no self complemention), then the conformational change must be read directly as Signal[1]. If n is 2 or more, then self-complementation, Bn, is the initiator of Signal[1]. The spacing between the repeat epitope, 1-0-1, must permit self-complementation, a stringent requirement of this assumption. If self-complementation obtains (n>1), then increased spacing of epitopes, 1-0-0-1-0-0-1, would lead to an antagonist of both activation and inactivation; the presence or absence of eTh would play no role if Signal[1] were blocked. If n=1 and the U→B conformational change is read directly, then the spacing between epitopes that resulted in loss of eTh-independent activation would engender inactivation obligatorily.
The spacing between epitopes on a linear polymer is critical in determining whether the monoclonal antigen-receptor will bind to it intramolecularly (“bivalent” binding) or intermolecularly (cross linking). “Bivalent” binding leads to a linear aggregate of Ig. Most of the eTh-independent polysaccharides probably have epitopes spaced such that there is some ratio of intra- to inter-molecular binding thereby allowing a two dimensional patch to be formed on the surface. This could well be essential to delivering Signal[3].
A monomeric site-filling ligand should be tolerigenic (i.e., initiate delivery of Signal[1]). The data are lacking on this question; the generally accepted assumption that i-cells are blind to monomers is still based on absence of evidence.
What about polyclonal activators?
We have not discussed polyclonal activators such as LPS. These are usually referred to as TI-1 antigens, a misnomer in that they are rarely studied as antigens. These substances appear to be Signal[2] substitutes, requiring Signal[1] from usually cryptic or sometimes revealed sources of antigen. Signal[3] is not one ‘nonspecific’ Signal[2], but rather is an activating interaction with the antigen-receptor complex of the i-cell. The eTh-independent antigens that we have been discussing are not polyclonal activators.
Some unexplained phenomena
The early literature contains studies showing that pneumococcal polysaccharide is immunogenic for mouse and human but not for rabbit and horse. For these latter species, the polysaccharide must be coupled to a carrier to become immunogenic. This would be interpreted today to indicate that for mouse and human the polysaccharide is an eTh-independent antigen whereas for rabbit and horse, it is an eTh-dependent antigen.
An analogous situation can be generated either by mutation or by differentiation. The Xid mutant has a defect in response to eTh-independent antigens. Similarly, the two lineages B1 and B2 appear to respond differentially to eTh-independent antigens.
While these phenomena are not all or none; they can be categorized as due to degrees of inability to read Signal[3] as a consequence of either a species difference, a mutational defect or a stage in the ontogeny of different lineages of B-cell.
Activation of iT versus iB
Thus far the discussion has concentrated almost entirely on the humoral immune system, in particular, the activation of iB-cells. The cell-mediated system responds only to peptide (P)-MHC restricting element (R) complexes. The iT-cell is blind to the original protein antigen. What property then would lead to eTh-independent activation of iT-cells? The hint comes from a special case; the response to allo-MHC (i.e., allo-R) may be eTh-independent. An allochimera made in a SCID mouse responds to allo-MHC but not to antigens under control of restrictive recognition (5). In this case, the recognition of allo-R must be peptide-unspecific. It is not our intention to develop this here as it is part of a discussion to be published elsewhere. Rather, here only the eTh-independence of iT activation will be considered.
Three threshold densities of PR complex can be envisaged:
High density as exemplified by allo-R, interactions with which permit the TCR to deliver Signal[3].
Intermediate density as exemplified by NS complexes to which the animal responds (eTh-dependently) and S complexes to which it is tolerant. This density range is capable of delivering Signal[1].
Low density that escapes recognition resulting in activation (i.e., is unable to initiate Signal[1]).
In cases where the response to a PR complex is eTh-dependent, the complex is in the intermediate density range and delivers Signal[1]. The iT and eTh interact on the surface of the APC to deliver Signal[2] resulting in the activation of iT.
By contrast, allo-R is viewed by the iT-cell to be present in high density on the APC or other target cell because the recognition is peptide-unspecific. This difference between intermediate density and high density engagement of iT-cell antigen-receptors is translated into eTh-dependence or eTh-independence in roughly the same way as described for iB-cells. In other words, a given peptide presented in high density (comparable to allo-R) would be predicted to activate iT-cells specific for that peptide, eTh-independently.
A given high density PR complex is viewed as being a “polymer” by the T-cell antigen-receptor, which is aggregated to deliver Signal[3] that activates eTh-independently. The same rules apply here as do for the iB-cell, namely the S-NS discrimination must be germline encoded. A given intermediate density PR complex is viewed as being a “monomer” by the T-cell antigen-receptor and requires Signal([1]+[2]) to activate iT (i.e., activation is eTh-dependent). It appears, therefore, that the allosteric model proposed here for the B-cell antigen-receptor is also applicable to the T-cell antigen-receptor.
Footnotes
This work was supported by a grant (RR07716) from the National Center For Research Resources at the National Institutes of Health.
This paper was written while Melvin Cohn was a Scholar-in-Residence at the Fogarty International Center for Advanced Study in the Health Sciences, National Institutes of Health, Bethesda, Maryland, USA.
REFERENCES
- 1.Mond JJ, Lees A, Snapper CM. T cell-independent antigens type 2. Annu Rev Immunol. 1995;13:655–92. doi: 10.1146/annurev.iy.13.040195.003255. [DOI] [PubMed] [Google Scholar]
- 2.Snapper CM, Mond JJ. A model for induction of T cell-independent humoral immunity in response to polysaccharide antigens. J Immunol. 1996;157:2229–33. [PubMed] [Google Scholar]
- 3.Dintzis HM, Dintzis AZ, Vogelstein B. Molecular determinants of immunogenicity: the immunon model of immune response. Proc Natl Acad Sci. USA. 1976;73:3671–75. doi: 10.1073/pnas.73.10.3671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Langman RE, Cohn M. Has immunoglobulin come to a sticky end? Scand J Immunol. 1991;33:99–109. doi: 10.1111/j.1365-3083.1991.tb03739.x. [DOI] [PubMed] [Google Scholar]
- 5.Lin Y, Langman R, Cohn M. The priming of cytotoxic T-cell precursors is strictly heolpter T cell-dependent. Scan J Immunol. 1992;35:621–626. doi: 10.1111/j.1365-3083.1992.tb03262.x. [DOI] [PubMed] [Google Scholar]