

Abstract
The outer leaflet of the outer membrane of the Gram-negative bacterium serves as a permeability barrier and is composed of lipopolysaccharide, also known as endotoxin. The membrane anchor of lipopolysaccharide is lipid A, the biosynthesis of which is essential for cell viability. The first committed step in lipid A biosynthesis is catalyzed by UDP-(3-O-(R-3-hydroxymyristoyl))-N-acetylglucosamine deacetylase (LpxC), a zinc-dependent deacetylase. Here we report the crystal structure of LpxC from Aquifex aeolicus, which reveals a new α+β fold reflecting primordial gene duplication and fusion, as well as a new zinc-binding motif. The catalytic zinc ion resides at the base of an active-site cleft and adjacent to a hydrophobic tunnel occupied by a fatty acid. This tunnel accounts for the specificity of LpxC toward substrates and inhibitors bearing appropriately positioned 3-O-fatty acid substituents. Notably, simple inhibitors designed to target interactions in the hydrophobic tunnel bind with micromolar affinity, thereby representing a step toward the structure-based design of a potent, broad-spectrum antibacterial drug.
Septic shock is the most common cause of death in hospital intensive care units and results from the severe hypotension and multiple organ failure that often accompany sepsis. Approximately 400,000 cases of sepsis and 200,000 cases of septic shock are diagnosed annually in the United States, causing an estimated 100,000 deaths per year (1, 2). Gram-negative bacterial sepsis arises from the systemic response to infection, mainly the overexpression of cytokines and inflammatory mediators in response to macrophage activation by endotoxin (also known as lipopolysaccharide or LPS) (3–8). Approximately 2 × 106 LPS molecules assemble to form the outer leaflet of the outer membrane of the Gram-negative bacterium (3, 7), thereby serving as a permeability barrier that protects the bacterium from many antibiotics, such as erythromycin (9–11). Accordingly, the LPS barrier is a daunting obstacle confronting the research and development of safe and effective antibiotics targeting Gram-negative bacteria.
Each LPS molecule contains three main components: an immunodominant and highly variable repeating oligosaccharide known as the O-antigen, a core polysaccharide, and lipid A (refs. 3–8 and Fig. 1). The hydrophobic anchor of LPS is lipid A, a phosphorylated, β(1→6)-linked glucosamine disaccharide hexaacylated with N-linked and O-linked fatty acids. Lipid A is essential for LPS assembly in the outer membrane and consequently the viability of the Gram-negative bacterium. Bacterial strains containing defects in lipid A biosynthesis are remarkably hypersensitive to antibiotics (9–11). Given that lipid A is the toxic component of LPS and is essential for bacterial survival (3–8), inhibitors of enzymes in the lipid A biosynthetic pathway may comprise antibacterial agents that target Gram-negative bacteria and manage Gram-negative sepsis more effectively.
Fig. 1.

The reaction catalyzed by LpxC is the first committed step of lipid A biosynthesis. Lipid A is the hydrophobic anchor of LPS, also known as endotoxin, which forms the outer leaflet of the outer membrane of a Gram-negative bacterium.
UDP-(3-O-(R-3-hydroxymyristoyl))-N-acetylglucosamine deacetylase (LpxC) catalyzes the first committed step of lipid A biosynthesis (refs. 12–15; Fig. 1) and is essential for bacterial growth and virulence (16). Although LpxC is a zinc metalloenzyme (17), the lack of characteristic zinc-binding motifs (18) in its amino acid sequence suggests that this enzyme represents a class of zinc-dependent deacetylases (17). Recent mutagenesis studies (19) with LpxC enzymes from Escherichia coli and Aquifex aeolicus indicate residues important for catalysis and zinc binding. To date, inhibitors developed against LpxC enzymes from different Gram-negative bacteria contain hydroxamate or phosphonate zinc-binding motifs and some exhibit potent antibacterial properties (20–25). LpxC is thus validated as a target for the development of antibacterial agents selective against Gram-negative bacteria. To accelerate the structure-based design of antibiotics, we now report the three-dimensional structure of recombinant LpxC from A. aeolicus (24), and we report a series of hydrophobic compounds that bind to the enzyme with micromolar affinity.
Materials and Methods
Expression and Purification. Initial crystallization experiments with wild-type LpxC did not yield good results, so a variant was designed for subsequent crystallization trials. Recombinant C193A/ΔD284-L294 LpxC was prepared from the plasmid pAaLpxC (pET21a) containing the cloned lpxC gene from A. aeolicus (24) as detailed in Supporting Text, which is published as supporting information on the PNAS web site, www.pnas.org [the numbering convention used for the E. coli enzyme is adapted for the A. aeolicus enzyme (19)]. The plasmid was transformed into E. coli strain BL21(DE3)pLysS (Novagen) for overexpression and cultures were grown by following established protocols (19). Enzyme was purified as described (19) with the addition of a Superdex 200 step before concentration to 2.2 mg/ml; the activity of this variant is comparable to that of the wild-type enzyme measured under standard conditions (unpublished results).
Crystallization and Structure Determination. For crystallization at 21°C, a sitting drop containing 5.0 μl of protein solution [2.2 mg/ml LpxC, 25 mM Hepes (pH 7.0), 50 mM NaCl, 10 mM magnesium acetate, and 0.5 mM ZnSO4] was equilibrated against a 500-μl reservoir of 0.8 M NaCl/0.1 M Hepes (pH 7.0). Crystals of dimensions 0.3 × 0.1 × 0.05 mm3 appeared in 5–7 days; larger crystals of dimensions 0.6 × 0.2 × 0.2 mm3 were obtained by macroseeding. Crystals diffracted X-rays to 2.0-Å resolution and belonged to space group P61 with unit cell dimensions a = b = 101.66 Å, c = 125.10 Å. With two molecules in the asymmetric unit, VM = 3.0 Å3/Da (59% solvent content).
The structure of LpxC was solved by multiwavelength anomalous dispersion (MAD) (26). X-ray diffraction data were collected at beamline F-2 of the Cornell High Energy Synchrotron Source (Ithaca, NY) at wavelengths corresponding to the absorption peak and edge of zinc as well as a remote reference wavelength. Seven zinc sites were located in the asymmetric unit containing two monomers by using software routines implemented in CNS and initial 2.0-Å phases were improved by solvent flipping (27). Electron density maps were fit by using O (28) and the structure was refined by using CNS (27). Strict noncrystallographic symmetry constraints were used initially and relaxed into appropriately weighted restraints as guided by Rfree. Zinc ion sites were confirmed by their anomalous signals, and water molecules were added sequentially to electron density peaks ≥3σ that had appropriate hydrogen bonding partners. The fatty acid in the hydrophobic tunnel was interpreted as myristic acid (C14) and built into the electron density map at the end of refinement. Alternatively, this fatty acid could have been interpreted as a disordered palmitic acid (C16); MS analysis actually suggests a mixture of both myristic and palmitic acids (data not shown). Three residues at the C terminus of each monomer are disordered (L281-R283). The final model has excellent geometry with 85.3% and 14.3% of residues adopting most favored and additionally allowed conformations, respectively. Data collection and refinement statistics appear in Table 1. Atomic coordinates and structure factor amplitudes have been deposited in the Protein Data Bank with accession code 1P42.
Table 1. Data collection, phasing, and refinement statistics.
| Remote | Edge | Peak | |
|---|---|---|---|
| Data collection and phasing | |||
| Wavelength, Å | 1.2565 | 1.2832 | 1.2825 |
| Resolution, Å | 2.0 | 2.0 | 2.0 |
| No. of total reflections | 497,657 | 364,430 | 299,731 |
| No. of unique reflections* | 97,852 | 97,565 | 96,091 |
| Completeness, % | |||
| Overall | 100.0 | 99.9 | 98.5 |
| Outer 0.1-Å shell | 100.0 | 99.9 | 93.1 |
| Rmerge† | |||
| Overall | 0.059 | 0.060 | 0.055 |
| Outer 0.1-Å shell | 0.329 | 0.342 | 0.269 |
| Mean figure of merit for MAD phasing‡ | 0.633 | ||
| Refinement statistics | |||
| No. of reflections, working set/testing set | 92,811/4,815 | ||
| Data cutoff, σ | 0.0 | ||
| R/Rfree§ | 0.199/0.211 | ||
| No. of protein atoms | 4,316 | ||
| No. of zinc ions | 7 | ||
| No. of ligand atoms | 32 | ||
| No. of water molecules | 309 | ||
| rms deviations | |||
| Bond lengths, Å | 0.006 | ||
| Bond angles, ° | 1.3 | ||
| Proper dihedral angles, ° | 23.5 | ||
| Improper dihedral angles, ° | 0.7 |
Friedel mates are treated as unique reflections for data reduction in MAD experiments
Rmerge = 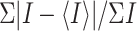 ,
where I is the observed intensity and
,
where I is the observed intensity and 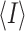 is the
average intensity calculated for replicate data
is the
average intensity calculated for replicate data
Mean figure of merit =
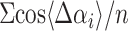 , where
Δαi is the error in the phase angle for
reflection i, and n is the number of reflections
, where
Δαi is the error in the phase angle for
reflection i, and n is the number of reflections
R =
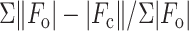 ,
where
,
where 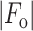 and
and 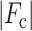 are
the observed and calculated structure factor amplitudes, respectively.
Rfree is calculated in the same manner for reflections in
the test set excluded from refinement
are
the observed and calculated structure factor amplitudes, respectively.
Rfree is calculated in the same manner for reflections in
the test set excluded from refinement
Inhibitor Affinity. Inhibitor binding was assayed by isothermal titration calorimetry (29). Inhibitors were either synthesized as described in Supporting Text or purchased from Sigma-Aldrich. Experiments were performed at 30°C on an isothermal microcalorimeter from Microcal (Northampton, MA). A. aeolicus LpxC was stripped of all metal ions by dialysis against 1.0 mM EDTA in 25 mM Hepes (pH 7.0)/0.1 M NaCl at room temperature for ≥4 h. The EDTA was then removed by extensive dialysis against EDTA-free buffer and the enzyme was reconstituted to a 1:1 Zn2+:LpxC ratio by the addition of ZnSO4. A colorimetric assay employing 4-(2-pyridylazo)-resorcinol (PAR) was used to determine Zn2+ concentrations (17) and verify the preparation of apo and 1:1-reconstituted LpxC. The calorimeter cell contained either ≈40 or ≈60 μM enzyme, and the syringe contained 250 or 400 μM aliphatic compound. A series of 30 injections (8-μl each) were performed at 180-sec intervals. Titrations of aliphatic compounds into buffer were also performed as control experiments by using identical conditions. Data were fit to a single binding-site model by using ORIGIN V. 2.9 (Microcal). A representative titration curve can be seen in Fig. 6, which is published as supporting information on the PNAS web site. In cases where DMSO was necessary as a carrier solvent to facilitate solubilization of the aliphatic compound of interest, equal amounts of DMSO (volume percent) were included in the protein solution. In no case did the concentration of DMSO exceed 1.3% (vol/vol) of the solution. The following compounds were insufficiently soluble for study: myristic acid (C14), dodecylamine, dodecanal, dodecanethiol, dodecanesulfonamide, and dodecaneboronic acid.
Results and Discussion
Structure and Mechanism. Crystals of LpxC were grown by vapor diffusion in sitting drops and diffracted x-rays to 2.0-Å resolution. The crystal structure was solved using the anomalous dispersion of zinc. We suspected that the anomalous scattering of a single zinc ion bound to a polypeptide chain of 271 residues would be insufficient for the calculation of MAD phases. Therefore, we exploited the fact that LpxC, like many zinc proteases, is inhibited by excess zinc (17). We expected to find that the preparation of LpxC crystals in the presence of millimolar concentrations of Zn2+ would lead to the binding of additional zinc ions, which in turn would facilitate MAD phasing. This strategy proved highly effective, because a total of seven zinc ions bound to two LpxC monomers in the asymmetric unit.
The overall fold of LpxC belongs to the α+β class and its topology (Fig. 2A) is previously unobserved based on a DALI search (30). The tertiary structure is formed by two domains connected by a 16-residue linker; each domain consists of a five-stranded β-sheet and two principal α-helices, and the two domains assemble so that the β-sheets sandwich the α-helices (Fig. 2B). Each domain has identical topology of secondary structural elements despite insignificant amino acid sequence identity, indicative of primordial gene duplication, fusion, and evolutionary divergence. The active site is located at the interface of the two domains and is also flanked by two smaller subdomains: a βββ subdomain inserted between β-strand 4 and helix A, and a βαβ subdomain inserted between β-strand 4′ and helix A′.
Fig. 2.

(A) Topology diagram of LpxC. Locations of catalytic zinc
(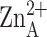 ) ligands on helices A
and A′ are indicated by filled circles. (B) Overall fold of
LpxC: blue → red correlates with N terminus → C terminus. Secondary
structural elements are labeled as in A, and zinc ions appear as gray
spheres.
) ligands on helices A
and A′ are indicated by filled circles. (B) Overall fold of
LpxC: blue → red correlates with N terminus → C terminus. Secondary
structural elements are labeled as in A, and zinc ions appear as gray
spheres. 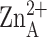 is the catalytic
zinc ion and
is the catalytic
zinc ion and 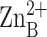 is the
inhibitory zinc ion.
is the
inhibitory zinc ion. 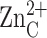 cross-links H62 and H200, thereby stabilizing the βαβ
subdomain (β10-helix C-β11), and
cross-links H62 and H200, thereby stabilizing the βαβ
subdomain (β10-helix C-β11), and
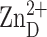 makes an interlattice
contact. Myristic acid (black) extends through the βαβ
subdomain and coordinates to
makes an interlattice
contact. Myristic acid (black) extends through the βαβ
subdomain and coordinates to
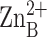 .(C) Active-site
cleft of LpxC; for clarity, only
.(C) Active-site
cleft of LpxC; for clarity, only
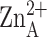 and
and
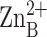 are shown, and the
zinc-bound solvent molecule appears as a partially occluded red sphere. Note
the hydrophobic tunnel adjacent to
are shown, and the
zinc-bound solvent molecule appears as a partially occluded red sphere. Note
the hydrophobic tunnel adjacent to
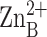 , which is framed by the
βαβ subdomain and occupied by myristic acid (green).
(D) Model of the LpxC-substrate complex. The nucleophilic zinc-bound
solvent molecule appears as a red sphere.
, which is framed by the
βαβ subdomain and occupied by myristic acid (green).
(D) Model of the LpxC-substrate complex. The nucleophilic zinc-bound
solvent molecule appears as a red sphere.
The van der Waals surface of the enzyme reveals an ≈20-Å-deep,
conical active-site cleft at the bottom of which is a binuclear zinc cluster
in the zinc-inhibited enzyme (Fig.
2C). Unexpectedly, a separate ≈15-Å-long
hydrophobic tunnel also leads to the zinc cluster
(Fig. 2C). This
hydrophobic tunnel is framed by the βαβ subdomain and is lined
by several conserved aliphatic residues. Surprisingly, a saturated fatty acid
molecule interpreted as myristic acid occupies this tunnel and its carboxylate
group coordinates to the inhibitory zinc ion. The
3-O-(R-3-hydroxymyristoyl) substituent of the substrate
presumably binds in this tunnel during catalysis. Previous biochemical studies
(17) of LpxC from E.
coli indicate that this substituent substantially affects binding and
catalysis: the kcat/KM for
deacetylation of UDP-N-acetylglucosamine (which lacks the
3-O substituent) catalyzed by the E. coli enzyme is
diminished 5 × 106-fold due in part to a 104-fold
increase in the KM value. Moreover, there is no detectable
deacetylation of UDP-N-acetylglucosamine by the A. aeolicus
enzyme (unpublished results). Given that enzyme-inhibitor affinity also
depends on the presence and length of an O3 fatty acid substituent
(24), and given that the O3
fatty acid substituent of the substrate analogue inhibitor TU-514
(24) binds in the hydrophobic
tunnel after 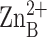 is
dissociated (unpublished results), we conclude that the
3-O-(R-3-hydroxymyristoyl) substituent of the substrate
makes key interactions within the hydrophobic tunnel required for optimal
binding and catalysis, e.g., by stabilizing the conformation of the
βαβ subdomain that forms one wall of the active-site cleft. A
model of the LpxC-substrate complex is found in
Fig. 2D. Because
UDP-N-acetylglucosamine is situated at a biosynthetic branchpoint
leading to either lipid A or peptidoglycan formation
(14), the lack of significant
LpxC activity against a substrate lacking an O3 fatty acid substituent
preserves cellular pools of UDP-N-acetylglucosamine for peptidoglycan
biosynthesis.
is
dissociated (unpublished results), we conclude that the
3-O-(R-3-hydroxymyristoyl) substituent of the substrate
makes key interactions within the hydrophobic tunnel required for optimal
binding and catalysis, e.g., by stabilizing the conformation of the
βαβ subdomain that forms one wall of the active-site cleft. A
model of the LpxC-substrate complex is found in
Fig. 2D. Because
UDP-N-acetylglucosamine is situated at a biosynthetic branchpoint
leading to either lipid A or peptidoglycan formation
(14), the lack of significant
LpxC activity against a substrate lacking an O3 fatty acid substituent
preserves cellular pools of UDP-N-acetylglucosamine for peptidoglycan
biosynthesis.
The binuclear zinc cluster of zinc-inhibited LpxC
(Fig. 3) is comparable to
clusters observed in the active sites of zinc proteases similarly inhibited by
excess zinc (31,
32). The catalytic
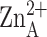 ion is more deeply
situated in the active site than the inhibitory
ion is more deeply
situated in the active site than the inhibitory
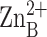 ion and is coordinated by
H79 of helix A, H238 and D242 of helix A′, and a solvent molecule with
tetrahedral geometry. Notably, the direct zinc ligands are located on
topologically equivalent α-helices at the domain–domain interface.
Also notable is the fact that helix A only contributes one ligand; typically,
the first and second zinc ligands in the amino acid sequences of zinc enzymes
are separated only by a short polypeptide segment of 1–3 residues
(18). Histidine ligands to the
catalytic zinc ion were correctly identified in recent mutagenesis studies
(19). The
HKX(L,F)D sequence motif is found in all currently available
LpxC sequences (boldface, direct zinc ligands; X, aliphatic residues M, I, L,
V, T, and A). We conclude that this sequence represents an alternate
zinc-recognition motif.
ion and is coordinated by
H79 of helix A, H238 and D242 of helix A′, and a solvent molecule with
tetrahedral geometry. Notably, the direct zinc ligands are located on
topologically equivalent α-helices at the domain–domain interface.
Also notable is the fact that helix A only contributes one ligand; typically,
the first and second zinc ligands in the amino acid sequences of zinc enzymes
are separated only by a short polypeptide segment of 1–3 residues
(18). Histidine ligands to the
catalytic zinc ion were correctly identified in recent mutagenesis studies
(19). The
HKX(L,F)D sequence motif is found in all currently available
LpxC sequences (boldface, direct zinc ligands; X, aliphatic residues M, I, L,
V, T, and A). We conclude that this sequence represents an alternate
zinc-recognition motif.
Fig. 3.

Binuclear metal cluster in zinc-inhibited LpxC
(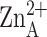 -
-
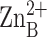 separation = 3.4
Å). (A) Omit electron density maps in which zinc ions (magenta,
contoured at 15σ) or zinc ligands (green, contoured at 4σ) were
omitted from the structure factor calculation. (B) Metal-coordination
interactions, measured in angstroms. Because the positions of nucleophilic
zinc-bound solvent molecules remain nearly unchanged on the binding of
inhibitory zinc ions to the zinc proteases carboxypeptidase A and thermolysin
(31,
32), the position of the
metal-bridging solvent molecule (red sphere) observed in zinc-inhibited LpxC
likely corresponds to the position of the nucleophilic
separation = 3.4
Å). (A) Omit electron density maps in which zinc ions (magenta,
contoured at 15σ) or zinc ligands (green, contoured at 4σ) were
omitted from the structure factor calculation. (B) Metal-coordination
interactions, measured in angstroms. Because the positions of nucleophilic
zinc-bound solvent molecules remain nearly unchanged on the binding of
inhibitory zinc ions to the zinc proteases carboxypeptidase A and thermolysin
(31,
32), the position of the
metal-bridging solvent molecule (red sphere) observed in zinc-inhibited LpxC
likely corresponds to the position of the nucleophilic
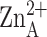 -bound solvent molecule in
the active enzyme.
-bound solvent molecule in
the active enzyme.
Site-directed mutagenesis studies of LpxC from E. coli and A.
aeolicus indicate that invariant residues E78 and H265 are important for
catalysis; moreover, the decreased susceptibility of E78 variants to
inhibition by zinc suggests that E78 coordinates to an inhibitory zinc ion
(19). The crystal structure
confirms that E78, H265, a solvent molecule, and the carboxylate of myristic
acid coordinate to inhibitory
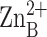 with tetrahedral geometry
(Fig. 3). X-ray crystal
structures of the zinc proteases thermolysin and carboxypeptidase A reveal
that inhibitory zinc ions interact with conserved glutamate residues E166 and
E270, respectively (31,
32). These residues serve as
general bases in the corresponding peptidase reactions
(33,
34), and by analogy we propose
that E78 of LpxC serves as a general base in the deacetylase reaction
(Fig. 4), as considered by
Jackman et al. (19).
In thermolysin, the inhibitory zinc ion is also liganded by Y157 and H231
(31) and these residues serve
as electrostatic catalysts to stabilize the negative charge of the tetrahedral
intermediate and its flanking transition states
(33). By analogy, we propose
that H265 of LpxC similarly serves as an electrostatic catalyst. The
imidazolium side chain of H265 donates a hydrogen bond to the invariant and
essential (19) carboxylate
side chain of D246; this interaction may ensure that H265 is properly
positioned and positively charged for effective electrostatic catalysis.
Alternatively, H265 could serve as a proton donor to the leaving amino group
in catalysis, but it appears that E78, after protonation in the first step of
catalysis, is better positioned for this role.
with tetrahedral geometry
(Fig. 3). X-ray crystal
structures of the zinc proteases thermolysin and carboxypeptidase A reveal
that inhibitory zinc ions interact with conserved glutamate residues E166 and
E270, respectively (31,
32). These residues serve as
general bases in the corresponding peptidase reactions
(33,
34), and by analogy we propose
that E78 of LpxC serves as a general base in the deacetylase reaction
(Fig. 4), as considered by
Jackman et al. (19).
In thermolysin, the inhibitory zinc ion is also liganded by Y157 and H231
(31) and these residues serve
as electrostatic catalysts to stabilize the negative charge of the tetrahedral
intermediate and its flanking transition states
(33). By analogy, we propose
that H265 of LpxC similarly serves as an electrostatic catalyst. The
imidazolium side chain of H265 donates a hydrogen bond to the invariant and
essential (19) carboxylate
side chain of D246; this interaction may ensure that H265 is properly
positioned and positively charged for effective electrostatic catalysis.
Alternatively, H265 could serve as a proton donor to the leaving amino group
in catalysis, but it appears that E78, after protonation in the first step of
catalysis, is better positioned for this role.
Fig. 4.

Proposed mechanism for LpxC. The catalytic zinc ion polarizes a bound solvent molecule, thereby assisting general base E78 in promoting the nucleophilic attack of a water molecule at the scissile amide linkage of the substrate, and it subsequently stabilizes the oxyanion intermediate and its flanking transition states.
A few other invariant or highly conserved residues are found in the active site of LpxC. Residues T191 and F192 in the invariant R190-F192 segment form a small hydrophobic niche that may accommodate the methyl group of the substrate acetyl group, as indicated in Fig. 4. Invariant residue K239 is contained in the HKX(L,F)D zinc-binding motif discussed earlier and its side chain protrudes into the active site. Preliminary x-ray crystallographic studies of substrate analogue binding (unpublished results) and modeling of the enzyme–substrate complex (Fig. 2D) suggest that K239 interacts with the UDP-glucose portion of the substrate. Additionally, the segment G(V,I,T)G(I,L,V)HXG beginning with G15 is located at the mouth of the hydrophobic tunnel; invariant residue H19 may also interact with the substrate, because catalytic activity is compromised but not abolished by the H19A substitution (19).
The crystal structure of another metal-requiring deacetylase, a histone deacetylase homologue from A. aeolicus that deacetylates histones in vitro (35), reveals some interesting contrasts in comparison with LpxC. Histone deacetylase adopts a fold topologically distinct from that of LpxC (but identical to that of the binuclear manganese metalloenzyme arginase; ref. 36), its metal ion is liganded by one histidine and two aspartate residues, and an active-site histidine residue is proposed as a general base (35). Accordingly, there is no evidence of convergent evolution in the mechanisms of the metal-dependent deacetylation reactions catalyzed by histone deacetylase and LpxC.
Structure-Based Inhibitor Design and the Hydrophobic Tunnel. To be useful against the LpxC enzymes from a broad range of Gram-negative bacteria, an inhibitor should target interactions with conserved features and surfaces in the enzyme active site. The primary determinant of LpxC-inhibitor recognition is zinc coordination, and some of the most potent inhibitors known to date exploit a hydroxamate functionality for this task (20–25). Secondary determinants of recognition are conserved surfaces in the enzyme active site, perhaps the most important of which is the hydrophobic tunnel. Given that an inhibitor bearing the longer 3-O-myristic acid substituent is more potent (IC50 = 7.0 μM) than an inhibitor bearing the shorter 3-O-hexanoic acid substituent or no substituent at all (IC50 > 3,100 μM; ref. 24), van der Waals interactions between the long-chain aliphatic group of an inhibitor and the hydrophobic tunnel must contribute substantially to inhibitor binding. Can a potent inhibitor be designed to target exclusively the hydrophobic tunnel?
To address this question, we determined the affinities of saturated
C6–C12 aliphatic compounds capable of binding in
the hydrophobic tunnel and simultaneously coordinating to
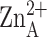 using isothermal
titration calorimetry (Fig. 5).
Of the compounds examined, those with chain lengths of C10 or
greater exhibited significant binding affinity for A. aeolicus LpxC.
The tightest binding compounds exhibited Kd values of 0.3
μM and contained C12 aliphatic chains with carboxylate or
phosphonate head groups. Strikingly, complex isoxazoline inhibitors bearing
carboxylate or phosphonate head groups, but lacking an aliphatic substituent
to interact with the hydrophobic tunnel, bind more weakly to LpxC from E.
coli with IC50 values of >450 μM and 4 μM,
respectively (25). Therefore,
targeting the hydrophobic tunnel exclusively yields inhibitor leads of simpler
design and higher affinity. Given the increasing occurrence of antibiotic
resistance in current day therapy, and given that LpxC is validated as a
target for the design of potent antibacterials
(20–25),
we advance that the next step in this program, the structure-based design of
broad spectrum LpxC inhibitors targeting the hydrophobic tunnel in the LpxC
active site, may represent a new and effective strategy for the successful
treatment of Gram-negative sepsis.
using isothermal
titration calorimetry (Fig. 5).
Of the compounds examined, those with chain lengths of C10 or
greater exhibited significant binding affinity for A. aeolicus LpxC.
The tightest binding compounds exhibited Kd values of 0.3
μM and contained C12 aliphatic chains with carboxylate or
phosphonate head groups. Strikingly, complex isoxazoline inhibitors bearing
carboxylate or phosphonate head groups, but lacking an aliphatic substituent
to interact with the hydrophobic tunnel, bind more weakly to LpxC from E.
coli with IC50 values of >450 μM and 4 μM,
respectively (25). Therefore,
targeting the hydrophobic tunnel exclusively yields inhibitor leads of simpler
design and higher affinity. Given the increasing occurrence of antibiotic
resistance in current day therapy, and given that LpxC is validated as a
target for the design of potent antibacterials
(20–25),
we advance that the next step in this program, the structure-based design of
broad spectrum LpxC inhibitors targeting the hydrophobic tunnel in the LpxC
active site, may represent a new and effective strategy for the successful
treatment of Gram-negative sepsis.
Fig. 5.

Structure-affinity relationships for aliphatic LpxC inhibitors that target zinc coordination and binding in the hydrophobic tunnel. For chain lengths with n ≤ 6, no binding was observed in isothermal titration calorimetry experiments; therefore, the Kd values indicated represent the minimum detection threshold.
Supplementary Material
Acknowledgments
We thank the Cornell High Energy Synchrotron Source for beamline access. This work was supported by the National Institutes of Health.
Abbreviations: LPS, lipopolysaccharide; LpxC, UDP-(3-O-(R-3-hydroxymyristoyl))-N-acetylglucosamine deacetylase; MAD, multiwavelength anomalous dispersion.
Data deposition: The atomic coordinates for Aquifex aeolicus LpxC have been deposited in the Protein Data Bank, www.rcsb.org (PDB ID code 1P42).
References
- 1.Parillo, J. E. (1990) Ann. Intern. Med. 113 227-242. [DOI] [PubMed] [Google Scholar]
- 2.Parillo, J. E. (1993) N. Engl. J. Med. 328 1471-1477. [DOI] [PubMed] [Google Scholar]
- 3.Raetz, C. R. H. (1986) Annu. Rev. Genet. 20 253-295. [DOI] [PubMed] [Google Scholar]
- 4.Raetz, C. R. H. (1990) Annu. Rev. Biochem. 59 129-170. [DOI] [PubMed] [Google Scholar]
- 5.Ulevitch, R. J. & Tobias, P. S. (1995) Annu. Rev. Immunol. 13 437-457. [DOI] [PubMed] [Google Scholar]
- 6.Raetz, C. R. H. (1993) J. Bacteriol. 175 5745-5753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rick, P. D. & Raetz, C. R. H. (1999) in Endotoxin in Health and Disease, eds. Brade, H., Opal, S. M., Vogel, S. N. & Morrison, D. C. (Dekker, New York), pp. 283-304.
- 8.Wyckoff, T. J. O., Raetz, C. R. H. & Jackman, J. E. (1998) Trends Microbiol. 6 154-159. [DOI] [PubMed] [Google Scholar]
- 9.Nikaido, H. & Vaara, M. (1985) Microbiol. Rev. 49 1-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vuorio, R. & Vaara, M. (1992) Antimicrob. Agents Chemother. 36 826-829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vaara, M. (1993) Antimicrob. Agents Chemother. 37 354-356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Anderson, M. S., Bulawa, C. E. & Raetz, C. R. H. (1985) J. Biol. Chem. 260 15536-15541. [PubMed] [Google Scholar]
- 13.Anderson, M. S., Robertson, A. D., Macher, I. & Raetz, C. R. H. (1988) Biochemistry 27 1908-1917. [DOI] [PubMed] [Google Scholar]
- 14.Anderson, M. S., Bull, H. G., Galloway, S. M., Kelly, T. M., Mohan, S., Radika, K. & Raetz, C. R. H. (1993) J. Biol. Chem. 268 19858-19865. [PubMed] [Google Scholar]
- 15.Young, K., Silver, L. L., Bramhill, D., Cameron, P., Eveland, S. S., Raetz, C. R. H., Hyland, S. A. & Anderson, M. S. (1995) J. Biol. Chem. 270 30384-30391. [DOI] [PubMed] [Google Scholar]
- 16.Beall, B. & Lutkenhaus, J. (1987) J. Bacteriol. 169 5408-5415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jackman, J. E., Raetz, C. R. H. & Fierke, C. A. (1999) Biochemistry 38 1902-1911. [DOI] [PubMed] [Google Scholar]
- 18.Vallee, B. L. & Auld, D. S. (1993) Acc. Chem. Res. 26 543-551. [Google Scholar]
- 19.Jackman, J. E., Raetz, C. R. H. & Fierke, C. A. (2001) Biochemistry 40 514-523. [DOI] [PubMed] [Google Scholar]
- 20.Onishi, H. R., Pelak, B. A., Gerckens, L. S., Silver, L. L., Kahan, F. M., Chen, M.-H., Patchett, A. A., Galloway, S. M., Hyland, S. A., Anderson, M. S. & Raetz, C. R. H. (1996) Science 274 980-982. [DOI] [PubMed] [Google Scholar]
- 21.Chen, M.-H., Steiner, M. G., de Laszlo, S. E., Patchett, A. A., Anderson, M. S., Hyland, S. A., Onishi, H. R., Silver, L. L. & Raetz, C. R. H. (1999) Bioorg. Med. Chem. Lett. 9 313-318. [DOI] [PubMed] [Google Scholar]
- 22.Kline, T., Andersen, N. H., Harwood, E. A., Bowman, J., Malanda, A., Endsley, S., Erwin, A. L., Doyle, M., Fong, S., Harris, A. L., et al. (2002) J. Med. Chem. 45 3112-3129. [DOI] [PubMed] [Google Scholar]
- 23.Clements, J. M., Coignard, F., Johnson, I., Chandler, S., Palan, S., Waller, A., Wijkmans, J. & Hunter, M. G. (2002) Antimicrob. Agents Chemother. 46 1793-1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jackman, J. E., Fierke, C. A., Tumey, L. N., Pirrung, M., Uchiyama, T., Tahir, S. H., Hindsgaul, O. & Raetz, C. R. H. (2000) J. Biol. Chem. 275 11002-11009. [DOI] [PubMed] [Google Scholar]
- 25.Pirrung, M. C., Tumey, L. N., Raetz, C. R. H., Jackman, J. E., Snehalatha, K., McClerren, A. L., Fierke, C. A., Gantt, S. L. & Rusche, K. M. (2002) J. Med. Chem. 45 4359-4370. [DOI] [PubMed] [Google Scholar]
- 26.Hendrickson, W. A. (1991) Science 254 51-58. [DOI] [PubMed] [Google Scholar]
- 27.Brünger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J.-S., Kuszewski, J., Nilges, M., Pannu, N. S., et al. (1998) Acta Crystallogr. D 54 905-921. [DOI] [PubMed] [Google Scholar]
- 28.Jones, T. A., Zou, J.-Y., Cowan, S. W. & Kjeldgaard, M. (1991) Acta Crystallogr. A 47 110-119. [DOI] [PubMed] [Google Scholar]
- 29.Fisher, H. F. & Singh, N. (1995) Methods Enzymol. 259 194-221. [DOI] [PubMed] [Google Scholar]
- 30.Holm, L. & Sander, C. (1993) J. Mol. Biol. 233 123-138. [DOI] [PubMed] [Google Scholar]
- 31.Holland, D. R., Hausrath, A. C., Juers, D. & Matthews, B. W. (1995) Protein Sci. 4 1955-1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gomez-Ortiz, M., Gomis-Rüth, F. X., Huber, R. & Avilés, F. X. (1997) FEBS Lett. 400 336-340. [DOI] [PubMed] [Google Scholar]
- 33.Matthews, B. W. (1988) Acc. Chem. Res. 21 333-340. [Google Scholar]
- 34.Christianson, D. W. & Lipscomb, W. N. (1989) Acc. Chem. Res. 22 62-69. [Google Scholar]
- 35.Finnin, M. S., Donigian, J. R., Cohen, A., Richon, V. M., Rifkind, R. A., Marks, P. A., Breslow, R. & Pavletich, N. P. (1999) Nature 401 188-193. [DOI] [PubMed] [Google Scholar]
- 36.Kanyo, Z. F., Scolnick, L. R., Ash, D. E. & Christianson, D. W. (1996) Nature 383 554-557. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.