

Abstract
The hepatotoxicity of bromobenzene (BB) derives from its reactive metabolites (epoxides and quinones) which arylate cellular proteins. Application of proteomic methods to liver proteins from rats treated with an hepatotoxic dose of [14C]-BB has identified more than 40 target proteins, but no adducted peptides have yet been observed. Because such proteins are known to contain bromophenyl- and bromodihydroxyphenyl derivatives of cysteine, histidine and lysine, the failure to observe modified peptides has been attributed to the low level of total covalent binding and to the “dilution” effect of multiple metabolites reacting at multiple sites on multiple proteins. In this work glutathione transferase, a well known and abundant BB-target protein, was isolated from liver cytosol of rats treated with 14C-BB using a GSH-agarose affinity column and further resolved by reverse phase HPLC into subunits M1, M2, A1, A2 and A3. The subunits were identified by a combination of SDS-PAGE, whole-molecule mass spectrometry and peptide mass mapping and found to contain radioactivity corresponding to 0.01 - 0.05 adduct per molecule of protein. Examination of tryptic digests of these subunits by MALDI-TOF and ESI-MS again failed to reveal any apparent adducted peptides despite observed sequence coverages up to 87%. However, using HPLC-LTQ-FTMS to search for predicted modified tryptic peptides revealed peaks corresponding, with a high degree of mass accuracy, to a bromobenzoquinone adduct of peptide 89-119 in both GSTA1 and A2. The identity of these adducts and their location at Cys-111 was confirmed by MS-MS. No evidence for the presence of any putative BB-adducts in GST M1, M2 or A3 was obtained. This work highlights the challenges involved in the unambiguous identification of reactive metabolite adducts formed in vivo.
Introduction
The covalent binding of xenobiotic compounds to liver proteins was first described in the 1940s in connection with studies of the disposition of carcinogenic azo dyes in rats. It wasn't until the early 1970s that protein covalent binding of reactive metabolites of chemicals (e.g. bromobenzene (1)) and drugs (e.g. acetaminophen (2, 3)) was directly associated with cytotoxicity and target organ damage. In the intervening years many different pathways by which unreactive chemicals can be converted to chemically reactive metabolites have been described (4); this knowledge now forms much of the basis for “structural alerts” that influence the design of safety testing protocols for potential drug candidates.
In the case of bromobenzene, the structures of its stable metabolites (phenols, dihydrodiols and mercapturic acids) implicated as a common precursor an arene oxide that could, potentially, also react with and covalently modify protein nucleophiles (1, 5). Later in vitro studies indicated that in addition to undergoing conjugation and excretion, bromophenols could undergo further oxidation to quinones potentially capable of protein covalent binding (6). Direct support for the covalent binding of both epoxide and quinone metabolites of bromobenezene was provided by the isolation and structural characterization of modified amino acids after chemical degradation of unfractionated liver protein (7-9). However, in vivo studies with bromophenol (10), and parallel studies with the meta isomer of acetaminophen (11, 12) showed that while both undergo metabolic activation and covalent binding, toxicity does not ensue. Thus, not all covalent binding is equally significant toxicologically, especially when measured by an index as crude as total radioactivity bound to total precipitable protein.
By the 1990s it was recognized that perhaps the question of which specific proteins became covalently modified might be more important than the question of which specific metabolite was responsible for the binding. Application of protein separation technology to proteins labeled covalently by reactive metabolites showed that binding was somewhat selective and not entirely random (13-15). Some of the first proteins identified as targets for reactive metabolites were common abundant proteins such as glutathione transferases, calreticulin, microsomal carboxylesterase, carbonic anhydrase III, protein disulfide isomerase, Grp78 and even certain cytochrome P450 enzymes (16). However, none of these early targets seemed to be acutely critical for cell survival, such that their covalent modification could have directly and immediately compromised cell viability. Furthermore, at the levels of covalent binding commonly observed for agents like bromobenzene or acetaminophen (typically 1-6 nmol-equiv/mg protein, (17)), covalent binding only occurs to a relatively small extent for any particular protein. For example, for a 25 kDa protein labeled to an average extent of 4 nmol-equiv./mg protein, only one protein molecule in ten carries an adduct moiety. Thus, direct inhibition of vital enzymes is unlikely to be a general mechanism for reactive metabolite-induced acute toxicity. On the other hand, chemical modification of even a small subpopulation of target molecules might constitute a signal for down-stream events finally leading to cell death (18-20).
In the past decade the advent of proteomic approaches based on two-dimensional gel electrophoretic separations coupled with mass spectrometric methods for protein identification has led to a rapid increase in the number of proteins identified as targets for various reactive metabolites (16). However, while there is substantial knowledge about different types of reactive metabolites that can form, and the list of target proteins continues to grow, the identification of a specific modification on a specific protein at a specific residue position remains a very significant challenge. Here we report that treatment of rats with bromobenzene results in the site-specific modification of Cys-111 in cytosolic glutathione transferase subunits A1 and A2 by bromobenzoquinone.
Experimental Procedures
Materials
[14C]-Bromobenzene (5.17 Ci/mol) was prepared in our laboratory, stored and handled as described previously (21). Trypsin, sequencing grade, from bovine pancreas was obtained from Roche (Nutley, NJ). 4-Vinylpyridine was obtained from Sigma and was distilled and stored under nitrogen at −20 °C. Sequenal grade urea was obtained from Pierce (Rockford, IL). Dithiothreitol (DTT), phenylmethylsulfonylfluoride (PMSF), glutathione (GSH), S-hexylglutathione and prepacked glutathione-agarose affinity columns (2.5 ml) were obtained from Sigma (St. Louis, MO). Electrophoresis supplies, Bradford reagent and Precision Protein Standards were obtained from Bio-Rad (Hercules, CA). Brominated RNase was available from earlier work in our laboratory (22). HPLC grade solvents and analytical grade inorganic salts were obtained from Fisher (Fair Lawn, NJ). Deionized water (resistivity >17 MΩ/cm) was used for preparation of all buffers.
Treatment of animals and preparation of liver cytosol
Male Sprague Dawley rats (160-180 g, Charles River Laboratories, Wilmington, MA) were housed in a temperature and humidity controlled room with a 12 hr light/dark cycle and ad libitum access to food and water. After acclimating for at least three days, animals were given 3 daily ip injections of sodium phenobarbital (80 mg/kg) in 0.9% saline (4.0 mL/kg). After the third dose the animals weighed 190-210 g. Food was withheld overnight and the next morning the rats were injected ip with [14C]-bromobenzene (2 mmol/kg) in corn oil (1 mL/kg). PB-control animals received only phenobarbital treatment and no bromobenzene. After 4 h the rats were killed by decapitation under CO2 narcosis. The livers were immediately removed, chilled in ice-cold buffer A (50 mM potassium phosphate, pH 7.4, containing 0.15 M KCl, 5 mM EDTA, 1 mM PMSF), minced on an ice-cold glass plate, and homogenized in a single batch in ice-cold buffer A (3 mL/g tissue) using a motor-driven Teflon-glass homogenizer. All subsequent steps were carried out at 4 °C. The homogenate was successively centrifuged at 6500 ×gave. (10 min), 25,000 ×gave. (20 min) and 100,000 ×gmax. (60 min). The 100,000 ×g supernatant (cytosol) fractions were combined and clarified by recentrifugation at 100,000 ×g. The resulting PB-control cytosol (22 mg protein/ml) was then aliquoted and stored at −70 °C. Cytosol from animals treated with [14C]-bromobenzene (18.7 mg protein/mL) was further dialyzed against 20 mM potassium phosphate, pH 7.4, containing 0.5 mM DTT (4 × 60 volumes), to remove reversibly bound radioactivity, and stored identically. The six PB-treated rats dosed with [14C]-bromobenzene furnished a total of 43 g liver, from which 170 mL of final homogenate was made. Dialysis of the final 100,000 ×g supernatant afforded 69 mL of clear cytosol containing 18.7 mg protein/mL and 3.9 nmol-equiv. BB/mg protein.
Determination of covalently bound radiolabel
Aliquots of the subcellular fractions were precipitated with 10% trichloroacetic acid, and protein precipitates were successively washed with acetone, methanol/water (80:20 v/v, 3 times), acetone and diethyl ether, dried by rotary evaporation, dissolved in 1 M NaOH and neutralized with 1 M HCl, after which the radioactivity was measured by scintillation counting and protein determined by Bradford assay (Bio-Rad).
Affinity Isolation of Glutathione Transferase
All steps were carried out at 4 °C, substantially as described by Nerland et al. (23). A portion of dialyzed cytosol from the livers of PB-control rats containing 46 mg protein was diluted to 10 mL with buffer C (10 mM Tris, pH 7.8, containing 1 mM EDTA and 0.2 mM DTT) and applied to a prepacked glutathione-agarose affinity column pre-equilibrated with buffer C. After recirculating the loading solution through the column overnight (0.1 mL/min), the column was washed sequentially with buffer C, buffer C containing 200 mM NaCl, and finally buffer C containing 200 mM NaCl plus 5 mM S-hexylglutathione (see Table 1A for volumes). Protein elution was monitored by A280 and GST activity as described below. Fractions of the eluate containing significant GST activity were combined, dialyzed twice against 100 volumes of water, and lyophilized to give a dry white powder. A portion of the cytosol from the livers of PB-pretreated rats given [14C]-bromobenzene (204 mg protein, 3.59 μCi 14C), was processed similarly (Table 1B).
Table 1.
Affinity Isolation of Cytosolic Glutathione Transferase.
| A. PB-Controla | Volume (mL) | Protein (mg) | Activity (U)b |
|---|---|---|---|
| Load | 10.5 | 140 | 34.8 |
| FT + Wash | 25 | 121 | 8.5 |
| NaCl Wash | 14 | 2.0 | 0.3 |
| Hex-SG | 13.5 | 7.7 | 6.2c |
| B. [14C]-BBa | Volume (mL) | Protein (mg) | 14C (nCi) |
| Load | 11.8 | 204 | 3431 |
| FT + Wash | 49 | 197 | 3141 |
| NaCl Wash | 17 | 3.7 | 21 |
| Hex-SG | 8.6 | 9.8 | 95 |
Refer to Experimental Procedures for details of column loading and elution.
One unit is 1 μmol product/min/mg protein at room temperature.
May be underestimated because of inhibition by residual S-hexyl-glutathione.
GST Activity Assay
The enzymatic activity in each fraction was assayed using 1 mM 2,4-dinitrochlorobenzene (DNCB) and 1 mM GSH in buffer B (100 mM phosphate buffer, pH 6.5) according to the method of Habig et al. (24). For the assay, DNCB solution (25 mM in ethanol, 120 μL) was added to 2.65 mL buffer B in a 3 mL cuvette. Glutathione solution (25 mM in buffer B, 120 μL) was then added, and after rapid mixing, a 100 μL aliquot of an appropriate dilution of enzyme preparation was added, the cuvette was shaken and its absorbance monitored at 340 nm for 2 min at room temperature. Activities were calculated as μmol product/min/mg protein using Δε = 9.6 mM−1cm−1.
HPLC Separation of GST Subunits
Samples of lyophilized GST were reconstituted in water (0.25 mL/mg protein) and combined with an equal volume of solvent A (40% acetonitrile in 0.1% aqueous TFA) prior to injection. GST subunits were separated essentially as described by Nerland et al. (23) using a Vydac 201 TP54 C18 reverse phase column (4.6 × 250 mm) eluted at 1 mL/min with the following gradient of solvent B (90% acetonitrile in 0.1% aqueous TFA) in solvent A: 5-10% B in 8 min, 10-15% B in 6 min, 15-35% B in 16 min, 35-70% B in 2 min, 70% B for 2 min, 70-5% B in 5 min, with UV detection at 214 nm. Peaks were collected manually for liquid scintillation analysis and for examination by mass spectrometry.
Electrophoresis
Separations were performed using a Mini-PROTEAN II cell equipped with a 1000/500 Power Supply (Bio-Rad). Purified GST, flow-through, and the original liver cytosol were separated by Tricine-SDS-PAGE according to a reported method (25). The separating gel and stacking gel contained 16.5% and 4% acrylamide, respectively. The gels were run at 30 V for 25 min or until the protein had concentrated into a thin band at the top of the separating gel; the voltage was then increased to 90 V for another 3-4 h. Proteins were visualized using 0.025% Coomassie brilliant blue R250 in a mixture of methanol/acetic acid/water (50:10:40 v/v/v), and destained first in the same solvent without dye (3 changes) and then in methanol/acetic acid/water (5:5:90 v/v/v, 3 changes) until a clear gel was obtained.
Protein mass spectrometry
ESI mass spectra were acquired on a Q-Tof2 (Micromass Ltd., Manchester, UK) hybrid mass spectrometer operated in MS mode and acquiring data with the time of flight analyzer. The instrument was operated for maximum sensitivity with all lenses optimized while infusing a sample of lysozyme. The cone voltage was 60eV and Ar was admitted to the collision cell. Spectra were acquired at 11364 Hz pusher frequency covering the mass range 800 to 3000 amu and accumulating data for 3 seconds per cycle. Time to mass calibration was made with CsI cluster ions acquired under the same conditions. Samples (35-60 μg) were desalted on a short column (3 cm × 1 mm ID) of polymeric HPLC resin (PRP1, Hamilton, Reno, Nevada) by loading the sample in 1% HOAc and eluting protein with 95% MeOH, 0.08% formic acid at 25 μl/min directly into the source.
Protein digestion and peptide mass mapping
Individual GST subunits collected from HPLC fractionation of PB-control and BB-treated rat liver cytosol were run on SDS-PAGE gels and the individual bands were excised from the gel, cut into small pieces, destained with 200 mM NH4HCO3/acetonitrile (1:1, v/v), reduced with DTT, alkylated with 4-vinyl pyridine and digested with sequencing grade trypsin as described previously(14). Peptides in the digest mixture were concentrated in a C-18 ZipTip, eluted with 2 μL of a saturated solution of α-cyano-4-hydroxy-trans-cinnamic acid in 50% aqueous acetonitrile containing 0.1% trifluoroacetic acid and applied to a MALDI sample plate. The samples were analyzed on a Voyager-DE STR MALDI-TOF mass spectrometer (Perseptive Biosystems, Framingham, MA) operated in positive reflector mode with accelerating voltage 20 kV, mirror voltage ratio 1.12, and extraction delay 180 ns. Data acquisition was performed over the m/z range 700-3000. Mass spectra were externally calibrated using a standard mixture of known peptides covering the entire mass range, and where possible the calibration was verified using internal m/z peaks arising from trypsin autolysis. Searches of the Swiss-PROT database were performed using MS-Fit (http://prospector.ucsf.edu/ucsfhtml4.0/msfit.htm). The maximum mass error allowed was set to 50 ppm with one missed cleavage allowed.
For in-solution digestion, individual GST subunits collected from HPLC fractionation of liver cytosol from rats treated with phenobarbital and [14C]-bromobenzene (ca. 70 μg each as a lyophilized white solid) were dissolved in 20 μL of digestion solution (aqueous 8 M urea and 0.4 M NH4HCO3). After 15 min sonication, 5 μL DTT solution (54 mM) was added and the mixture was incubated at 50 °C for 15 min. After cooling to room temperature 4-vinyl pyridine solution (5 μL, 100 mM in water) was added and the mixture was incubated at room temperature for 20 min in the dark. Water (30 μL) and trypsin solution (20 μL, 0.1 mg/ml) were added and the mixture was incubated at 37 °C for 18h. The reactions were chilled on ice and stored frozen at −20 °C for further analysis. Tryptic peptides were separated on a RP-HPLC column (0.32 mm ID × 5 CM Zorbax SBC18, 300 Å pore size, 3.5 μm particles (packed by Micro-Tech Scientific, Vista, CA) with a linear methanol/water gradient (20 to 95 vol-% methanol in 65 minutes), with a constant 0.08% (v/v) formic acid modifier, at a flow rate of 10 μL/min. The chromatograph was an Ultra Plus II MicroLC system (Micro-Tech Scientific, Vista, CA). Peptides were eluted directly into the source of a Q-Tof2 mass spectrometer (Micromass, Ltd., Manchester, UK) and data was acquired in TOF mode limiting spectra to 350-2500 u in 5 second acquisition cycles.
Capillary LC-MS of Tryptic Digests
The GST isoforms digested with trypsin were introduced into a LTQ-FT hybrid linear quadrupole ion trap Fourier transform ion cyclotron resonance (FT-ICR) mass spectrometer (ThermoFinnigan, Bremen, Germany) by capillary LC. Peptides were separated on a reverse-phase column (0.3 × 150 mm, Pepmap C18) at a flow rate 5 μl/min with a linear gradient raising from 5 to 50% (v/v) acetonitrile in 0.06% (v/v) aqueous formic acid over a period of 45 min and from 50 to 95% during 5 min using an LC Packings Ultimate Chromatograph (Dionex, Sunnyvale, CA). In order to identify cysteine-containing peptides we first performed an experiment using dynamic exclusion with survey MS scans in the range m/z 300-2000, acquired in the FT-ICR using full MS target automated gain control (AGC) value of 5×105 with resolution 25,000 at m/z 400. The two most intense ions in the survey scan were selected and sequentially fragmented in the ion trap using an AGC value of 2×103 or an ion accumulation time of 200 ms. The ion selection threshold was 2000 counts. The second MS experiment, adapted from Olsen and Mann (26), allowed us to expand the dynamic range of the instrument and as a result to detect low-abundance adducts of Cys-containing peptides. The survey LTQ-MS1 scan was acquired over a 250 u range around the expected m/z for a modified Cys-containing peptide using an AGC value of 1×104 or an ion accumulation time 400 ms. Ions corresponding to anticipated adducted peptides were isolated for both a 10 u mass range FT-ICR scan (ion accumulation time 1 s, AGC value 1×105) and an MS/MS scan in the LTQ (400 ms) using a list of anticipated masses. LTQ-MS/MS scans were acquired with isolation window of 3.0 m/z.
Results
Isolation and characterization of rat liver glutathione transferases
The results of a typical isolation of glutathione transferase enzymes from PB-control cytosol by affinity chromatography are summarized in Table 1A. The majority of the cytosolic protein passed through the column in the loading volume, flow-through and early washes. Elution with S-hexylglutathione brought off 7.7 mg protein or about 5.5% of that applied. The apparent GST activity of this fraction was only 17% of that applied to the column, but this value is undoubtedly low because, as shown by HPLC analysis (see below), dialysis consistently failed to remove all of the S-hexylglutathione ligand, and the latter is a potent inhibitor of GST activity. Total protein recovery was 94%. SDS-PAGE analysis revealed the affinity purified material to contain three closely spaced bands of apparent molecular weight around 25 kDa (Figure 2A), which is very consistent with previous observations (24, 23). Direct analysis of this mixture by ESIMS indicated the presence of five major components having masses of 25185, 25465, 25514, 25568 and 25779 Da (see below).
Figure 2.
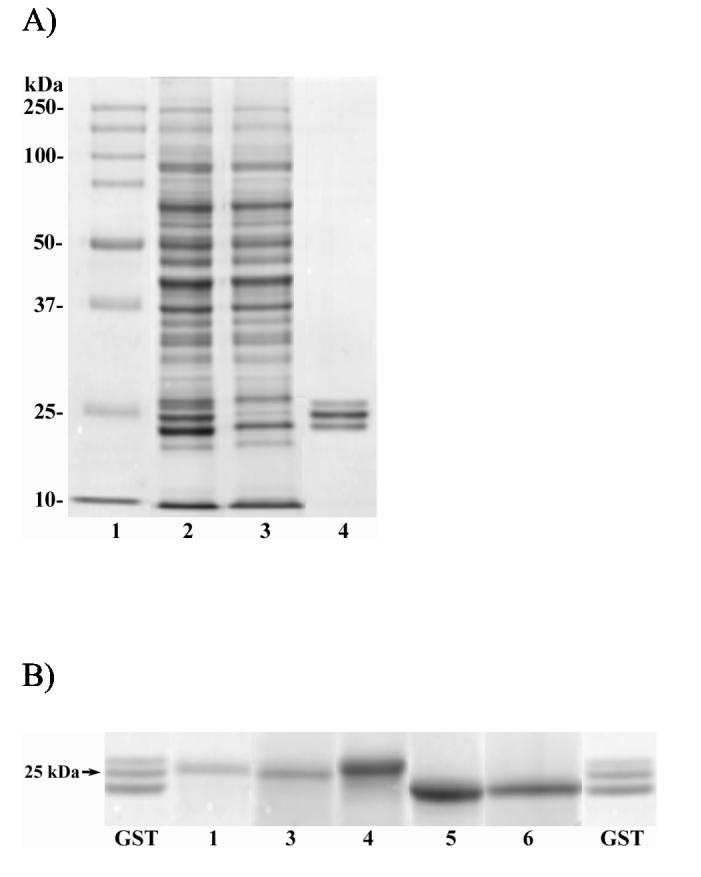
SDS-PAGE analysis of rat liver cytosol and affinity-isolated GSTs. Panel A: lane 1, molecular standards; lane 2, unfractionated cytosol from PB-control rat liver; lane 3, flow through fraction from the GSH-Agarose column; lane 4, affinity purified GST. Panel B: lanes marked GST, affinity purified GST from rats treated with [14C]-BB prior to HPLC (same as lane 4 above); lanes numbered 1-6, HPLC peaks numbered 1-6, respectively, in Figure 1.
When the same isolation procedure was applied to cytosol from [14C]-bromobenzene-treated rats a similar result was obtained (Table 1B). The affinity purified material contained ca. 5% of the protein but only 3% of the radioactivity applied to the column. The labeling density was 1.86 nmol-equiv. bromobenzene/mg protein, or about one-half of the average value for all cytosolic proteins. Since the molecular weight of the GSTs is ca. 25 kDa, this corresponds to a labeling density of 0.047 adduct/protein molecule, or to only one protein molecule out of 21 carrying some type of bromobenzene-derived adduct. SDS-PAGE analysis of the radioactive GST was deferred until after further separation by HPLC (see below). As with the GSTs from the PB-control rats, direct analysis of this mixture by ESIMS indicated the presence of five major components having masses essentially identical to those observed for GSTs from PB-control rats.
The affinity-isolated GSTs from both PB-control and BB-treated rats were next fractionated by reverse-phase HPLC essentially as described by Nerland et al. (23). Whereas both preparations showed the same five components by ESIMS, the PB-control GSTs resolved into six separate HPLC peaks (Figure 1A). In contrast, but in agreement with the ESIMS results, the GST fraction from the BB-treated rats resolved into only five peaks (Figure 1B) whose retention times (Table 2) were identical to five corresponding peaks in Figure 1A. In both chromatograms the large peak immediately after the solvent front has the same retention time as S-hexylglutathione, the ligand used to elute the affinity column. The same excellent resolution of all major peaks could be obtained at protein loadings up to at least 160 μg total protein per injection, but the separation was critically dependent on the use of the Vydac 201 TP54 C-18 column, as other C-18 columns, even other types from the same manufacturer, gave markedly inferior or no separation. ESIMS analysis of individual HPLC peaks from the PB-control GSTs (Table 3) corroborated the masses observed by direct analysis of the mixture prior to HPLC separation (see above), and allowed the tentative identification of individual isoforms based on their apparent masses. Assuming that all the protein injected emerged from the column as peaks, the relative amounts of the different GST subunits in the sample are as given in Table 2; the relative proportions of the five peaks vary from 12% to 32% of the total, but in general the profile looks remarkably similar to that reported by Nerland et al. (23). The specific radioactivity associated with each peak varied less than 3-fold.
Figure 1.
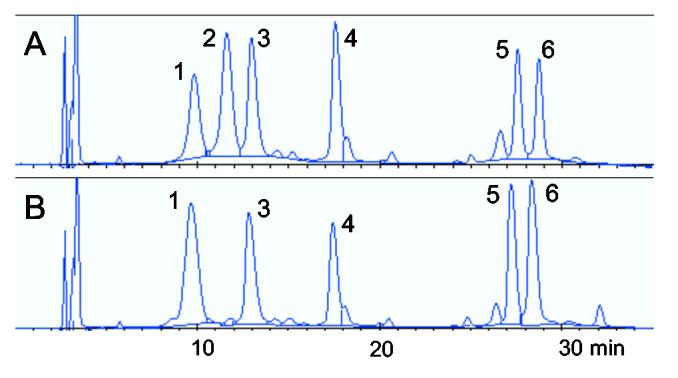
Reverse phase HPLC separation of GST subunits isolated by affinity adsorption. A) GST from liver cytosol of PB-control rats. B) GST from liver cytosol of BB-treated rats.
Table 2.
Relative Peak Areas and Radioactivity Associated with HPLC-separated Subunits of Glutathione Transferase.
Table 3.
Apparent Masses of HPLC-Separated GST Subunits from PB-Control Rats as Determined by ESI-MS.
| HPLC peak | GST subunit assigneda | Observed mass (Da) | Theoretical mass (Da) | Δ (Da)b |
|---|---|---|---|---|
| 1 | GSTM1 | 25786 | 25782 | 4 |
| 2 | GSTM1 | 25787 | 25782 | 4 |
| 3 | GSTM2 | 25611c | 25571 | 40d |
| 4 | GSTA3 | 25193 | 25188 | 5 |
| 5 | GSTA1 | 25522 | 25522e | 0 |
| 6 | GSTA2 | 25475 | 25470e | 5 |
See text for basis of subunit assignment.
Observed mass minus theoretical mass.
Determined by MALDI-TOF MS.
This isoform may be acetylated on its N-terminus, based on the mass difference.
Includes 42 Da for reported N-terminal acetylation on this isoform.
HPLC-separated GST peaks from the BB-treated animals were collected individually and submitted to analysis by SDS-PAGE. As shown in Figure 2B, the relative molecular weights of the individual HPLC peaks estimated by electrophoresis do not correspond exactly to the molecular weights observed by mass spectrometry, but are well within the tolerances usually observed for electrophoresis. Individual protein bands were excised from the gels, digested with trypsin and the resulting peptides analyzed by MALDI-TOF MS in order to establish the identity of the subunits with greater certainty. As shown in Table 4, various subunits were identified on the basis of 19-30 peptide mass matches at moderately high accuracy (50 ppm), sequence coverages of 43-72% and strong scores from the search program MS-Fit. These results agree with our initial assignments based on protein molecular weights (Table 3), and with assignments made by Nerland et al. (23). The only ambiguity concerns HPLC peak 5, which appears to be a mixture of GSTA1 and GSTA2, two very similar subunits which differ by only 9 out of 221 amino acids, but which differ significantly in molecular weight.
Table 4.
GST Subunit Identification by Peptide Mass Mapping.
| HPLC peak | GST subunit assigned | Number of peptide matchesa | Percent sequence coverage | MOWSE score |
|---|---|---|---|---|
| BB-Treated | ||||
| 1 | GSTM1 | 20 | 57 | 4.59 E+05 |
| 3 | GSTM2 | 30 | 72 | 2.26 E+10 |
| 4 | GSTA3 | 19 | 53 | 4.63 E+02 |
| 5 | GSTA2b | 23 | 52 | 4.92 E+05 |
| 5 | GSTA1b | 25 | 50 | 1.43 E+05 |
| 6 | GSTA2 | 20 | 56 | 1.03 E+04 |
| PB-Control | ||||
| 1 | GSTM1 | 6 | 30 | 4.59 E+01 |
| 2 | GSTM1 | 24 | 66 | 1.61 E+07 |
Peptide masses determined by MALDI-TOF MS.
Peak 5 appears to be a mixture of two isozymes; all but four matches are identical for subunits. Three of the matches are unique to GSTA1 and one is unique to GSTA2.
As noted above, HPLC peak 2 in the GST fraction from PB-control animals was not seen in the GST fraction from the BB-treated animals. Since this peak had the same apparent mass as peak 1, these two peaks were individually collected after HPLC and submitted to SDS-PAGE followed by in-gel digestion and peptide mass mapping (Table 4). Collectively, our results strongly suggest that these two HPLC peaks are the same GST subunit. It is not clear why two forms of the same protein should exist or be separable on HPLC as we reproducibly observe, nor why one of these peaks should be missing from the GSTs of BB-treated animals. Nerland et al. (23) treated male Sprague-Dawley rats with acrylonitrile, and observed in the cytosol from both control and treated animals the same five GST peaks as we observed in the cytosol of BB-treated animals.
In rat liver the major GST subunit targeted by acrylonitrile, a direct-acting alkylating agent, is GSTM1 (23), and this subunit was modified to the extent of 70%, almost entirely on residue Cys-86. Adduction was clearly observed by whole-protein mass spectrometry, as indicated by a mass increment of +53 for the Michael addition of CH2=CH-CN, as well as by the appearance of appropriately modified peptides in tryptic digests. Interestingly, while radioactivity measurements indicated that 30% of GSTM1 was unmodified, the native form was not observed by whole-molecule mass spectrometry, although both modified and unmodified peptides containing Cys-86 were observed. In our study the average level of adduction of the various GSTs was only 1-5%, despite the fact that a substantial dose of bromobenzene was used (2.0 mmmol/kg, comparable to the 2.2 mmol/kg dose of acrylonitrile used by Nerland et al.). There may be several reasons for the greatly reduced extent of GST modification after bromobenzene compared to acrylonitrile. First, the latter is a direct-acting alkylator, whereas bromobenzene requires bioactivation by one or more metabolic steps to become an alkylating agent (epoxide or quinone). Thus with bromobenzene, the time course of electrophile influx could give more opportunity for endogenous detoxication mechanisms to intercept reactive metabolites leading to less net protein modification overall. Another reason may be the selectivity of the electrophile per se toward different protein nucleophiles. Thus, acrylonitrile is highly selective for GSTM1 > GSTM2 > others, while bromobenzene metabolites collectively show relatively modest selectivity among GST subunits.
Given the very low average adduction of GST subunits by bromobenzene metabolites, it is not surprising that we could not observe mass shifts corresponding to any known (7, 8) adduct types (i.e. bromophenyl-, dihydroxyphenyl- or bromodihydroxyphenyl-) at the whole protein level. In the course of our peptide mass mapping studies using in-gel digestion and MALDI-TOF MS we observed a total of 314 peptides by careful manual inspection. Of these, 102 masses matched predicted masses of tryptic peptides from GST subunits (and therefore could not have contained bromine), while another 212 masses matched neither predicted masses for unmodified peptides, nor predicted masses for peptides modified by any of the known bromobenzene metabolites. The absence of a bromine isotope pattern among any of the mass spectra we examined was striking.
Since our prior experience with analysis of brominated peptides (22) indicated that the bromine isotope pattern is quite easily detected, even at very low levels of analyte (as in the tails of HPLC peaks, for example), and that the greatest number of brominated peaks was detected when we used in-solution digestion followed by LC/ESIMS, we submitted the GSTM1 subunit from BB-treated rats to in-solution digestion and analyzed the digest by both MALDI-TOF MS and HPLC-ESIMS. In the former case we observed 23 matches giving 85% sequence coverage, but saw no evidence of brominated compounds in the spectra. In the latter case we observed a total of 33 peptide matches giving 87% coverage, plus another 241 masses that did not match expected tryptic peptides from GSTM1. Several of the eluting peaks showed MS data with isotope clusters suggesting possible bromine content, but the results were not conclusive.
We next examined these same digests using an HPLC coupled to an LTQ-FTMS instrument run under typical operating conditions (i.e. observe m/z 300-2000) but this too failed to reveal convincing evidence from either MS1 or MS2 for the presence of bromine-containing peptides in the digest. This was surprising since the absolute sensitivity of this instrument was quite impressive, i.e. brominated peptides were easily observed in 50 fmol of a digest of brominated RNase (data not shown). We therefore changed our strategy to search specifically for anticipated adducts, i.e., bromophenyl- and bromodihydroxyphenyl derivatives of expected cysteine-containing tryptic peptides. Since the xenobiotic-adducted peptides were expected to be minor components in the digests, we sampled only narrow mass ranges (250 u) around the m/z values of potential interest in order to enhance the ratio of adduct ions to other ions in the LTQ in each sampling cycle (which is very fast on the HPLC time scale). For MS2 experiments precursor ion selection was based on a window of 3 u to include all isotopomers of the parent. For concurrent FTMS experiments, precursor ion selection in MS1 was based on a 10 u window. For reference purposes we also searched for and examined related peptides containing pyridylethylcysteine.
Applying this selective search approach to the digest of GSTA2 we observed an HPLC peak having a broad isotope cluster at m/z 1235.247 (Figure 3), corresponding to a bromobenzoquinone adduct of tryptic peptide 89-119 (structure 2a in Figure 4). This observation is in excellent agreement with the value of calculated m/z (monoisotopic) for the MH33+ form of this peptide (i.e., 1235.248). The identity of this adducted peptide was further confirmed by the MS2 analysis of this ion (Figure 5), which showed 24 of 30 possible b-ions and 22 of 30 possible y-ions. Finally, all y-ions larger than y8 showed a mass shift corresponding to the addition of bromobenzoquinone to Cys-111 of peptide 89-119. For comparison the corresponding pyridylethylated peptide (2b) had observed and calculated masses of 1208.293 and 1208.290, respectively. Similar examination of the digest of GSTA1 produced evidence for the analogous peptide 1a (m/z 1236.572 observed; 1236.571 calculated for MH33+). In MS2 this ion showed 12 b-ions and 16 y-ions, and all y-ions larger than y8 were shifted by the mass of bromobenzoquinone. The corresponding pyridylethylated peptide 1b was also clearly observed at m/z 1209.607 (vs. 1209.612 calculated for MH33+). Thus we can confidently claim the presence of peptide 1a and 2a, respectively, in the digests of GSTA1 and GSTA2 isolated from the livers of bromobenzene-treated rats. Cysteine 17, the only other cysteine in GSTA1 and A2, is also known to be reactive toward hydrophobic electrophiles (27). We did not observe BB-derived adducts at this position, although we did observe vinylpyridine adducts of Cys-17 (specifically, peptides 13-19 and 15-32).
Figure 3.

Observed mass spectra of tryptic peptide 89-119 from GSTA2 modified at Cys-111 by vinylpyridine (panel A) or bromobenzoquinone (panel C). For comparison, the respective calculated spectra are shown below in panels B and D. See Figure 5 for MS2 spectrum of the BBQ adduct of peptide 89-119.
Figure 4.

Sequences of tryptic peptides 89-119 derived from GSTA1 (1) and GSTA2 (2) and their modifications at Cys-111.
Figure 5.
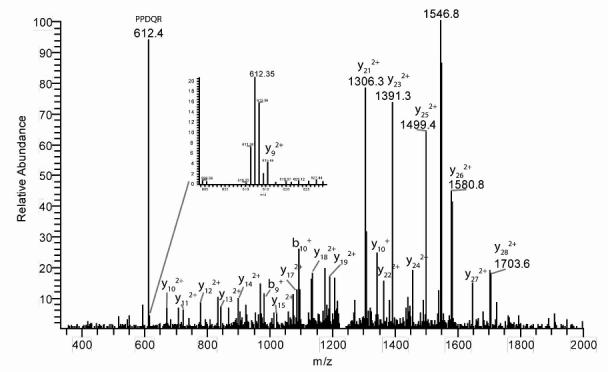
MS2 fragmentation patern of GSTA2 tryptic peptide 89-119 adducted at Cys-111 by bromobenzoquinone. See Figure 3C for mass spectrum of parent ion. The inset shows the small y9 fragment ion at the correctly-shifted mass.
Given the very small samples available, the mass accuracy of the peptide mass measurements stated above is remarkable. This feature, together with abundant (if slightly less mass-accurate) MS2 data is esential to the unambiguous identification of adducted peptides among the other materials in the digests. For example, in the digest of GSTA1 we observed a peak with a broad isotope pattern at m/z 1225.600, close to the monoisotopic value of 1225.907 expected for the MH33+ form of the bromophenyl adduct of peptide 89-119 (i.e. peptide 1c in Figure 4). For this peak we observed 17 assignable y- and b-ions in MS2, and all y-ions larger than y8 appeared to be shifted by the mass of bromobenzyne (C6H3Br), suggesting it could be a bromophenyl adduct such as 1c. Unfortunately, these observations can not support the presence of peptide 1c in the digest because the observed m/z of the triply-protonated parent ion means that the actual peptide (in the unprotonated form) would be too light by essentially one mass unit, which is unreasonable for an instrument of this mass accuracy. Secondly, while the average mass discrepancy (observed minus theoretical) for the doubly-protonated y-ions from this parent was -0.2 u, those for the y-ions of peptides 1a, 1b, 2a and 2b mentioned above were all around +0.2 u. This not only shows the power of the LTQ-FTMS instrument for characterizing tiny samples of adducted peptides, it shows that mass measurements of very high mass accuracy are required to avoid making incorrect assignments.
Finally, we also applied this specific-search method to the digests of subunits GSTA3 and GSTM2, but in these cases we observed no true matches to anticipated adducts. Curiously, in GSTM2, as in GSTA1, we again observed a peak lighter by 1 amu than an anticipated bromophenyl adduct, both in MS1 and MS2, but we have no explanation of what this material might be. No matches to any anticipated adducts were found for GSTA3.
Discussion
The cytotoxicity of many xenobiotic organic chemicals is strongly correlated with their metabolic conversion to electrophilic metabolites and the covalent binding of the latter to cellular proteins. Numerous studies have shown that this binding often involves different sites on different proteins, and in some cases even multiple reactive metabolites from a single parent compound. On the other hand, binding is not completely random and can be quite selective in some cases (13-15). A comprehensive survey of reactive metabolite target proteins (16) shows that many are enzymes, but in the few cases studied, inhibition of these enzymes is modest and seems unlikely to contribute to cell death. In the case of GSTA1 and A2, a progression from the alkylation of Cys-111, to a loss of enzymatic activity, to the development of toxicity seems particularly unlikely for several reasons. First, although the effect of Cys-111 alkylation on the activity of GSTA1 and A2 is not known, the distance from this residue to the active site suggests that its modification would not be likely to affect catalysis adversely. Second, even if the Cys-111-alkylated species were completely inactive, the fact that cells have a great overcapacity of GSTs (28) effectively removes loss of catalytic function of these enzymes as a contributing factor in bromobenzene-induced hepatotoxicity. Growing evidence suggests that this type of post-translational modification may activate signaling pathways that culminate in the cumulative biochemical and cytostructural changes recognized as oncotic and/or apoptotic cell death.
For some toxic compounds, protein covalent binding is an obligatory step in the chain of events leading to toxicity(1-3), but for others it is equally clear that covalent binding does not lead to toxicity (11, 10, 12). Thus, not all covalent binding events are equally consequential for the cell. Even for compounds that require metabolic activation and covalent binding to elicit toxicity, some of the covalent binding events that occur may be inconsequential. Thus to understand the signaling mechanisms it becomes important to elucidate the detailed chemistry of the adduction process.
In previous work toward this goal we isolated and characterized a dozen bromophenyl- and bromodihydroxyphenyl derivatives of cysteine, histidine and lysine from the livers of bromobenzene-treated rats (7-9). In previous publications (29, 14, 15) and in related unpublished work1 we have identified more than 40 proteins that become adducted by bromobenzene metabolites in rat liver (29, 13, 14). Until now, however, these two types of adduct information have not been united by the identification of specific adducts at specific residue positions in specific proteins after in vivo exposure of an animal to a hepatotoxin. The current finding that a bromoquinone metabolite of bromobenzene arylates Cys-111 in rat GSTA1 and GSTA2 is a step toward the desired detailed view of post-translational modification of target proteins by reactive metabolites.
We focused on GSTs in this work for several reasons. First, cytosolic GSTs are well known targets for reactive metabolites of bromobenzene (30, 31, 13) and acetaminophen (32), as well as the intrinsically reactive chemical acrylonitrile (23). They are also relatively abundant proteins in the liver, and it is relatively easy to separate and isolate the major subunits in substantially pure form. In addition, crystal structures for some of GSTs are available, and this makes it feasible to observe how the local environment around nucleophilic target sites influences their reactivity toward electrophilic metabolites. For example, Nerland et al. (23) found that when administered to rats, acrylonitrile reacted extensively yet selectively with Cys-86 of GSTM1; it reacted to a lesser extent with Cys-86 of GSTM2, and did not label α class GSTs such as GSTA1 and GSTA2. Examination of the crystal structure of GSTM2 revealed that Cys-86 may be activated by the nearby basic imidazole side chain of His-84. In GST π, Lys-54 interacts with Cys-47, lowers its pKa, and increases its reactivity toward electrophiles (33).
In rat GSTA1 both Cys-17 and Cys-111 are reactive toward a variety of hydrophobic alkylating agents (27). Rat GSTA2 and mouse GSTA1 differ from rat GSTA1 in only 9 and 14 positions, respectively, out of a total of 221 residues. The high-resolution crystal structure of mouse GSTA1 (34) shows Cys-111 on the wall of a large groove formed between the two monomers (Figure 6). This groove has largely hydrophobic walls and is a major binding site for non-substrate hydrophobic ligands (27). As shown in Figure 6C, the epsilon-amino group of Lys-119 is immediately adjacent to the sulfhydryl group of Cys-111 and is thus in position to activate it. In addition, as shown in Figure 6, Lys-119, Leu-122, Met-104 and Gln-107 comprise a hydrophobic pocket which could favor pre-equilibrium non-covalent binding of bromobenzoquinone near Cys-111 in a way that enhances the probability of its covalent reaction with the nucleophilic sulfur. Both factors could thus contribute to an elevated reactivity of Cys-111 in GSTA1 and A2.
Figure 6.

Structures of GSTA1. Upper left: Space filling model of the homodimer (PDB 1F3A) showing the hydrophobic non-substrate ligand-binding groove at the top center of the molecule. The upper right structure was produced by removing the A-chain of the homodimer and rotating the structure slightly about its vertical axis to show better the location of Cys-111 (yellow sphere near the top of the molecule). Bottom panel: Stereo view of the environment around Cys-111 showing the nitrogen of Lys-119 poised immediately adjacent to the sulfur of Cys-111, which is at the back of a hydrophobic pocket created by the side chains of Leu-122, Met-104 and Gln-107. This figure was made using PyMol (http://pymol.sourceforge.net/). Color scheme: carbon, green; nitrogen, blue; oxygen, red; sulfur, yellow.
This work illustrates some of the challenges and difficulties involved in adduct identification from in vivo samples, especially in cases where there is a low overall extent of adduction, and where the formation of multiple electrophilic metabolites and the presence of multiple target nucleophiles cause “dilution” of each specific adduct within a large pool of many possible structures. Such is the case with bromobenzene (22). Even after extensive purification of a single protein, and even with the isotopic signature furnished by the bromine atom, unambiguous identification of adduct peptides required high sensitivity along with high mass accuracy, good agreement of observed and calculated isotope ratios in MS1 and numerous assignable fragment ions in MS2 to locate adduct position within the peptide. Only using the combined stringency of all these criteria was it possible to rule out some otherwise “close” matches and avoid errors in adduct identification.
The toxicological relevance of observing that an abundant enzyme is covalently modified to a small extent (≤1 molecule in 20) is an open question. The best-known function of the GSTs, catalyzing the conjugation of glutathione to a variety of endogenous and xenobiotic electrophiles, can be inhibited, sterically, by the alkylation of residues near the active sites (33, 35, 36). More recently, GSTs have also been shown to be involved in signaling pathways involving JNK, TNF and Ask1 (18, 37, 38, 19, 39) and thereby to impact intracellular processes including proliferation, differentiation and apoptosis. Omega, Pi, Mu and Theta class GSTs also modulate signaling pathways by metabolizing cyclopentenone prostanoid ligands that affect gene expression triggered by PPARγ, Nrf2 and NF-κB (19). It is interesting to speculate that these reported interactions between GSTs and signaling pathways could exemplify a broader pattern of cellular reaction to the introduction of post-translational modifications by electrophilic metabolites of xenobiotic compounds. For example, proteins of the endoplasmic reticulum such as protein disulfide isomerase, GRP78 and calreticulin are both targets for damage by reactive electrophiles and signaling molecules involved in regulating or modifying cell death pathways (40). Even oxidant-induced cell injury is mediated not only through effects on macromolecules but also through downstream signal transduction pathways (41-43). These reports suggest that the apparently simple alkylation of proteins by reactive electrophiles can have far-reaching consequences propagated through complex signaling mechanisms. Much additional effort will be required to reveal these mechanisms and learn to control them.
Acknowledgment
This work was supported by NIH grant GM-27184. The Q-Tof2 was purchased with support from KSTAR, the University of Kansas and the Kansas-administered NSF EPSCoR program. Funds for the purchase of the LTQ/FTMS were provided by the University of Kansas Center for Research, Inc. We thank Dr. Weijun Huang for preparation of Figure 6.
Footnotes
Courier address: KU-KDCM 4048 Malott 1251 Wescoe Hall Drive Lawrence, KS 66045-7582
Manuscript in preparation.
References
- 1.Brodie BB, Reid WD, Cho AK, Sipes G, Krishna G, Gillette JR. Possible mechanism of liver necrosis caused by aromatic organic compounds. Proc. Nat. Acad. Sci. USA. 1971;68:160–164. doi: 10.1073/pnas.68.1.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jollow DJ, Mitchell JR, Potter WZ, Davis DC, Gillette JR, Brodie BB. Acetaminophen-induced hepatic necrosis. II. Role of covalent binding in vivo. J. Pharmacol. Exp. Therap. 1973;187:195–202. [PubMed] [Google Scholar]
- 3.Mitchell JR, Jollow DJ, Potter WZ, Davis DC, Gillette JR, Brodie BB. Acetaminophen-induced hepatic nectosis. I. Role of drug metabolism. J. Pharmacol. Exp. Therap. 1973;187:185–194. [PubMed] [Google Scholar]
- 4.Kalgutkar AS, Gardner I, Obach RS, Shaffer CL, Callegari E, Henne KR, Mutlib AE, Dalvie DK, Lee JS, Nakai Y, O'Connell JP, Boer J, Harriman SP. A comprehensive listing of bioactivation pathways of organic functional groups. Current Drug Metab. 2005;6:161–225. doi: 10.2174/1389200054021799. [DOI] [PubMed] [Google Scholar]
- 5.Zampaglione N, Jollow DJ, Mitchell JR, Stripp B, Hamrick M, Gillette JR. Role of detoxifying enzymes in bromobenzene-induced liver necrosis. J. Pharmacol. Exp. Therap. 1973;187:218–227. [PubMed] [Google Scholar]
- 6.Hesse S, Wolff T, Mezger M. Involvement of phenolic metabolites in the irreversible protein-binding of [14C]-bromobenzene catalysed by rat liver microsomes. Arch. Toxicol. Suppl. 1980;4:358–362. doi: 10.1007/978-3-642-67729-8_77. [DOI] [PubMed] [Google Scholar]
- 7.Bambal RB, Hanzlik RP. Bromobenzene-3,4-oxide alkylates histidine and lysine side chains of rat liver proteins in vivo. Chem. Res. Toxicol. 1995;8:729–735. doi: 10.1021/tx00047a013. [DOI] [PubMed] [Google Scholar]
- 8.Slaughter DE, Hanzlik RP. Identification of epoxide- and quinone-derived bromobenzene adducts to protein sulfur nucleophiles. Chem. Res. Toxicol. 1991;4:349–359. doi: 10.1021/tx00021a015. [DOI] [PubMed] [Google Scholar]
- 9.Slaughter DE, Zheng J, Harriman S, Hanzlik RP. Identification of covalent adducts to protein sulfur nucleophiles by alkaline permethylation. Anal. Biochem. 1993;208:288–295. doi: 10.1006/abio.1993.1048. [DOI] [PubMed] [Google Scholar]
- 10.Monks TJ, Hinson JA, Gillette JR. Bromobenzene and p-bromophenol toxicity and covalent binding in vivo. Life Sci. 1983;30:841–848. doi: 10.1016/0024-3205(82)90598-7. [DOI] [PubMed] [Google Scholar]
- 11.Matthews AM, Hinson JA, Roberts DW, Pumford NR. Comparison of covalent binding or acetaminophen and the regioisomer 3′-hydroxyacetanilide to mouse liver protein. Toxicology Letters. 1997;90:77–82. doi: 10.1016/s0378-4274(96)03831-3. [DOI] [PubMed] [Google Scholar]
- 12.Roberts SA, Jollow DJ. Acetaminophen structure-toxicity relationships: Why is 3-hydroxyacetanilide not hepatotoxic? Pharmacologist. 1978;20:259. [Google Scholar]
- 13.Dennehy MK, Richards KAM, Wernke GR, Shyr Y, Liebler DC. Cytosolic and nuclear protein targets of thiol-reactive electrophiles. Chem. Res. Toxicol. 2006;19:20–29. doi: 10.1021/tx050312l. [DOI] [PubMed] [Google Scholar]
- 14.Koen YM, Hanzlik RP. Identification of seven proteins in the endoplasmic reticulum as targets for reactive metabolites of bromobenzene. Chem. Res. Toxicol. 2002;15:699–706. doi: 10.1021/tx0101898. [DOI] [PubMed] [Google Scholar]
- 15.Koen YM, Williams TD, Hanzlik RP. Identification of three protein targets for reactive metabolites of bromobenzene in rat liver cytosol. Chem. Res. Toxicol. 2000;13:1326–1335. doi: 10.1021/tx000165l. [DOI] [PubMed] [Google Scholar]
- 16. A searchable, annotated compilation of protein targets for reactive metabolites of drugs and chemicals can be found at http://tpdb.medchem.ku.edu:8080/protein_database/
- 17.Evans DC, Watt AP, Nicoll-Griffith DA, Baillie TA. rug–protein covalent adducts: An industry perspective on minimizing the potential for drug bioactivation in drug discovery and development. Chem. Res. Toxicol. 2004;17:3–16. doi: 10.1021/tx034170b. [DOI] [PubMed] [Google Scholar]
- 18.Adler V, Yin Z, Fuchs SY, Benezra M, Rosario L, Tew KD, Pincus MR, Sardana M, Henderson CJ, Wolf CR, Davis RJ, Ronai Z. Regulation of Jnk signaling by GSTp. EMBO Journal. 1999;18:1321–1334. doi: 10.1093/emboj/18.5.1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hayes JD, Flanagan JU, Jowsey IR. Glutathione transferases. Ann. Rev. Pharmacol. Toxicol. 2005;45:51–88. doi: 10.1146/annurev.pharmtox.45.120403.095857. [DOI] [PubMed] [Google Scholar]
- 20.Lewis MD, Roberts BJ. Role of CYP2E1 activity in endoplasmic reticulum ubiquitination, proteasome association, and the unfolded protein response. Arch. Biochem. Biophys. 2005;436:237–245. doi: 10.1016/j.abb.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 21.Weller PE, Hanzlik RP. Synthesis of substituted bromobenzene derivatives via bromoanilines. A moderately selective ortho-bromination of [14C]-aniline. J. Labelled Comp. Radiopharm. 1988;25:991–998. [Google Scholar]
- 22.Yue W, Koen YM, Williams TD, Hanzlik RP. Use of isotopic signatures for mass spectral detection of protein adduction by chemically reactive metabolites of bromobenzene: Studies with model proteins. Chem. Res. Toxicol. 2005;18:1748–1754. doi: 10.1021/tx050199z. [DOI] [PubMed] [Google Scholar]
- 23.Nerland DE, Cai J, Pierce WM, Benz FW. Covalent binding of acrylonitrile to specific rat liver glutathione s-transferases in vivo. Chem. Res. Toxicol. 2001;14:799–806. doi: 10.1021/tx010002c. [DOI] [PubMed] [Google Scholar]
- 24.Habig WH, Pabst MJ, Jakoby WB. Glutathione S-transferases. J. Biol. Chem. 1974;249:7130–7139. [PubMed] [Google Scholar]
- 25.Schägger H, Jagow von G. Tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa. Anal. Biochem. 1987;166:368–379. doi: 10.1016/0003-2697(87)90587-2. [DOI] [PubMed] [Google Scholar]
- 26.Olsen JV, Mann M. Improved peptide identification in proteomics by two consecutive stages of mass spectrometric fragmentation. Proc. Nat. Acad. Sci. USA. 2004;101:13417–13422. doi: 10.1073/pnas.0405549101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vargo MA, Colman RF. Affinity labeling of rat glutathione S-transferase isozyme 1-1 by 17-beta-iodoacetoxy-estradiol-3-sulfate. J. Biol. Chem. 2001;276:2031–2036. doi: 10.1074/jbc.M008212200. [DOI] [PubMed] [Google Scholar]
- 28.Rinaldi R, Eliasson E, Swedmark S, Morgenstern R. Reactive intermediates and the dynamics of glutathione transferases. Drug Metab. Disposition. 2002;30:1053–1058. doi: 10.1124/dmd.30.10.1053. [DOI] [PubMed] [Google Scholar]
- 29.Rombach EM, Hanzlik RP. Identification of a rat liver microsomal esterase as a target protein for bromobenzene metabolites. Chem. Res. Toxicol. 1998;11:178–184. doi: 10.1021/tx970076h. [DOI] [PubMed] [Google Scholar]
- 30.Aniya Y, McLenithan JC, Anders MW. Isozyme selective arylation of cytosolic glutathione S-transferase by [14C]-bromobenzene metabolites. Biochem. Pharmacol. 1988;37:251–257. doi: 10.1016/0006-2952(88)90726-5. [DOI] [PubMed] [Google Scholar]
- 31.Monks TJ, Lau SS, Gillette JR. Diffusion of reactive metabolites out of hepatocytes: Studies with bromobenzene. J. Pharmacol. Exp. Therap. 1984;228:393–399. [PubMed] [Google Scholar]
- 32.Wendel A, Cikryt P. Binding of paracetamol metabolites to mouse liver glutathione S-transferases. Res. Commun. Chem. Pathol. Pharmacol. 1981;33:463–473. [PubMed] [Google Scholar]
- 33.Vega MC, Walsh SB, Mantle TJ, Coll M. The three-dimensional structure of Cys-47-modified mouse liver glutathione S-transferase p1-1. J. Biol. Chem. 1998;273:2844–2850. doi: 10.1074/jbc.273.5.2844. [DOI] [PubMed] [Google Scholar]
- 34.Gu Y, Singh SV, Zinhua J. Residue R216 and catalytic efficiency of a murine class alpha glutathione S-transferase toward benzo[a]pyrene 7(R),8(S)-diol 9(S),10(R)-epoxide. Biochemistry. 2000;39:12552–12557. doi: 10.1021/bi001396u. [DOI] [PubMed] [Google Scholar]
- 35.Lemercier J-N, Meier BW, Gomez JD, Thompson JA. Inhibition of glutathione S-transferase p1-1 in mouse lung epithelial cells by the tumor promotor 2,6-di-tert-butyl-4-methylene-2,5-cyclohexadieneone (BHT-quinone methide): Protein adducts investigated by electrospray mass spectrometry. Chem. Res. Toxicol. 2004;17:1675–1683. doi: 10.1021/tx049811x. [DOI] [PubMed] [Google Scholar]
- 36.van Zanden JJ, Hamman OB, van Irsel MLPS, Boeren S, Cnubben NHP, Lo Bello M, Vervooort J, van Bladeren PJ, Reitjens IMCM. Inhibition of human glutathione S-transferase P1-1 by the flavonoid quercetin. Chem.-Biol. Interactions. 2003;145:139–145. doi: 10.1016/s0009-2797(02)00250-8. [DOI] [PubMed] [Google Scholar]
- 37.Ketterer B. A bird's eye view of the glutathione transferase field. Chem.-Biol. Interactions. 2001;138:27–42. doi: 10.1016/s0009-2797(01)00277-0. [DOI] [PubMed] [Google Scholar]
- 38.Udomsinprasert R, Bogoyevitch MA, Ketterman AJ. Regulation of glutathione S-transferase spliceforms and the drosophila c-Jun N-terminal kinase pathway components. Biochem.l J. 2004;383:483–490. doi: 10.1042/BJ20040519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang Y, Singh R, Lefkowitch JH, Rigoli RM, Czaja MJ. Tumor necrosis factor-induced toxic liver injury results from Jnk2-dependent activation of caspase-8 and the mitochondrial death pathway. J. Biol. Chem. 2006;281:15258–15267. doi: 10.1074/jbc.M512953200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cribb AE, Peyrou M, Muruganandan S. The endoplasmic reticululum in xenobiotic toxicity. Drug Metab. Rev. 2005;37:405–442. doi: 10.1080/03602530500205135. [DOI] [PubMed] [Google Scholar]
- 41.Liu H, Lo CR, Czaja MJ. Nf-κB inhibition sensitizes hepatocytes to Tnf-induced apoptosis through a sustained activation of Jnkl and c-Jun. Hepatology. 2002;35:772–778. doi: 10.1053/jhep.2002.32534. [DOI] [PubMed] [Google Scholar]
- 42.Wang Y, Schattenberg JM, Rigoli RM, Storz P, Czaja MJ. Hepatocyte resistance to oxidative stress is dependent on protein kinase c-mediated down-regulation of c-Jun/Ap-1. J. Biol. Chem. 2004;279:31089–31097. doi: 10.1074/jbc.M404170200. [DOI] [PubMed] [Google Scholar]
- 43.Xu Y, Bialik S, Jones BE, Iimuro Y, Kitsis RN, Srinivasan A, Brenner DA, Czaja MJ. (275) Nf-κB inactivation converts a hepatocyte cell line Tnf-a response from proliferation to apaptosis. Am. J. Physiol. 275:C1058–C1066. doi: 10.1152/ajpcell.1998.275.4.C1058. [DOI] [PubMed] [Google Scholar]