

Abstract
Water molecules at the surface of DNA are critical to its equilibrium structure, DNA–protein function, and DNA–ligand recognition. Here we report direct probing of the dynamics of hydration, with femtosecond resolution, at the surface of a DNA dodecamer duplex whose native structure remains unperturbed on recognition in minor groove binding with the bisbenzimide drug (Hoechst 33258). By following the temporal evolution of fluorescence, we observed two well separated hydration times, 1.4 and 19 ps, whereas in bulk water the same drug is hydrated with time constants of 0.2 and 1.2 ps. For comparison, we also studied calf thymus DNA for which the hydration exhibits similar time scales to that of dodecamer DNA. However, the time-resolved polarization anisotropy is very different for the two types of DNA and clearly elucidates the rigidity in drug binding and difference in DNA rotational motions. These results demonstrate that hydration at the surface of the groove is a dynamical process with two general types of trajectories; the slowest of them (≈20 ps) are those describing dynamically ordered water. Because of their ultrafast time scale, the “ordered” water molecules are the most weakly bound and are accordingly involved in the entropic (hydration/dehydration) process of recognition.
Hydration of DNA plays important role in its structure, conformation, and function. Of significance to the function is the selective recognition by DNA of small molecules (ref. 1 and references therein). X-ray crystallography, NMR, dielectric relaxation, and molecular dynamics simulation studies have shown that a significant amount of water molecules are bound to DNA (for reviews, see refs. 2–6). For example, measurements of dielectric relaxation caused by water molecules bound to DNA in mixed water-ethanol solutions have found that 18–19 water molecules per nucleotide are present in B-DNA, but only 13–14 water molecules are bound in A-DNA (5). The study also suggested that a structural transition of poly(dG-dC)·poly(dG-dC) DNA from its B to Z form takes place on the removal of the bound water molecules, preferentially from the phosphate groups.
The molecular picture of hydration in the minor groove of B-DNA is unique. An x-ray crystallographic investigation (7) followed by solution NMR study (8) on a model dodecamer B-DNA duplex (for the sequence of A/T tracts, CGCGAATTCGCG) showed that the minor groove is hydrated in an extensive and regular manner, with a zigzag “spine” of first- and second-shell hydration along the floor of the groove. In contrast, hydration within the major groove is principally confined to a monolayer of water molecules. The conformational energy calculation suggested that the presence of the spine of hydration is the prime reason for the further narrowing of minor groove (9).
The influence of drug binding on DNA hydration is striking. Acoustic and densimetric studies have shown that a fraction (not total) of the water molecules is released on recognition (10, 11). Hence, the balance between enthalpic and entropic changes in determining the overall free energy of recognition. The degree of order in water dynamics is determined by the time scale of the motion and is critical to the hydrophobic contribution. Recently, studies of solvation dynamics have been reported for an extrinsic chromogenic probe, inserted into DNA either by covalent adduction of coumarin dye (12) or hydrophobic intercalation of acridine dye (13). The fluorescence results (12) give two relaxation time constants of 300 ps (47%) and 13 ns (53%), both measured with 100-ps time resolution and attributed to the local reorganization (by DNA and/or water) in the modified DNA. The results from femtosecond-resolved transient absorption (13) suggest an ultrafast (within 200 fs) “repolarization” of nuclear degrees of freedom of the DNA pocket. However, the lack of information on structures for both complexes keeps unknown the extent of perturbation on DNA. Moreover, neither study gave the hydration dynamics in the DNA grooves. Using the natural DNA bases to probe hydration is hampered by their ultrafast lifetimes (ref. 14 and references therein).
In this article, we present our study of DNA hydration dynamics, with femtosecond resolution, in the minor groove (Fig. 1), using the dodecamer B-DNA duplex d(CGCAAATTTGCG) whose x-ray structure without and with the drug Hoechst 33258 (H33258) has been reported (15, 16); the site for recognition is the minor groove and remains unchanged on binding. We follow the time evolution of the Stokes shift of the fluorescence of the drug, bound to DNA, and obtain the decay of the hydration correlation function. The function reflects the rotational and translational motions of water molecules in the minor groove. The drug, which is in a class of antimicrobial agents (17), has high affinity to DNA (Kd ≈ 10-8 M); its fluorescence intensity increases when complexed to DNA, relative to that in aqueous solution, and is accompanied by a significant blue shift (18–20). We also report our study of calf thymus DNA (CT-DNA) on binding to the same drug (H33258) to compare with the dodecamer DNA. Finally, to probe the rigidity of the drug and the time scale of its motion with DNA we examined the time-resolved polarization anisotropy, r(t).
Fig. 1.
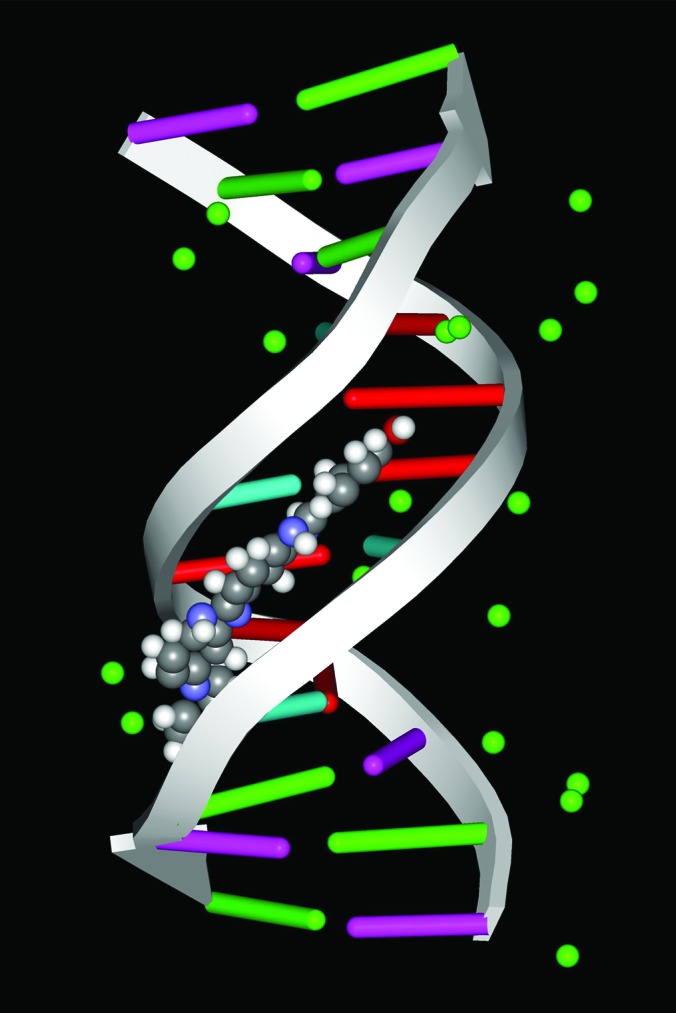
The x-ray structure of the drug Hoechst 33258 with the dodecamer duplex DNA. The structure was downloaded from the Protein Data Bank (ID code 264D) and handled with the program weblab viewerlite (see ref. 16). In the absence of the drug there are 71 water molecules with the dodecamer (15), and in the presence of the drug only 18 water molecules (green balls) are present. The location with respect to the groove is more clear in Fig. 6.
Experimental Procedures
Time-Resolved Studies. All of the transients were obtained by using the femtosecond-resolved fluorescence up-conversion technique. The detailed experimental setup is described elsewhere (21). For the studies reported here, a fs excitation pulse (200 nJ) was used at 350 nm. To measure time-resolved anisotropy, r(t), and to construct hydration correlation function, C(t), we followed the procedure detailed in our previous publications (21–23).
Sample Preparation. CT-DNA (Sigma) and Hoechst 33258 (Molecular Probes, 99% pure) were used without further purification. The drug was in the form of trihydrochloride, pentahydrate, and at neutral pH it only has one positive charge (see below, Scheme 1). The dodecamer DNA with sequence CGCAAATTTGCG was synthesized, purified by reverse-phase cartridge, annealed to make duplex, and supplied by Gene Link (Hawthorne, NY). The purity of the duplex was checked by urea PAGE; essentially one band was characterized by the supplier. Aqueous DNA solutions were prepared in a Tris buffer (0.01 M Tris/0.001 M EDTA) at pH 7.4 in water from a Nanopure (Dubuque, IA) purification system.
Scheme 1.
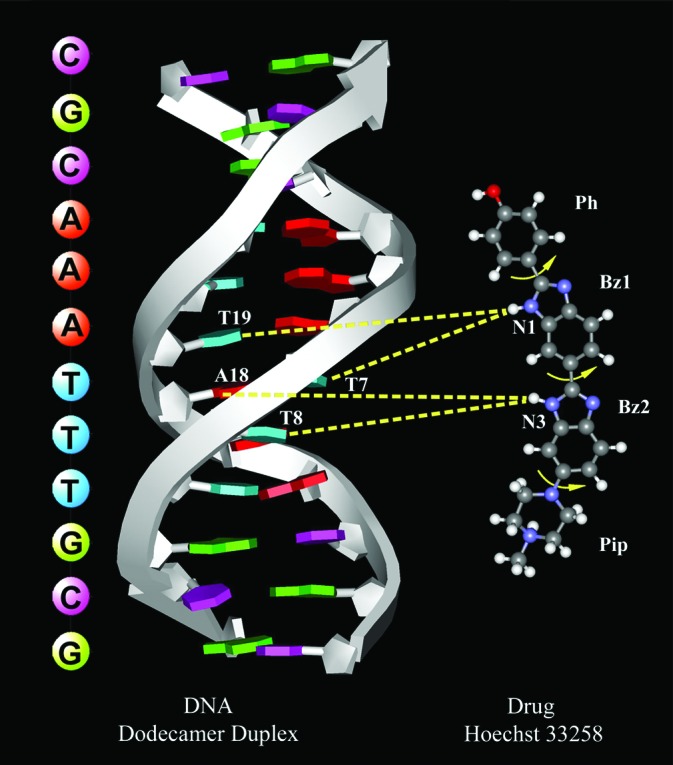
Molecular structures of the drug (Hoechst 33258) and its interaction with the dodecamer DNA. Four moieties are indicated in the drug structure: a piperazine (nonaromatic) ring (Pip), two benzimidazole (aromatic) rings (Bz1 and Bz2), and a phenyl (aromatic) ring (Ph). Possible free rotations about the connecting bonds are indicated with arrows. Two three-centered hydrogen bonds between the drug (two N—H bonds of N1 and N3) and DNA bases are shown in accord with crystal structure (16). At neutral pH the drug molecule (trihydrochloride form) has only one positive charge on the terminal nitrogen atom of the Pip ring (44). In the view presented, the drug is projected out from the minor groove (see Fig. 1); the atom colors for the drug are red for oxygen, white for hydrogen, blue for nitrogen, and gray for carbon.
The DNA–drug complexes were prepared by mixing the drug H33258 (96 μM) with the duplex (144 μM) in the aqueous buffer solutions with continuous stirring for 4 h. The yield of the complexes is >99% (1:1 complex) at our drug concentration, because the equilibrium constant is ≈108 M-1 (24). The procedure for making CT-DNA aqueous solution is similar to that of ref. 20. Solid CT-DNA (1 mg/ml) was dissolved into buffer solutions. The DNA solutions were sonicated for 30 s to reduce the DNA chain length and stirred for 1 h. The ratio of absorption at 260 and 280 nm for the DNA solutions gave a value 1.84, in accord with the limit of 1.8–1.9 for highly purified preparation of DNA (25).
For the dodecamer duplexes, the complexes are 1:1, given the equilibrium constant and the relative concentrations. For CT-DNA, the drug sites are far apart because of the concentration used. The nucleotide concentration was determined by absorption spectroscopy of CT-DNA using the average extinction coefficient per nucleotide of CT-DNA (6,600 M-1·cm-1 at 260 nm) (20) and found to be 2.5 mM, i.e., 1.25 mM for base pairs. A known concentrated drug solution was added dropwise to the CT-DNA solutions with continuous stirring for 2 h to achieve a final concentration of 50 μM for the drug. Accordingly, we estimate that 25 bp of CT-DNA are available for each drug molecule.
Results
Drug in Bulk Water. The steady-state fluorescence spectra of the drug in three neat solvents are presented in Fig. 2 Upper. The spectra show a large red solvatochromic effect. The fluorescence maximum changes from 436 nm in dioxane to 477 nm in ethanol and to 510 nm in aqueous buffer solutions. The effect of DNA binding on the fluorescence intensity and maxima is shown in Fig. 2 Lower.
Fig. 2.

(Upper) Normalized steady-state fluorescence spectra of the drug H33258 in three solvents: dioxane, ethanol, and water. For water, both absorption and emission are shown. The excitation wavelength (350 nm) is indicated by an arrow. (Lower) Fluorescence spectra of DNA–drug complexes in aqueous solutions. For comparison we also include the fluorescence spectrum of the drug H33258 in water (buffer) solution. The three spectra correspond to solutions with the same total concentration of the drug.
In water, the fluorescence is quenched dramatically; the quantum yield is 0.015 in water and increases to 0.5 in ethanol (19). It is known (20, 26) that two major processes are involved in the deactivation of the excited state. At high pH (>7.0) the excited molecule undergoes intramolecular proton transfer from the phenol (see below) to the closest benzimidazole nitrogen, resulting in a keto structure. In polar solvents, the fluorescence intensity decreases and is dominated by emission from the keto form. The deactivation at low pH values (<7.0), and on the complexation with DNA, mainly involves rotation along the bisbenzimide axis (20), similar to other cases involving charge transfer (27). The solvent's pH, polarity, and rigidity determine the wavelength and yield of emission. We have studied the lifetime of the drug in bulk water and found it to be multiple exponentials (ps and ns ranges) and to depend on its concentration. The enhancement of the ps component, characteristic of high pH decay (20) at high concentration, may suggest the presence of ground-state conformations whose equilibrium changes with concentrations and/or aggregation; a subset of those conformations is more poised for twisting/proton transfer. In contrast, in DNA the fluorescence decay is dominated by ns components and does not change with concentration of DNA studied (μM to mM); DNA/drug remains at 1.5.
Femtosecond-resolved fluorescence up-conversion transients of the drug in bulk water (buffer) are presented in Fig. 3 Upper Left for three characteristic wavelengths; we have studied at least 13 of these transients at different wavelengths. The fs transients are typical of those observed for other chromophores in water (22, 23). On the blue edge of the spectrum, the signal is seen to decay (≈1.5 ps), whereas on the red edge it rises on a similar time scale. The signal decays with time constants of ≈40 and 500 ps in the time window studied, up to 200 ps. From the transients it is clear that the contribution of ultrafast hydration is well separated from the nonradiative processes. Note that hydration is essentially independent of details of the solute fluorophore (see, e.g., refs. 21–23 and references therein).
Fig. 3.

(Upper Left) Femtosecond-resolved fluorescence at three characteristics wavelengths for H33258 in water (buffer). The excitation wavelength was 350 nm. (Upper Right) The corresponding normalized TRES. In water, the shift from the initial t = 0 spectrum to 5-ps spectrum completes the process of hydration. The spectrum at equilibrium (steady-state) is shown for comparison. (Lower) Hydration correlation function, C(t), of the drug H33258 in water. (Lower Inset) The time-resolved anisotropy, r(t), of the probe drug in water (buffer) at emission wavelength 510 nm with excitation at 400 nm using single-photon counting. Note the difference in time scales between C(t) and r(t).
From this family of transients we constructed the time-resolved emission spectra (TRES) shown in Fig. 3 Upper Right and the hydration correlation function C(t) given in Fig. 3 Lower. C(t) shows an apparent biexponential decay with time constants of 195 fs (33%) and 1.2 ps (67%). This behavior is typical of hydration of a molecular probe in bulk water (22, 28, 29). The overall spectral shift we observed is 3,184 cm-1; any sub-100-fs component in the dynamics would be unresolved. To complete the picture regarding bulk dynamics of the drug in water and in a less polar solvent (for comparison with DNA environment), we made similar studies of the drug in ethanol (data not shown). In this case, decays are slower than those in water and consistently show the time scale of solvation for ethanol [the average solvation time is 16 ps (30)].
To elucidate the time scale for the rotational diffusion of the drug and its complex we measured the time-resolved anisotropy, r(t), using single-photon counting techniques (Fig. 3 Lower Inset). At t = 0, r(t) ≈0.4 and decays to the base line (properly corrected for polarization/intensities) with τ = 530 ps. According to the Stokes–Einstein–Debye hydrodynamics theory, and assuming a prolate shape for the drug molecule (31, 32), the rotational times in water range from 1 to 5 ns, considering the boundary conditions. Our measured value of 530 ps must include the twisting process, which changes the anisotropy. Note that the r(t) decay in water is on a much longer time scale than that of hydration. Thus rotational diffusion and conformational changes are separable in their time scales from that of hydration. Vibrational relaxation is insignificant given the calculated 0—0 transition in water (316.5 nm) and for the reasons given in ref. 23.
Drug–DNA Dodecamer Complex. Fig. 4 Upper Left shows three characteristic transients obtained from up-conversion experiments on the drug–DNA dodecamer complex in aqueous buffer solutions; other wavelengths are not shown. On the blue edge of the spectrum the signals decay on different time scales depending on wavelengths, whereas on the red edge the signal is seen to rise. The constructed TRES are shown in Fig. 4 Upper Right, indicating that hydration is complete within 100 ps; the spectrum at 100 ps reaches the equilibrium spectrum obtained from steady-state fluorescence.
Fig. 4.
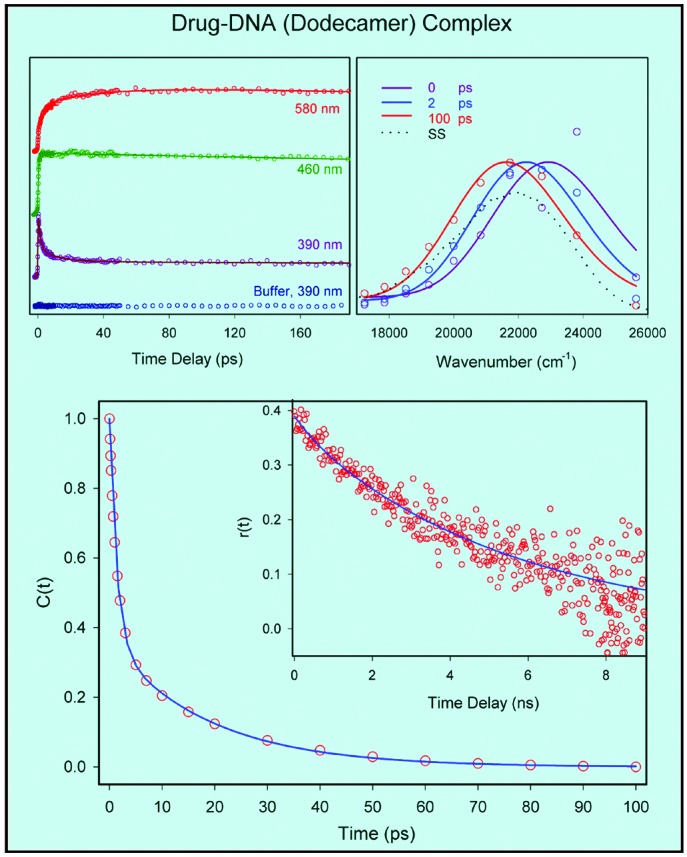
(Upper Left) Femtosecond-resolved fluorescence at three characteristics wavelengths for the drug–dodecamer DNA complex. The excitation wavelength was 350 nm. (Upper Right) The corresponding normalized TRES. The dotted line shows the steady-state emission spectrum of the complex. (Lower) Hydration correlation function, C(t), of the complex. (Lower Inset) The time-resolved anisotropy, r(t), of the probe drug in the complex at emission wavelength 510 nm, with excitation at 400 nm using single-photon counting. Note the difference in time scales between C(t) and r(t).
The C(t) function, as shown in Fig. 4 Lower, is a sum of two exponentials with the time constants of 1.4 ps (64%) and 19 ps (36%); any sub-100-fs components in these dynamics are unresolved. The net spectral shift observed is 1,304 cm-1, consistent with the behavior depicted in Fig. 2. To ascertain the degree of orientational rigidity of the drug in the complex we obtained the fluorescence anisotropy, r(t), decay at 510 nm by using single-photon counting. The r(t) is observed (Fig. 4 Lower Inset) to decay with a time constant of ≈5.5 ns (the estimated hydrodynamic rotational relaxation times of the complex are in the range 10–50 ns). The one order of magnitude lengthening of the drug anisotropy decay in dodecamer, compared with that in the bulk, is consistent with a rigid drug binding in the minor groove.
Drug–CT-DNA Complex. In Fig. 5 Upper Left, we show the up-conversion signals at three characteristic wavelengths; other wavelengths are not shown. The time scales of the decay in the blue edge and corresponding rise in the red edge are similar to those observed in the dodecamer duplex. The constructed TRES in Fig. 5 Upper Right indicate that at 100 ps the equilibrium of the fluorescence, as observed from steady-state emission spectrum, has been reached.
Fig. 5.

(Upper Left) Femtosecond-resolved fluorescence at three characteristics wavelengths for the drug–CT-DNA complex. The excitation wavelength was 350 nm. (Upper Right) The corresponding normalized TRES. The dotted line shows the steady-state emission spectrum of the complex. (Lower) Hydration correlation function, C(t), of the complex. (Lower Inset) The time-resolved anisotropy, r(t), of the probe drug in the complex at emission wave-length 510 nm, with excitation at 400 nm using single-photon counting. Note the difference in time scales between C(t) and r(t).
The C(t) function for the CT-DNA in Fig. 5 Lower can be fitted to a biexponential decay with time constants of 1.1 ps (60%) and 19 ps (40%); any sub-100-fs components in these dynamics are unresolved. The net spectral shift observed is 1,582 cm-1. The time constants along with their contributions are similar to those observed in the dodecamer duplex. But, the fluorescence anisotropy at 510 nm decays with a different time constant: ≈55 ns (major component), compared with that of ≈5.5 ns in the duplex. This finding is consistent with the fact that natural CT-DNA is much longer than the duplex DNA (12 pairs of bases only), but the effect of binding to sites of genomic DNA is another factor to consider.
Discussion
Summarizing our observations we can make the following points: (i) Hydration dynamics in the minor grooves of both types of DNA (dodecamer duplex and calf thymus) are similar but differ substantially from those in bulk water; (ii) for both types of DNA, and within our time resolution, a “bimodal” hydration behavior with two distinct time constants, ≈1 ps (60%) and ≈20 ps (40%), was observed, reflecting the presence of two types of water, bulk-type, labile water and weakly bound, ordered water; and (iii) rotational diffusion of the drug–DNA complexes is much slower than that of the drug in bulk water (530 ps), but is different for the two types of DNA studied; 5.5 ns for dodecamer duplex and 55 ns for calf thymus. These results indicate the rigidity caused by binding in the minor grooves and the disparity in time scales of hydration and rotational diffusion.
The observed bimodality (Fig. 6) in surface hydration of DNA(s) are in fact consistent with our previous reports (21–23) on hydration dynamics at protein surfaces. The DNA hydration time of 19 ps (40%) for the weakly bound water is in line with those observed for the protein surface hydration: Subtilisin Carlsberg (38 ps, 39%), Monellin (16 ps, 54%), and α-chymotrypsin (28 ps, 10%). Relating these times of the correlation functions to residence times in the water layer identifies the effect of first-shell polarization caused by the restricted motions of water molecules by rotational and translational diffusion (33). These residence times are for a layer of nm thickness (22) and for the weakest-bound water because our measurements span the earliest possible (fs) time scale. The persistence of time scales in DNA and proteins is consistent with solvation by water (not internal groups), as discussed elsewhere (23).
Fig. 6.

Comparison of the hydration correlation functions for the drug–DNA (dodecamer and calf thymus) complexes in aqueous buffer solutions. We also include the result for the drug in bulk water (buffer) for comparison. (Inset) The structured water (green balls) with the drug in the minor groove (see Fig. 1 and text), according to x-ray studies (16). The time scales given denote τBW (bulk water), τDOW (dynamically ordered water), τCC (conformational change water), and τSW (structural water); see text.
Considering the equilibrium between water molecules and the DNA sites, a residence time of minor grooves with koff ≈ 5 × 1010 s-1 and diffusion controlled kon ≈ 1010 M-1·s-1 (34) gives Kd ≈ 5 M, so that each accessible site on the average would be in contact with water for >90% of the time; the site has very high occupancy at equilibrium. Dynamically speaking, however, the site is in exchange with bulk, destroying the order on the ps time scale, and this is the origin of bimodality (22, 33).
The residence time for ordered water relative to other time constants of DNA is of significance to the stability and recognition. First, we must consider the time scale of making and breaking bonds of the dynamically ordered water, τDOW, relative to that of structural conformational changes, τCC, by bending and twisting (ref. 35 and references therein). The value of τCC is important to, for example, the change of the B form of DNA to A and Z forms whose relative stabilities depend on the water content and sequence; B form predominates in aqueous solution. If τDOW is shorter than τCC then recognition is an effective process with structural integrity. The loss of order on the ps time scale is significant in increasing the entropy and it is possible that this contribution to the free energy is governed by the change in the rotations of water molecules. It is interesting that for this drug and DNA sequence, the entropic contribution is indeed dominant (36). Repeating these experiments for different drugs or sequences should correlate the thermodynamics (37–39) with the dynamics.
Second, it is important to compare the residence time of weakly bound water, τDOW, with the time of breaking/making hydrogen bonds, τHB, in bulk water. With a few kcal/mol barrier, kinetically τHB is on the order of a few picoseconds, and for an effective recognition, τDOW should not be orders of magnitude longer than the value of τHB, so that the efficiency becomes optimum. If τDOW/τHB ≈ 1, then the degree of order is that of the bulk. Lastly, the time scale for the motion of the drug in the groove, by orientational diffusion, τOD, relative to that of τDOW. For the drug studied here, τOD is much longer than τDOW for both types of DNA, as evidenced by the anisotropy, ensuring a well defined geometry, certainly on the time scale of dynamically ordered water.
The robustness of the range of values for the hydration times in DNA (and proteins) is indicative of the nature of the layer, being ordered on the molecular scale even in the presence of the drug. This picture is consistent with the results of NMR study (34) on the hydration and solution structure of the duplex sequence we have used and its complex with a minor groove binding drug, propamidine. It was found that complexation with the drug has little effect on the residence times for water molecules bound either in the major groove or at the sites in the minor groove. The range of residence times was found to be ≈0.2–0.4 ns for surface water at grooves; the residence times of water molecules in the major groove are an order of magnitude shorter than for the most long-lived waters in the minor groove.
From the above discussion two points should be emphasized. Our above-mentioned results of hydration dynamics in the two DNA systems studied were obtained by using the time window from zero and up to 200 ps. Longer-time Stokes shifts may be present, reflecting the influence of more rigid water structure on the time scale indicated by NMR studies (4, 40). However, the resemblance of the evolving spectra to the maximum of steadystate fluorescence spectrum in 100 ps [νmax(∞)] indicates that most of the dynamics are complete within our time window, but a fraction of strongly bound water may still be present with a time scale of sub-ns or longer [we measured the lifetime of the drug in the dodecamer and found it to be ≈1.5 and 4 ns, consistent with the values in the literature (20)]. We note that the ns decays are similar for all DNA concentrations studied, which indicates that the dodecamer remains as a double strand, and not as a hairpin that is possible for central AT and terminal GC bases of a single strand; NMR confirms the double-strand structure at 1.1 mM (34).
Because the ordered water is probed here around the drug in the grooves, one must not ignore such water in the study of recognition processes and drug design strategies (36). In fact, studies using densimetric and ultrasonic measurements have shown that a minor groove binding drug, netropsin, displaces, depending on the base sequence, a significant number of water molecules on complexation with a DNA duplex (10). We note that thermodynamic measurements of release of water is concerned with essentially all waters occupied in the grooves whereas the dynamics experiments can detect only a small subpopulation of a certain net of interfacial water molecules that contribute to ligand binding. The dynamics of the ordered water and its loss by, for example, rotational diffusion contribute to the entropic process involved.
From a structural point of view, the drug studied here (Scheme 1) binds in the minor groove covering the sequence AATTT of the central A tract, with the piperazine group close to one of the GC region. It makes two three-centered hydrogen bonds from the nitrogen atoms of benzimidazole rings to the N (A18) and O (T7, T8, T19) atoms of the DNA bases. This hydrogen bonding (and electrostatic/dispersion interactions) is facilitated by the presence of ordered water (entropic) around the drug; if ordered water is involved in direct binding of the drug then enthalpic contributions must be included. Hydrogen bonding is also possible for the drug (Scheme 1; nitrogens opposite to N1 and N3) with water near the surface of the groove, which is more of a bulk type in our bimodal distribution of hydration. The most weakly bound water molecules are of critical importance to biological function (41) and are unlikely to be seen in crystal structures or by NMR (41). They are part of total hydration, which influences structural and biological activities (42, 43). The residence times of weakly bound water are only an order of magnitude different from that of bulk, and they are the ones that have to be probed with femtosecond resolution.
In conclusion, the study presented here characterizes, with femtosecond resolution, the dynamics of hydration/dehydration at the DNA surface of known local structure and with a drug in the minor groove. The fact that the water is dynamically ordered at the surface of DNA without spatial averaging or position inhomogeneity of the drug (known structure) allow us to observe the earliest processes of hydration dynamics. Recognition of minor grooves by charge and shape complementarities, and using directional hydrogen bonds, cannot be fully understood from a static structure without including the role of water and the time scale for the loss of the order. It may turn out that this dynamically ordered water is also crucial for interfacial recognition, not only of drugs but also between macromolecules.
Acknowledgments
We thank Drs. Jorge Peon and Spencer Baskin for helpful discussion and Profs. Kenneth J. Breslauer and Peter B. Dervan for careful reading of the manuscript and helpful suggestions. This work was supported by the National Science Foundation.
Abbreviations: CT-DNA, calf thymus DNA; TRES, time-resolved emission spectra.
References
- 1.Dervan, P. B. (2001) Bioorg. Med. Chem. 9 2215-2235. [DOI] [PubMed] [Google Scholar]
- 2.Feig, M. & Pettitt, B. M. (1998) Biopolymers 48 199-209. [DOI] [PubMed] [Google Scholar]
- 3.Schneider, B., Cohen, D. & Berman, H. M. (1992) Biopolymers 32 725-750. [DOI] [PubMed] [Google Scholar]
- 4.Halle, B. & Denisov, V. P. (1998) Biopolymers 48 210-233. [DOI] [PubMed] [Google Scholar]
- 5.Umehara, T., Kuwabara, S., Mashimo, S. & Yagihara, S. (1990) Biopolymers 30 649-656. [DOI] [PubMed] [Google Scholar]
- 6.Duan, Y., Wilkosz, P., Crowley, M. & Rosenberg, J. M. (1997) J. Mol. Biol. 272 553-572. [DOI] [PubMed] [Google Scholar]
- 7.Drew, H. R. & Dickerson, R. E. (1981) J. Mol. Biol. 151 535-556. [DOI] [PubMed] [Google Scholar]
- 8.Leipinsh, E., Otting, G. & Wuthrich, K. (1992) Nucleic Acids Res. 20 6549-6553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chuprina, V. P. (1987) Nucleic Acids Res. 15 293-311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chalikian, T. V., Plum, G. E., Sarvazyan, A. P. & Breslauer, K. J. (1994) Biochemistry 33 8629-8640. [DOI] [PubMed] [Google Scholar]
- 11.Chalikian, T. V., Volker, J., Srinivasan, A. R., Olson, W. K. & Breslauer, K. J. (1999) Biopolymers 50 459-471. [DOI] [PubMed] [Google Scholar]
- 12.Brauns, E. B., Madaras, M. L., Coleman, R. S., Murphy, C. J. & Berg, M. A. (1999) J. Am. Chem. Soc. 121 11644-11649. [Google Scholar]
- 13.Hess, S., Davis, W. B., Voityuk, A. A., Rosch, N., Michel-Beyerle, M. E., Ernsting, N. P., Kovalenko, S. A. & Lustres, J. L. P. (2002) Chemphyschem. 3 452-455. [DOI] [PubMed] [Google Scholar]
- 14.Pal, S. K., Peon, J. & Zewail, A. H. (2002) Chem. Phys. Lett. 363 57-63. [Google Scholar]
- 15.Edwards, K. J., Brown, D. G., Spink, N., Skelly, J. V. & Neidle, S. (1992) J. Mol. Biol. 226 1161-1173. [DOI] [PubMed] [Google Scholar]
- 16.Vega, M. C., Saez, I. G., Aymami, J., Eritja, R., Marel, G. A. V. D., Boom, J. H. V., Rich, A. & Coll, M. (1994) Eur. J. Biochem. 222 721-726. [DOI] [PubMed] [Google Scholar]
- 17.Denham, D. A., Suswillo, R. R., Rogers, R., McGreevy, P. B. & Andrew, B. J. (1976) J. Helminthol. 50 243-250. [DOI] [PubMed] [Google Scholar]
- 18.Latt, S. A. & Stetten, G. (1976) J. Histochem. Cytochem. 24 24-33. [DOI] [PubMed] [Google Scholar]
- 19.Gorner, H. (2001) Photochem. Photobiol. 73 339-348. [DOI] [PubMed] [Google Scholar]
- 20.Cosa, G., Focsaneanu, K.-S., McLean, J. R. N., McNamee, J. P. & Scaiano, J. C. (2001) Photochem. Photobiol. 73 585-599. [DOI] [PubMed] [Google Scholar]
- 21.Peon, J., Pal, S. K. & Zewail, A. H. (2002) Proc. Natl. Acad. Sci. USA 99 10964-10969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pal, S. K., Peon, J. & Zewail, A. H. (2002) Proc. Natl. Acad. Sci. USA 99 1763-1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pal, S. K., Peon, J. & Zewail, A. H. (2002) Proc. Natl. Acad. Sci. USA 99 15297-15302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Haq, I., Ladbury, J. E., Chowdhry, B. Z., Jenkins, T. C. & Chaires, J. B. (1997) J. Mol. Biol. 271 244-257. [DOI] [PubMed] [Google Scholar]
- 25.Gallagher, S. R. (1994) in Current Protocols in Molecular Biology, eds. Ausubel, F. M., Brent, R., Kingston, K. E., Moore, D. D., Seidman, J. G., Smith, J. A. & Struhl, K. (Greene and Wiley-Interscience, New York), Suppl. 28, Appendix 3D.
- 26.Kalninsh, K. K. & Pestov, D. V. (1994) J. Photochem. Photobiol. A 83 39-47. [Google Scholar]
- 27.Zhong, D., Pal, S. K. & Zewail, A. H. (2001) Chemphyschem. 2 219-227. [DOI] [PubMed] [Google Scholar]
- 28.Jarzeba, W., Walker, G., Johnson, A. E., Kahlow, M. A. & Barbara, P. F. (1988) J. Phys. Chem. 92 7039-7041. [Google Scholar]
- 29.Jimenez, R., Fleming, G. R., Kumar, P. V. & Maroncelli, M. (1994) Nature 369 471-473. [Google Scholar]
- 30.Horng, M. L., Gardecki, J. A., Papazyan, A. & Maroncelli, M. (1995) J. Phys. Chem. 99 17311-17337. [Google Scholar]
- 31.Hu, C. M. & Zwanzig, R. (1974) J. Chem. Phys. 60 4354-4357. [Google Scholar]
- 32.Baskin, J. S. & Zewail, A. H. (2001) J. Phys. Chem. A 105 3680-3692. [Google Scholar]
- 33.Pal, S. K., Peon, J., Bagchi, B. & Zewail, A. H. (2002) J. Phys. Chem. B 106 12376-12395. [Google Scholar]
- 34.Lane, A. N., Jenkins, T. C. & Frenkiel, T. A. (1997) Biochim. Biophys. Acta 1350 205-220. [DOI] [PubMed] [Google Scholar]
- 35.Millar, D. P., Robbins, R. J. & Zewail, A. H. (1982) J. Chem. Phys. 76 2080-2094. [Google Scholar]
- 36.Haq, I. (2002) Arch. Biochem. Biophys. 403 1-15. [DOI] [PubMed] [Google Scholar]
- 37.Jin, R. & Breslauer, K. J. (1988) Proc. Natl. Acad. Sci. USA 85 8939-8942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Breslauer, K. J., Remeta, D. P., Chou, W., Ferrante, R., Curry, J., Zaunczkowski, D., Snyder, J. G. & Marky, L. A. (1987) Proc. Natl. Acad. Sci. USA 84 8922-8926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marky, L. A. & Breslauer, K. J. (1987) Proc. Natl. Acad. Sci. USA 84 4359-4363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Denisov, V. P., Carlstrom, G., Venu, K. & Halle, B. (1997) J. Mol. Biol. 268 118-136. [DOI] [PubMed] [Google Scholar]
- 41.Rand, R. P. (1992) Science 256 618-618. [DOI] [PubMed] [Google Scholar]
- 42.Mrevlishvili, G. M., Carvalho, A. P. S. M. C., da Silva, M. A. V. R., Mdzinarashvili, T. D., Razmadze, G. Z. & Tarielashvili, T. Z. (2001) J. Therm. Anal. Cal. 66 133-144. [Google Scholar]
- 43.Robinson, C. R. & Sligar, G. (1993) J. Mol. Biol. 234 302-306. [DOI] [PubMed] [Google Scholar]
- 44.Teng, M., Usman, N., Frederick, C. A. & Wang, A. H. J. (1988) Nucleic Acids Res. 16 2671-2690. [DOI] [PMC free article] [PubMed] [Google Scholar]